

Abstract
Recent reports suggest that botulinum neurotoxin (BoNT) A, which is widely used clinically to inhibit neurotransmission, can spread within networks of neurons to have distal effects, but this remains controversial. Moreover, it is not known whether other members of this toxin family are transferred between neurons. Here, we investigate the potential distal effects of BoNT/A, D, and tetanus toxin, using central neurons grown in microfluidic devices. Toxins acted upon the neurons that mediated initial entry, but all three toxins were also taken-up via an alternative pathway, into non-acidified organelles that mediated retrograde transport to the somato-dendritic compartment. Toxins were then released into the media where they entered, and exerted their effects upon, upstream neurons. These findings directly demonstrate that these agents undergo transcytosis and interneuronal transfer in an active form, resulting in long distance effects.
Graphical abstract

Introduction
The clostridial neurotoxins (CNTs), comprising tetanus (TeNT) and seven serologically distinct botulinum neurotoxins (BoNT), A-G, are among the deadliest agents known, with BoNT/A having an estimated LD50 of 1 ng/kg body weight (Gill, 1982). Due to their potential use as biological weapons, the CDC designated the BoNTs as tier 1 select agents. Paradoxically, BoNT/A (onabotulinumtoxinA, abobotulinumtoxinA, incobotulinumtoxinA) and BoNT/B (rimabotulinumtoxinB), are also used clinically. In addition to the well-known cosmetic uses of BOTOX®, both BoNT/A and BoNT/B are also used to treat numerous medical conditions, including cervical dystonia, strabismus, migraine headaches, overactive bladder (neurogenic and idiopathic), hyperhidrosis, upper limb spasticity, and blepharospasm (de Maio, 2008). They are also used ‘off label’ to treat a variety of additional conditions that include chronic lower back pain, traumatic brain injury, cerebral palsy, achalasia, voice abnormalities, and various additional dystonias (Scott, 1980, Schantz and Johnson, 1992, Silberstein et al., 2000, Foster et al., 2001, Jankovic, 1994). According to Allergan’s 2014 Annual Report, more than half of all patients who receive toxin injections do so for medical, rather than aesthetic, reasons. Given their extreme potency, wide-spread medical use, and potential use as bioterrorism agents, the CNTs are the subject of intensive investigation.
The CNTs are produced by anaerobic, spore forming bacteria of the genus Clostridium (Popoff and Bouvet, 2013). Each CNT is composed of a heavy (HC) and light (LC) chain linked via a disulfide bond. First step in the action of these agents involves high affinity interactions with neurons, mediated by their HCs. Binding occurs via a dual receptor mechanism, where the receptors are composed of polysialic gangliosides in conjunction with proteins. For most of these toxins, protein receptors are presented by recycling synaptic vesicles (SV). Upon exocytosis, the luminal domains of SV proteins are exposed to the extracellular milieu; BoNT/B, G, and a naturally occurring D-C chimera, bind to the intraluminal tail of the SV proteins synaptotagmin 1 and 2 (Nishiki et al., 1994, Dong et al., 2003, Rummel et al., 2007, Peng et al., 2012), while BoNT/A, D, E, and TeNT bind to synaptic vesicle protein 2 (SV2) (Dong et al., 2006, Dong et al., 2008, Yeh et al., 2010, Mahrhold et al., 2006, Mahrhold et al., 2013, Peng et al., 2011, Fu et al., 2009, Rummel et al., 2009b, Benoit et al., 2014, Yao et al., 2016). BoNT/F was also found to bind to SV2, however it is not clear whether SV2 serves as a functional protein receptor for this toxin as primary hippocampal neurons lacking SV2 show no changes in sensitivity to BoNT/F (Fu et al., 2009, Rummel et al., 2009a, Peng et al., 2012, Yeh et al., 2010). The identity of the protein receptor for BoNT/C remains to be established. Upon SV endocytosis, the drop in luminal pH triggers the transformation of the HC into a translocation machine (Fischer, 2013, Montal, 2010, Williamson and Neale, 1994, Fu et al., 2002, Puhar et al., 2004, Galloux et al., 2008, Pirazzini et al., 2013); interestingly, the ability to sense low pH requires the interaction of the HC with the ganglioside co-receptor (Sun et al., 2011). The HC then translocates the LC into the cytosol, where it cleaves neuronal soluble N-ethylmaleimide attachment receptors (SNAREs), which form the core of a conserved membrane fusion complex (Rothman, 1994). Cleavage thereby inhibits neurotransmission. BoNT/C cleaves the target membrane SNAREs syntaxin and SNAP-25, BoNT/A and E cleave SNAP-25, and BoNT/D, B, F, G, and TeNT cleave the vesicular SNARE, synaptobrevin/VAMP (Rossetto et al., 2014, Schiavo et al., 2000, Jahn and Niemann, 1994, Montecucco and Schiavo, 1995). Recently, two additional putative toxin receptors have been identified: fibroblast growth factor receptor 3 (FGFR3) for BoNT/A, and nidogens 1 and 2 for TeNT (Bercsenyi et al., 2014, Jacky et al., 2013). How these proteins act in conjunction with the primary protein receptor, SV2, remains unknown.
It is widely believed that BoNT/A confers its medicinal effects by inhibiting synaptic transmission near the site of injection; i.e. that this toxin has only local effects at the neuromuscular junction (NMJ), resulting in flaccid paralysis. However, this idea has been called into question by physicians utilizing this agent in human patients. For example, after peripheral injection of BoNT/A, reciprocal inhibition between agonist and antagonist muscles has been reported, raising the possibility that BoNT/A moves within networks of neurons to affect circuit function (Priori et al., 1995, Aymard et al., 2013, Marchand-Pauvert et al., 2013, CeballosBaumann et al., 1997, Giladi, 1997). Alternatively, the observed effects might be due to the compensatory reorganization and remodeling of neuronal networks upstream of the injection site, as a result of purely local effects on the initial uptake neurons (Berardelli and Curra, 2002, Curra et al., 2004, Gilio et al., 2000, Boroojerdi et al., 2003, Abbruzzese and Berardelli, 2006). A major goal in the field is to determine which of these models is correct.
What is known concerning the movement of the BoNTs? BoNT/A and E were recently shown to undergo retrograde axonal transport in cultured motor neurons, but putative transfer and action on upstream neurons was not addressed in these in vitro experiments (Restani et al., 2012a). For clarity, we note that these authors used the term ‘distal’ or ‘central’ effects, but this refers to the action of the toxins in the somatodendritic compartment (which can lie in the CNS in vivo) versus their action within pre-synaptic boutons, where they were initially taken-up. However, it is well established that the toxin light chain can diffuse and cleave SNAREs throughout neurons, so we use the term ‘distal effects’ to indicate the action of the toxins on neurons that are upstream from the neurons that mediate the initial uptake step. Interestingly, in whole animal experiments, BoNT/A was reported to have bona fide distal effects (i.e. effects on upstream neurons; (Antonucci et al., 2008, Restani et al., 2011)), but this could not be reproduced in an in vitro system based on cultured neurons (Lawrence et al., 2012). Furthermore, interpretation of data obtained from in vivo approaches can be confounded by myriad variables, including long axon collaterals that can make it appear as if distal effects are occurring. Collectively, the question of whether BoNT/A confers its medicinal effects indirectly (network remodeling) or directly (by interneuronal transfer and action of holotoxin) remains to be definitively addressed. The in vitro system described in the current study circumvents the caveats mentioned above and allowed us to resolve the controversy surrounding distal effects of CNTs.
The goal of the current study was to directly ascertain whether CNTs move from neuron to neuron, in an active form, resulting in cleavage of SNAREs in cells that are upstream from the initial uptake neurons. As earlier work focused on in vivo experiments (Antonucci et al., 2008, Restani et al., 2011, Restani et al., 2012a, Restani et al., 2012b), here we sought an in vitro experimental approach that can be used to directly visualize toxin action within networks of neurons, and which provides an experimentally amenable system to elucidate the mechanisms that mediate local versus distal effects. In our study we utilized cultured hippocampal neurons, grown in microfluidic devices, to directly compare our results with those of Antonnuci et al. (2008), who injected BoNT/A directly into the hippocampus of intact mice. The experiments reported here clearly establish that BoNT/A, BoNT/D, and TeNT undergo retrograde transport along axons, followed by cell-to-cell transfer of the intact holotoxins into upstream neurons where they cleave SNAREs. Additionally, we characterize the receptors and entry pathways that mediate interneuronal transfer of these agents. Together, these experiments demonstrate that CNTs interact with host cells in a more complex manner than was originally envisioned, prompting further re-evaluation of the clinical uses of BoNT/A.
Results
Visualization of interneuronal transfer and action of the CNTs
In order to address the question of whether the CNTs move within networks of neurons, we utilized compartmentalized microfluidic devices. Three toxin serotypes were examined: BoNT/A, which cleaves SNAP-25A and B (hereafter SNAP-25), and BoNT/D and TeNT, which cleave a number of synaptobrevin (syb) isoforms, with syb2 being the major isoform that mediates rapid exocytosis at many synapses (Montecucco and Schiavo, 1995, Turton et al., 2002, Taylor et al., 2005). The experimental layout is shown in Figure 1A. Rat hippocampal neurons are seeded in one macrochannel (i.e. the soma chamber). By 14 days in vitro, axons project through the microchannels to the opposing macrochamber (i.e. the axon chamber). Dendrites are unable to traverse the microchannel (Figure 1B); hence, only axons are present in the axon chamber (Taylor et al., 2005). The axon chamber was then loaded with a dye, Calcein-AM-green (Figure 1A), to label all of the axons and their corresponding cell bodies and processes residing within the soma chamber. Counterstaining of the soma side with Calcein-AM-red (Figure 1A), which labeled both projecting (to the axonal side) and non-projecting cells, revealed that only 24 ± 1.5 % of cells seeded in the soma side extend their axons to the axon chamber (green cells), and that over a half of these ‘projecting neurons’ (52 ± 2.9 %) reside in the proximal region of the chamber, close to the mouth of the microchannels. With rare exceptions (3 ± 0.6 %), neurons distal from the microgroove barrier do not extend axons to the axon chamber, creating a hierarchy of neuronal connectivity. Representative images of distal and proximal regions in the soma side, along with a proximal region in the axon side, are shown in Figure 1A (right panels). Microfluidic isolation is achieved and maintained by keeping the media volume in the soma chamber higher (300 μl) as compared to the axon chamber (200 μl). This volume difference prevents diffusion of molecules from the axon side to the soma side, as reported previously (Taylor et al., 2005, Wang et al., 2015, David et al., 2012); fluidic isolation is validated using fluorescent markers in Figure S1. Moreover, we routinely include fluorescent dyes in the microfluidics to ensure fluidic isolation is maintained and leak does not occur; when leak occurs these samples are excluded from further analysis (Figure S1; <5% of the devices fail to seal).
Figure 1. CNTs cleave substrates beyond the levels predicted by connectivity of neurons in compartmentalized microfluidic devices.

(A) Dissociated hippocampal neurons were seeded into the soma chamber (top, red) of a microfluidic device. The number of cells that extend axons through the microchannels to the axon chamber (projecting cells) was determined by differential Calcein-AM-red and -green staining, added to the soma (top) and axon (bottom) chambers, respectively. Projecting cells are green/yellow, non-projecting cells are red. The number of projecting neurons drops sharply from 52 ± 2.9% of cells residing proximal to the microchannels in the soma side, to 31 ± 3.5 % in the mid-proximal, 14 ± 2.5 % in mid-distal, and to just 3 ± 0.6 % in the most distal region. Representative image of Calcein-stained neurons; white boxes denote regions that are magnified in the right panel. (B) Dendrites fail to reach the axon side due to the length of the microgroove barrier, creating an isolated axonal compartment, as illustrated by staining with MAP2, to mark dendrites (red/orange) and tau1, to mark axons (blue). (C) BoNT/A was added to the soma (10 nM) or axon (30 nM) side of the microfluidic device. After 48 hours, cells in the soma chamber were assayed for substrate cleavage via immunoblot analysis. The horizontal dotted line represents the total fraction of cells projecting to the axon side (24% ± 1.5%). Cleavage of substrate was abolished by axotomy. Similarly, TeNT (D), or BoNT/D (E) were added to either the soma or axon side of microfluidic devices, and cleavage of syb2 was assayed by immunoblot analysis. All plotted values are averages ± SEM; experiments were carried out with 3-5 independent rat litters. Scale bars: 100 μm. See also Figures S1,2.
When the soma chamber was incubated with BoNT/A, D or TeNT, nearly complete cleavage of SNAP-25 or syb2, respectively, was observed at 48 hours (Figure 1C-E). Cleavage of SNAP-25 by BoNT/A was monitored by measuring the ratio of full-length to cleaved protein (c-SNAP-25); actin served as a loading control. Cleavage of syb2 by TeNT and BoNT/D was monitored by measuring the loss of substrate signal, and these values were normalized to the actin signals in each sample. Interestingly, when toxins were added to the axon side, the degree of cleavage observed in the soma side far exceeded the levels predicted by the fraction of neurons that project axons from the soma to the axon side (24 ± 1.5 %, as indicated by the dashed line in the bar graphs in Figure 1C-E). Namely, 48 hours after addition to the axon side, BoNT/A cleaved 49.6% of SNAP-25, while TeNT and BoNT/D cleaved 63.7% and 83.8% of syb2, respectively, on the soma side. Significant substrate protection was observed following axotomy in the axon chamber, demonstrating that axonal transport was required for efficient substrate cleavage in the soma chamber; these observations further rule out diffusion of toxin from one side of the device to the other. While we routinely used 10-30 nM toxin in the axon side, distal effects were also observed using lower, clinically relevant concentrations (pM) of BoNT/A and TeNT, though this required an extended incubation period (Figure S2); hence the remainder of the experiments were carried out using higher toxin concentration so that we could assay for toxin effects on short time scales (24-48 hours).
Next, we conducted experiments to more directly determine whether the observed substrate cleavage within soma chamber occurs in second-order neurons that do not project axons to the axon side. Due to the dramatic difference in connectivity of neurons between proximal and distal regions of the soma side we monitored substrate cleavage in those two regions via immunocytochemistry. For all of these experiments, Calcein-AM-green was added to the axon side to mark projecting neurons; representative images of this staining along with the corresponding immunocytochemistry are shown (Figure 2A). Each microfluidic was stained for MAP2 to mark all neurons and vGlut1 to mark synapses (similar results were obtained using vGAT, to mark inhibitory synapses; data not shown). Cleavage of SNAP-25 by BoNT/A was measured using an antibody that is specific for the cleaved form of this protein (c-SNAP-25). Only background signals for c-SNAP-25 were observed, proximally and distally, in control microfluidics, and the greatest degree of cleavage was observed in both regions when the soma chamber was directly treated with BoNT/A; these data are quantified in Figure 2B. We then treated the axon chamber with BoNT/A, and observed significant cleavage in the proximal region of the soma side, as expected, since over 50% of cells in this region project to the axon side. Importantly, we also observed efficient cleavage distally, in neurons that did not project axons to the axon side, as evidenced by the absence of Calcein-AM staining. As a control, we again performed axotomy, which diminished the cleavage of SNAP-25.
Figure 2. Cleavage of substrate by BoNT/A in upstream neurons.

(A) Representative Calcein-AM-green staining, after addition of dye to the axon side only, marking projecting neurons; proximal and distal regions of the soma side of the microfluidics are shown (regions of interest indicated by red boxes). Neurons growing in both proximal and distal regions stain positive for the dendritic marker, MAP2, and the synaptic vesicle marker, vGlut1; Calcein and MAP2 images were inverted and switched to grayscale for clarity. Neurons were treated with BoNT/A on the soma side (10 nM), or axon side (30 nM) with and without axotomy for 24 hours. Cleavage of SNAP-25 was assayed using an antibody that recognizes the BoNT/A cleaved form of this protein (c-SNAP-25). Only background signals for c-SNAP-25 were observed, both proximally and distally, in control microfluidics; robust cleavage was observed following soma treatment with BoNT/A, in both regions. After addition of BoNT/A to the axon side, c-SNAP-25 was observed in proximal as well as distal regions of the soma side, and cleavage was significantly reduced by axotomy. (B) The intensity of c-SNAP-25, normalized to vGlut1 was quantified. The statistical analysis was performed comparing proximal to proximal, or distal to distal, regions. All plotted values are averages ± SEM; minimum of 3-4 images from each region (proximal/distal), from 4-5 separate experiments. Scale bars: 100 μm.
To determine the generality of the findings obtained using BoNT/A, we tested both TeNT and BoNT/D using a similar experimental design. Representative images of Calcein-AM-green, along with vGlut and syb2 staining for each experiment, are shown (Figure 3A and C). In control microfluidics, syb2 and vGlut1 are co-localized, as expected. Soma treatment with TeNT and BoNT/D resulted in near complete cleavage of syb2 in both proximal and distal regions, as illustrated by the lack of staining following the application of either toxin, which was quantified in Figure 3B and D. Axon-side treatments with either toxin led to efficient cleavage in the soma side, both proximal to, and distal from, the microchannels; cleavage was inhibited by axotomy. Notably, BoNT/D had particularly strong distal effects; we observed almost complete cleavage of its substrate throughout the microfluidic device, including the reservoirs, which are millimeters away (data not shown).
Figure 3. Cleavage of substrate by BoNT/D and TeNT in upstream neurons.

(A) As in Figure 2, images of Calcein-AM-green stained neurons were inverted and switched to grayscale. Neurons in both proximal and distal regions of the soma side stain positive for the synaptic vesicle marker, vGlut1 (green), and syb2 (red); MAP2 staining is not shown. Microfluidics were treated with either (A) TeNT or (B) BoNT/D for 24 hours (10 nM toxin for soma side treatments, 30 nM for axon side treatments), fixed and assayed for syb2 cleavage via immunocytochemistry. In both cases cleavage of substrate was observed in proximal as well as distal, non-projecting neurons, and syb2 was protected from cleavage by axotomy. (C,D) Syb2 signals normalized to vGlut1. All plotted values are averages ± SEM; minimum of 3-4 images from each region (proximal/distal), from 4-5 separate experiments.
Holotoxins, released into the media from the initial uptake neuron, enter upstream neurons
The experiments above provide strong support for the idea that a fraction of BoNT/A, D and TeNT can in fact undergo retrograde transport and inter-neuronal transfer. In the next series of experiments we further explored this idea by determining whether substrate cleavage in the soma chamber could be blocked by adding excess toxin receptor binding domain (HC), to interfere with toxin-receptor recognition, or by adding anti-toxin antibodies that, in principle, should intercept toxin molecules as they leave the initial uptake neuron, thus preventing their action upon other neurons. Neutralizing anti-BoNT/A and anti-BoNT/D antibodies were tested previously (Bjornstad et al., 2014, Kalb et al., 2009); anti-TeNT antibody was validated by neutralization of TeNT in primary hippocampal neurons (data not shown). The scheme for these experiments is shown in Figure 4A.
Figure 4. BoNT/A, D and TeNT are released by the primary uptake neurons to enter upstream.
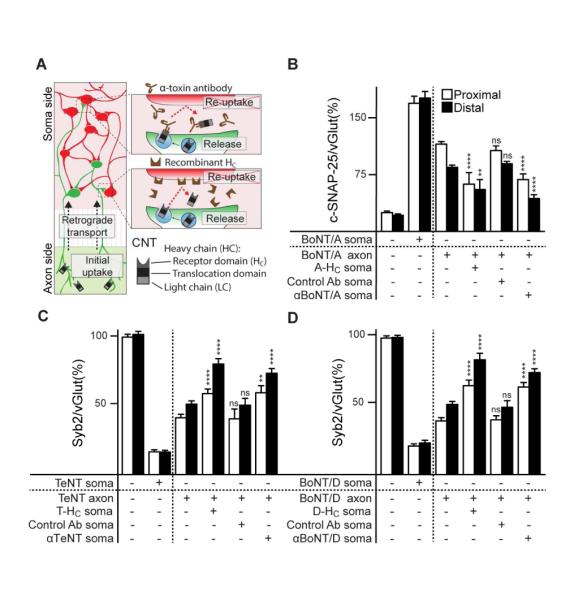
neurons. (A) Schematic representation of the experimental design. Soma chambers were pre-treated with either BoNT/A-, BoNT/D-, or TeNT-HC domains (300 nM for BoNT/A and TeNT, 100 nM for BoNT/D treatments) or anti-CNT antibodies specific for each toxin, followed by addition of holotoxin to the axon side of the microfluidic (30 nM of BoNT/A and TeNT, 10 nM of BoNT/D). After 24 hours, neurons were fixed and stained for vGlut1, MAP2, and c-SNAP-25 for BoNT/A (B), or syb2 for TeNT (C) and BoNT/D (D). Images were obtained from proximal and distal regions of soma side and analyzed as in Figures 2 and 3. In each case, inclusion of HC fragments, or antibodies, in the soma chamber, protected neurons from substrate cleavage, both proximally and distally for BoNT/A (B), TeNT (C), and BoNT/D (D), demonstrating that toxins physically leave uptake neurons and enter upstream neurons where they cleave SNAREs. All plotted values are averages ± SEM; minimum of 3-4 images from each region (proximal/distal), from 4-5 separate experiments.
Holotoxins were added to the axon side, while the soma side was pre-incubated with corresponding HC fragments, or anti-toxin antibodies. Substrate cleavage in both proximal and distal regions in the soma macrochannel was monitored via immunocytochemistry as described in Figure 2. For all three toxins, HC fragments and anti-toxin antibodies provided significant protection from cleavage within the soma side (Figure 4B-D). In each case there was a trend toward greater protection in the distal regions, as compared to the proximal regions, but since 48% of neurons in the proximal region do not project axons to the axon side of the device (see Figure 1A), some degree of protection, even in the proximal region, was expected if the toxins undergo interneuronal transfer. Together, these results indicate that after uptake on the axon side, the toxins are retrogradely transported to the soma side where they are released into the media; the toxins then enter upstream neurons.
An alternative, SV-independent, pathway underlies the distal effects of the CNTs
Following entry into neurons via recycling SVs, acidification of the vesicle lumen triggers the translocation of the LC into the cytosol. In order for the toxin to traffic to the soma side, and to be released in an active form to enter and affect upstream neurons, the LC must remain linked to the HC. This can be achieved if the toxin is located in a non-SV compartment that does not undergo significant luminal acidification (or acidifies slowly enough to first allow transport without translocation). Previous work reported that TeNT, as well as BoNT/A and E, indeed share a common retrograde transport organelle that does not acidify (Restani et al., 2012a). However, in contrast to BoNT/A, BoNT/E was shown not to exert distal effects (Restani et al., 2012b, Antonucci et al., 2008), and trafficking of BoNT/D has not been studied.
To directly monitor toxin co-trafficking, HC fragments derived from each of the three serotypes studied here were labeled with either Alexa488 or 568, and added to the axon chamber for 4-24 hours prior to imaging. Entry and trafficking of the labeled HC fragments was monitored, within the microchannels, ~350-400 μm away from the axon side (Figure 5A, red box). As before, microfluidic isolation was maintained with higher volume of media on the soma side throughout the entire length of the experiment to prevent diffusion of the HC domains to the soma side (see Figure S1). We observed that labeled HC domains derived from both BoNT/D and BoNT/A co-localized to the same transport organelles (77%, Figure 5E), which were observed moving retrogradely (Figure 5B). HC fragments from BoNT/D and TeNT were also co-localized in retrograde transport organelles (80%; Figure 5C and E), which do not acidify as evidenced by a lack co-localization of BoNT/D-HC with lysotracker (16%; Figure 5D and E; Movie S1). These observations fit the idea that a common carrier mediates the retrograde trafficking of these agents. The lack of acidification allows the holotoxins to remain intact so that when they are released they are able to bind, enter, and affect upstream neurons.
Figure 5. BoNT/A, TeNT and BoNT/D enter neurons via a SV2-independent pathway, and share a common transport organelle.

(A) Overlaid images of Calcein-AM-green added to the axon side with Calcein-AM-red added to soma side of a microfluidic device. Calcein-AM-green labels all axons, dendrites and cell bodies of neurons with axons that reached all the way to the axon side of the device. The red box indicates the image acquisition area. (B-E) Retrograde transport of toxin HC fragments was visualized using multi-channel live cell imaging. Representative kymographs show mobile vesicles containing the following Alexa-labelled-HC fragments: (B) BoNT/D with BoNT/A and (C) BoNT/D with TeNT. (D) Representative kymograph of axons treated with labeled BoNT/D- HC and subsequently loaded with Lysotracker prior to imaging (see also Movie S1). (E) Bar graph depicting average percentage of BoNT/D-HC positive organelles undergoing retrograde transport which were also positive for BoNT/A-HC, TeNT-HC or Lysotracker. Three to five independent rat litters were used; n = 57-98 particles for BoNT/D-HC. Values are presented as average ± SEM co-labeled retrograde transport particles. (F,H,J) Representative kymographs showing (F) BoNT/A HC-Qdot, (H) TeNT-HC-Qdot, and (J) BoNT/D-HC-Qdot retrograde transport in SV2A(−/+)B(−/−) and SV2A(−/−)B(−/−) mouse hippocampal neurons. Sale bar is 10 μm, the total time was 1 min. (G-K) Histograms of instantaneous speed (IS) measured from mobile HC-Qdots. Data were binned at 0.2 micron/sec increments by fractional frequency. Black bars are from SV2A(−/+)B(−/−) mouse (control) neurons, and red bars are from SV2A(−/−)B(−/−) mouse (SV2DKO) neurons. Two independent mouse litters were used; n > 300 particles for each HC. See also Figure S3.
TeNT, BoNT/A, and BoNT/D utilize SV2 as a protein receptor to enter cells via the SV recycling pathway (Dong et al., 2006, Yeh et al., 2010, Mahrhold et al., 2006, Peng et al., 2011). In the absence of this receptor, cleavage of substrate by all three toxins is significantly reduced, and KO animals lacking SV2A/B are resistant to the toxins. Of the three isoforms of SV2, the majority of hippocampal neurons exclusively express SV2A and SV2B (Dong et al., 2006, Janz and Sudhof, 1999, Bajjalieh et al., 1994, Peng et al., 2011), and only a small population of hippocampal neurons express SV2C (Peng et al., 2011, Dong et al., 2008). Therefore, neurons obtained from SV2AB double knock-out (DKO) animals serve as a desirable model to investigate whether the secondary, ‘non-productive’ pathway (i.e. fails to trigger translocation) occurs via an SV2 independent mechanism. To address this, hippocampal neurons from E15.5 WT and SV2A/B DKO mice were seeded into microfluidic devices. Toxin HC fragments, conjugated to quantum (Q) dots (Figure S3), were added to the axon side of 13-14 DIV cultures. After 6 hours, entry and trafficking of HC-Qdots was monitored, as described for the organic dyes above. Qdot labeled HC domains derived from all three toxins entered WT axons and underwent retrograde transport toward the soma side (Figure 5F-K). The speed of toxin HC transport is in agreement with data from earlier studies of TeNT and BoNT/A (Restani et al., 2012a), and is consistent with the speed reported for fast axon retrograde transport of endosomal carriers (Deinhardt et al., 2006, Deinhardt et al., 2007, Salinas et al., 2009). Importantly, all three HC fragments efficiently entered SV2A/B DKO neurons, where they also underwent processive, retrograde transport with speeds that were comparable to those found using WT neurons (Figure 5F-K, right panel; Movie S2). The entry and overlap in speed distribution profiles of these toxin fragments indicate that: 1) all three toxins share a retrograde trafficking route, and 2) sorting of the toxin into this organelle is independent of interactions with SV2, clearly establishing the existence of second receptor and uptake pathway for each toxin.
Assessing candidate receptors for the second CNT uptake pathway
Together, the results thus far reveal two distinct uptake/trafficking pathways for BoNT/A, D and TeNT: the canonical SV pathway, which leads to acidification and local effects, and a second, SV receptor independent pathway that routes the toxins to a common non-acidified carrier that mediates delivery to distal sites. As detailed above, receptors for the SV pathway have been established for most of the CNTs: SV2 serves as the SV receptor for all three toxins examined here. However, it has also been reported that fibroblast growth factor receptor 3 (FGFR3), and nidogen 1 and 2, might serve as receptors for BoNT/A and TeNT, respectively (Bercsenyi et al., 2014, Jacky et al., 2013). Namely, the 2,3 loop of FGFR3b (FGFR3L2,3) was shown to interfere with toxin-host cell interactions (Jacky et al. 2013) and peptides derived from nidogen 1 and 2 (N1 and N2) were shown to protect mice from the effects of TeNT (Bercsenyi et al, 2014). We tested these reagents in microfluidic devices by preincubating them with holotoxins and then adding them to the axon side, followed by immunocytochemistry to determine whether they prevented toxin action in proximal and distal regions in the soma chamber. The scheme for these experiments is shown in Figure 6A.
Figure 6. Receptor interference experiments.
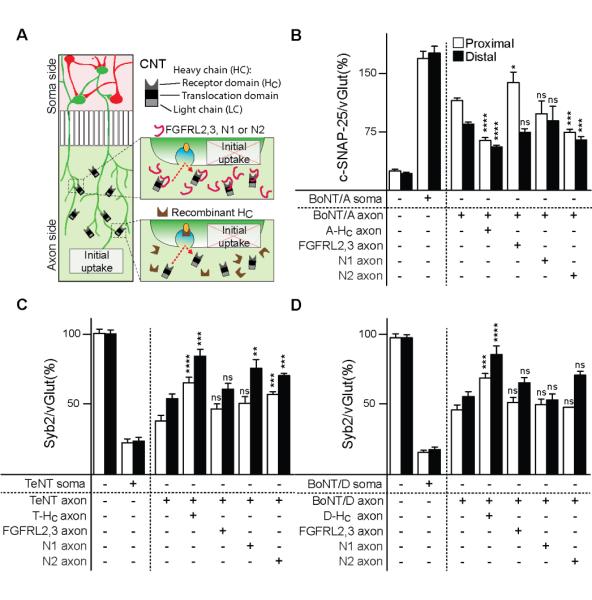
BoNT/A (30 nM), TeNT (30 nM), and BoNT/D (10 nM) holotoxins were pre-incubated with either their respective HC domains (300 nM for BoNT/A- HC and TeNT- HC, 100 nM for BoNT/D- HC), the FGFRL2,3 receptor fragment (300 nM for BoNT/A and TeNT, 100 nM for BoNT/D treatments), or N1/N2 peptide (20 μM), for 60 minutes prior to being added to the axon side of microfluidic devices. After 24 hours, SNARE cleavage was assayed by immunocytochemistry as described for Figures 2 and 3. (A) BoNT/A-mediated cleavage was inhibited by pre-incubation with excess A-HC. Pre-incubation with FGFRL2,3 had no effect on distal cleavage, but lead to increased proximal cleavage. The N2 peptide protected neurons both proximally and distally, while N1 was without an effect. (B) TeNT-mediated cleavage was inhibited by pre-incubation of holotoxin with T-HC and N2. Distal, but not proximal protection, was also observed with N1, and no effect was seen with the FGFRL2,3 fragment. (C) D-HC fragment protected syb2 from cleavage by BoNT/D both proximally and distally, however none of the remaining fragments tested had any effect on BoNT/D-mediated cleavage. All plotted values are averages ± SEM; minimum of 3-4 images from each region (proximal/distal), from 4-5 independent experiments. See also Figure S4.
As a control, and as expected, the A-HC fragment protected neurons in the soma macrochannel, when added together with BoNT/A on the axon side (Figure 6B). Surprisingly, inclusion of the FGFR3 loop 2,3, which was implicated in the action of BoNT/A (Jacky et al., 2013) but not any other serotype, resulted in enhanced cleavage of SNAP-25 in the proximal region of the soma macrochannel, and had no significant effect on cleavage in the distal region. Equally surprising was the finding that nidogen peptide N2 resulted in significant protection of distal neurons from the action of BoNT/A. This was unexpected as A-HC was found to bind nidogen-2 KO neurons just as efficiently as WT cells, and nidogens have been implicated only in the action of TeNT (Bercsenyi et al., 2014). The N1 peptide was without effect.
Similar experiments were carried out for TeNT. As expected, partial proximal and strong distal protection was observed on the soma side, when the axon side was co-treated with TeNT and T-HC (Figure 6C). FGFR3L2,3 was without effect, but both nidogen peptides, N1 and N2, reduced syb2 cleavage on the soma side in three out of four conditions; the only exception was N1, which failed to protect proximally. Together, these data suggest that nidogens might play a role in TeNT entry into, or trafficking within, the second uptake pathway.
Finally, we carried out the same experiments using BoNT/D, which has the most pronounced distal effects studied so far. While the D-HC fragment from this serotype resulted in protection from syb2 cleavage in both proximal and distal regions of the soma channel, none of the putative alternative receptor fragments had any demonstrable effect on the action of this toxin (Figure 6D). We note that none of the alternative receptor fragments affected the interaction of native SV2, from brain detergent extracts, with immobilized HC fragments derived from all three toxins, indicating that these agents do not interfere with the SV uptake pathway (Figure S4A-C). In addition, pre-incubation of toxins with alternative receptor fragments had no effect on toxin-mediated substrate cleavage when applied directly to neurons grown in mass culture (Figure S4D-F).
Discussion
Owing to the opposing clinical presentations of tetanus and botulism, which are associated with rigid and flaccid paralysis, respectively, it has been long assumed that TeNT and BoNTs, once they enter the nervous system at the neuromuscular junction (NMJ), have distinct itineraries. After TeNT is internalized, it is sorted into signaling endosomes that undergo retrograde transport into the spinal cord (Salinas et al., 2010). The holotoxin is then thought to undergo interneuronal transfer to upstream inhibitory neurons to cleave syb2 and block neurotransmission (Deinhardt et al., 2006, von Bartheld, 2004). This disinhibition results in the rigid paralysis characteristic of this agent (Montecucco and Schiavo, 1995, Turton et al., 2002). Since intoxication with BoNTs leads to flaccid paralysis, it was inferred that these toxins remain confined to the NMJ where they cleave their target SNAREs within presynaptic boutons of motor neurons. However, as discussed in the Introduction, recent studies raised the issue that BoNT/A might also undergo transcytosis and cell-to-cell transfer, but this remains controversial. Moreover, there is no evidence that any other BoNTs can move from neuron to neuron to exert distal effects, and numerous questions remain concerning the movement of TeNT from neuron to neuron.
In the current study, we used microfluidic devices to directly address the question of whether BoNT/A and TeNT, as well as the less studied serotype, BoNT/D can spread within networks of neurons to have distal effects. The microfluidic system used in these experiments provides a straightforward means to analyze toxin action within a hierarchy of neurons, which reflects the connectivity of neuronal networks that exist in vivo. We note that interpretation of in vivo experiments depends on absolute knowledge that there are no direct connections between the region injected with toxin and the distal region that is analyzed. This is circumvented in the microfluidics approach by the use of Calcein-AM to precisely map the connectivity of the entire network. Use of this dye made it possible to directly visualize distal effects of the CNTs, as substrate cleavage was observed in neurons that were not directly treated with toxins (Figures 2 and 3). The microfluidics also made it feasible to readily visualize toxin trafficking, and to carry out perturbation experiments that cannot be performed in whole animal studies. For example, it was possible to exploit SV2 KO neurons (as these mice are not viable as adults), to directly reveal the existence of an alternative, SV2-independent, uptake pathway of the three toxins examined here.
The retrograde trafficking of TeNT-containing signaling endosomes has been described in detail (Lalli and Schiavo, 2002, Caleo et al., 2009, Deinhardt et al., 2006), but the mechanisms that mediate the sorting and putative transcytosis from the initial uptake neuron to an upstream neuron remain somewhat obscure (Erdmann et al., 1981, Sverdlov and Alekseeva, 1966, Curtis and De Groat, 1968). One of the most convincing observations concerning distal effects of TeNT was the finding, more than three decades ago, that intraspinal injection of tetanus antitoxin, post peripheral injection of TeNT, prevents hind limb rigidity in rats (Erdmann et al., 1981). In the current study, we recapitulated these classical findings by showing that inclusion of an anti-TeNT antibody in the soma chamber of a microfluidic device provides protection from substrate cleavage in secondary neurons. We obtained similar results using the TeNT HC fragment. These results directly demonstrate that TeNT physically leaves the primary uptake neuron, is exposed to the extracellular milieu, and then binds and enters another neuron, where it cleaves its substrate. Hence, the reconstitution approach described here made it possible to directly visualize the distal action of TeNT. Parallel experiments using BoNT/A and BoNT/D revealed that both of these serotypes were also intercepted, during interneuronal transfer, by anti-toxin antibodies that prevent entry; protection from substrate cleavage was also observed using HC fragments. These findings demonstrate that these two serotypes also undergo cell-to-cell transfer to exert distal effects. Whether BoNT/B, C, G and F spread within networks of neurons are questions that have yet to be explored.
In the entry pathway that results in substrate cleavage, holotoxin is internalized by endocytosed SVs, and re-acidification of the vesicle lumen leads to the irreversible separation of the LC from the HC, as the LC is translocated into the cytosol. The LC is then trapped inside the cell, and cell-to-cell propagation of the holotoxin is no longer possible. Therefore, entry of the holotoxin into the retrograde pathway must either involve sorting away from recycling SVs before they acidify, or the holotoxins are internalized via a distinct pathway. In order to address this issue, we utilized neurons lacking the SV receptor, SV2. If SV2 serves as the only receptor for these toxins, entry would not be observed in the KO cells. However, Qdot labeled HC fragments from all three toxins were able to enter SV2 KO neurons, clearly establishing the existence of an alternative, SV2-independent, entry pathway. Moreover, after entry, each of the Qdot-HC conjugates underwent axonal retrograde trafficking with speeds consistent with the signaling organelle that Schiavo and co-workers have shown transports TeNT, BoNT/A, and BoNT/E (Restani et al., 2012a, Deinhardt et al., 2006). Additionally, this organelle does not acidify and hence allows for long distance transport of the holotoxin, in the absence of any translocation. We reiterate that BoNT/E undergoes retrograde traffic in the same compartment that transports TeNT, BoNT/A (Restani et al., 2012a), and BoNT/D (Figure 5). However, BoNT/E does not exert distal effects (Antonucci et al., 2008, Restani et al., 2012a). Thus, inclusion in this retrograde transport pathway does not always necessitate interneuronal transfer, a point we return to further below. We also note that previous work examined retrograde transport of BoNT/A and TeNT in primary motor neurons (Restani et al., 2012a) (trafficking of BoNT/D has not been studied before); the findings reported here extend these observations to central neurons.
The identification of an alternative uptake pathway, independent of recycling SVs, prompts the question of the identity of the receptors that mediate entry into this pathway. FGFR3, and nidogen 1 and 2, have been proposed to serve as alternative receptors for BoNT/A and TeNT, respectively (Jacky et al., 2013, Bercsenyi et al., 2014). We therefore made use of the toxin binding domains of all three of these putative secondary receptor molecules and determined whether these reagents inhibited distal effects in microfluidic devices. We found that the FGFR3 fragment failed to block distal effects of BoNT/A, indicating that interactions with this receptor are not required for retrograde trafficking or toxin transfer. Surprisingly, a greater degree of cleavage was observed in the proximal region of the soma channel in the presence of the receptor fragment. The reasons for this observation are unclear, but these results suggest that the FGFR3 fragment might enhance entry via recycling SVs, potentially by increasing the avidity of BoNT/A for the SV2-ganglioside co-receptor. The FGFR3 fragment had no discernable effect on the action of either TeNT or BoNT/D. We then turned our attention to the nidogen 1 and 2 peptides (Bercsenyi et al., 2014), which were shown to protect mice from the effects of TeNT. When added, in conjunction with TeNT, to the axon side, protection in the proximal and distal regions of the soma side was observed (the only case where this did not reach significance was for the N1 peptide in the proximal region of the soma channel). These findings are consistent with the idea that nidogens play a role in the distal effects of TeNT. Interestingly, the N2 peptide also protected SNAP-25 from cleavage by BoNT/A, proximally and distally, while the N1 peptide was without effect. Finally, neither nidogen peptide had any discernable effect on the action of BoNT/D. In conclusion, the CNTs studied here do not all enter the secondary SV2-independent uptake pathway via a single, common, alternative receptor.
While the experiments summarized above indicate convergent early sorting immediately following uptake via the secondary pathway, it should also be noted that after delivery to the somatodendritic compartment, the holotoxins are likely to undergo further sorting and routing to distinct destinations. For example, as mentioned above, BoNT/E is co-trafficked with TeNT and BoNT/A in the same organelle towards the soma (Restani et al., 2012a); however, BoNT/E does not exert distal effects (Antonucci et al., 2008), indicating that once it is delivered to the cell body it is sorted away from the interneuronal transfer pathway. Similarly, TeNT and cholera toxin have been reported to undergo co-transport back to the soma, where they are sorted apart from one-another to distinct destinations: cholera toxin is trafficked to the Golgi while TeNT is targeted to the plasma membrane for exocytosis (Schmieg et al., 2014). Putative sorting steps in the somato-dendritic compartment are appealing in the context of the CNTs due to the fact that TeNT causes rigid paralysis while BoNTs cause flaccid paralysis: these toxins might be routed to different release sites to affect distinct constellations of upstream neurons. However, it is also possible that BoNTs, akin to TeNT, can also cause disinhibition in the spinal cord; this might have gone undetected due to efficient inhibition of the NMJ by the same toxin. Electrophysiological recordings from motor neurons in animals challenged with BoNT/A will resolve this issue.
In summary, the experiments described here help to resolve the controversy regarding distal effects of CNTs by directly visualizing interneuronal transfer and distal action of tetanus toxin and botulinum toxins A and D in central neurons. An important next step in these studies will be to engineer the toxins such that they are able to enter only the local pathway, by mutating binding sites that mediate recognition of the alternative receptor. At present, these efforts can begin by focusing on interactions with nidogens for TeNT, and perhaps for BoNT/A, but this work will await the identification of the secondary receptor for BoNT/D. A key question will be whether such designer toxins are still efficacious in human patients. If so, off-target effects can be avoided; if not, then many of the clinical effects of these agents are in fact due to their distal effects, and this will require a re-evaluation of how these toxins are used in human patients.
Experimental Procedures
Ethics statement
All animal care and experiment protocols in this study were conducted under the guidelines set by the NIH Guide for the Care and Use of Laboratory Animals handbook. The protocols were reviewed and approved by the Animal Care and Use Committee (ACUC) at the University of Wisconsin - Madison (assurance number: A3368-01).
Cell culture
Rat or mouse hippocampal neurons were cultured as described previously (Yeh et al., 2010). Hippocampal neurons were cultured in standard neuron microfluidic devices (SND450, XONA microfluidic, Temecula, CA) mounted on glass coverslips as previously described (Taylor et al., 2005).
Treatments in microfluidic devices
For the experiments shown in Figures 1-3, toxins (30 nM) were applied to the axon side; axotomy was performed 2 hours before toxin treatment by rapid aspiration and reperfusion of the axon chamber. Calcein-AM, red or green, was added to either side of microfluidic devices at a final concentration of 1 μM; when treating the axon side, dye was incubated for 5 hours before imaging to allow the entire cell to become stained.
For the experiments shown in Figure 4, A-HC (300 nM), T-HC (300 nM), D-HC (100 nM), control Ab and anti-toxin antibodies were premixed as specified in the Figure Legend and added to the soma side prior to the axon treatment with respective toxins BoNT/A (30 nM), TeNT (30 nM), or BoNT/D (10 nM). For the experiments shown in Figure 6, toxins were premixed with either their respective HC fragments, or FGFR3bL2,3 (300 nM for BoNT/A and TeNT, 100 nM for BoNT/D), or nidogen N1 or N2 peptides (20 μM each), incubated for 60 min at 37°C , and then added to the axon side. After 24 hours, substrate cleavage was measured via immunocytochemistry.
For soma-side toxin treatments, neurons seeded on the soma side of a microfluidic device were incubated with 10 nM of either BoNT/A, D or TeNT, for 48 hours (for immunoblotting in Figure 1) or 24 hours (for immunocytochemistry in Figures 2, 3, 4 and 6).
For all treatments, preconditioned media was removed from the microfluidic chamber, mixed with an equal volume of fresh media, and used in the treatments to follow. All control microfluidics were sham treated with preconditioned media.
The fraction of neurons that project to the axon side (Figure 1 and S2) was calculated using data from 20 separate chambers obtained from 5 independent litters of rats.
Statistical methods
Statistical significance was by determined by performing the Student’s t-test for all immunocytochemistry data analysis, where: ns P > 0.05, * P≤0.05, ** P≤0.01, ***P ≤0.001, ****P≤0.0001. ANOVA with Dunnet’s post-hoc was used to analyze and compare immunoblot data, where p < 0.05.
Supplementary Material
Figure 7. Model depicting two distinct trafficking pathways for BoNT/A, D and TeNT.

Detailed model depicting the two distinct uptake pathways for BoNT/A, D and TeNT. Toxins can enter the SV2-dependent pathway, which leads to local effects and results in substrate cleavage throughout the cell (shown in green: pathway indicated by dashed lines, left side), or they can enter via an alternative receptor pathway (solid lines, right side), which leads to distal effects. Since the CNTs tested here do not all enter via the same secondary receptor, additional local sorting steps are required before the CNTs can converge into the common, non-acidified, retrogradely transported organelle [2]. The holotoxin is then delivered to the somato-dendritic compartment where it undergoes further sorting [3] followed by exocytosis [4]. Holotoxin is then released from the primary uptake neuron and is able to enter a secondary neuron (shown in red) via either pathway: the synaptic vesicle-dependent (by binding SV2), which leads to substrate cleavage, and the synaptic vesicle-independent pathway (by binding to alternative receptor(s)), which leads to further propagation of the holotoxin into upstream neurons.
Highlights.
BoNT/A, BoNT/D and TeNT enter neurons via two distinct pathways
Toxins undergo interneuronal transfer to affect networks of neurons
Microfluidic devices provide an amenable system to study toxin trafficking
In Brief.
Bomba-Warczak et al. demonstrate that BoNT/A, BoNT/D and TeNT enter neurons via two separate entry pathways, a canonical synaptic vesicle recycling pathway that leads to local effects, and a distinct secondary uptake pathway that directs these toxins to a common, non-acidified, retrograde carrier. This secondary pathway leads to distal effects in neurons upstream from the cells that mediate the initial uptake of these agents.
Acknowledgements
We thank M. Dong and all members of the Chapman lab and for discussions and critical reading of this study. Special thanks to C.S. Evans for HC labeling advice, and L.K. Roper for help with animal husbandry. This work was supported by a grant from the NIH (NINDS R21NS081549 to E.R.C.). E.A.J. acknowledges membership of and support (U54A157153) from the Region V "Great Lakes" Regional Center of Excellence in Biodefense and Emerging Infectious Diseases. E.R.C is an Investigator of the Howard Hughes Medical Institute. E.B.-W. was supported by Neuroscience Training Program grant (T32-GM007507). J. M. B. was an American Heart Association post-doctoral fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
E.B.-W., J.M.B., F.Y, J.D.V, and A.F.-B. and E.R.C. designed the experiments. E.B.-W. performed all dissections and plating, immunocytochemistry, Calcein-AM staining, fluorescent imaging of fixed cells, and analysis. E.B.-W., J.M.B., and J.D.V performed toxin treatments and maintained the SV2 KO mouse colony. J.D.V. and A.F.-B. performed Qdot experiments. J.D.V. conducted microfluidic isolation experiments, J.M.B. performed organic dye experiments, immunoblots of the soma chamber, and SV2 pull-down assays. J.M.B and J.D.V performed toxin long-term experiments. W.H.T. and E.A.J. provided BoNT/A and BoNT/D. E.R.C. and E.B.-W. wrote the manuscript, which was reviewed by all of the authors.
Literature Cited
- ABBRUZZESE G, BERARDELLI A. Neurophysiological effects of botulinum toxin type A. Neurotox Res. 2006;9:109–114. doi: 10.1007/BF03033927. [DOI] [PubMed] [Google Scholar]
- ANTONUCCI F, ROSSI C, GIANFRANCESCHI L, ROSSETTO O, CALEO M. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28:3689–96. doi: 10.1523/JNEUROSCI.0375-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AYMARD C, GIBOIN LS, LACKMY-VALLEE A, MARCHAND-PAUVERT V. Spinal plasticity in stroke patients after botulinum neurotoxin A injection in ankle plantar flexors. Physiol Rep. 2013;1:e00173. doi: 10.1002/phy2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAJJALIEH SM, FRANTZ GD, WEIMANN JM, MCCONNELL SK, SCHELLER RH. Differential Expression of Synaptic Vesicle Protein-2 (Sv2) Isoforms. Journal of Neuroscience. 1994;14:5223–5235. doi: 10.1523/JNEUROSCI.14-09-05223.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENOIT RM, FREY D, HILBERT M, KEVENAAR JT, WIESER MM, STIRNIMANN CU, MCMILLAN D, CESKA T, LEBON F, JAUSSI R, STEINMETZ MO, SCHERTLER GFX, HOOGENRAAD CC, CAPITANI G, KAMMERER RA. Structural basis for recognition of synaptic vesicle protein 2C by botulinum neurotoxin A. Nature. 2014;505:108. doi: 10.1038/nature12732. −+ [DOI] [PubMed] [Google Scholar]
- BERARDELLI A, CURRA A. Effects of botulinum toxin type A on central nervous system function. Naunyn-Schmiedebergs Archives of Pharmacology. 2002;365:R50–R50. [Google Scholar]
- BERCSENYI K, SCHMIEG N, BRYSON JB, WALLACE M, CACCIN P, GOLDING M, ZANOTTI G, GREENSMITH L, NISCHT R, SCHIAVO G. Tetanus toxin entry. Nidogens are therapeutic targets for the prevention of tetanus. Science. 2014;346:1118–23. doi: 10.1126/science.1258138. [DOI] [PubMed] [Google Scholar]
- BJORNSTAD K, TEVELL ABERG A, KALB SR, WANG D, BARR JR, BONDESSON U, HEDELAND M. Validation of the Endopep-MS method for qualitative detection of active botulinum neurotoxins in human and chicken serum. Anal Bioanal Chem. 2014;406:7149–61. doi: 10.1007/s00216-014-8170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOROOJERDI B, COHEN LG, HALLETT M. Effects of botulinum toxin on motor system excitability in patients with writer's cramp. Neurology. 2003;61:1546–1550. doi: 10.1212/01.wnl.0000095965.36574.0f. [DOI] [PubMed] [Google Scholar]
- CALEO M, ANTONUCCI F, RESTANI L, MAZZOCCHIO R. A reappraisal of the central effects of botulinum neurotoxin type A: by what mechanism? J Neurochem. 2009;109:15–24. doi: 10.1111/j.1471-4159.2009.05887.x. [DOI] [PubMed] [Google Scholar]
- CEBALLOSBAUMANN AO, SHEEAN G, PASSINGHAM RE, MARSDEN CD, BROOKS DJ. Botulinum toxin does not reverse the cortical dysfunction associated with writer's cramp - A PET study. Brain. 1997;120:571–582. doi: 10.1093/brain/120.4.571. [DOI] [PubMed] [Google Scholar]
- CURRA A, TROMPETTO C, ABBRUZZESE G, BERARDELLI A. Central effects of botulinum toxin type A: Evidence and supposition. Movement Disorders. 2004;19:S60–S64. doi: 10.1002/mds.20011. [DOI] [PubMed] [Google Scholar]
- CURTIS DR, DE GROAT WC. Tetanus toxin and spinal inhibition. Brain Res. 1968;10:208–12. doi: 10.1016/0006-8993(68)90123-6. [DOI] [PubMed] [Google Scholar]
- DAVID AT, SAIED A, CHARLES A, SUBRAMANIAN R, CHOULJENKO VN, KOUSOULAS KG. A Herpes Simplex Virus 1 (McKrae) Mutant Lacking the Glycoprotein K Gene Is Unable To Infect via Neuronal Axons and Egress from Neuronal Cell Bodies. Mbio. 2012:3. doi: 10.1128/mBio.00144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE MAIO M. Therapeutic uses of botulinum toxin: from facial palsy to autonomic disorders. Expert Opin Biol Ther. 2008;8:791–8. doi: 10.1517/14712598.8.6.791. [DOI] [PubMed] [Google Scholar]
- DEINHARDT K, REVERSI A, BERNINGHAUSEN O, HOPKINS CR, SCHIAVO G. Neurotrophins Redirect p75NTR from a clathrin-independent to a clathrin-dependent endocytic pathway coupled to axonal transport. Traffic. 2007;8:1736–49. doi: 10.1111/j.1600-0854.2007.00645.x. [DOI] [PubMed] [Google Scholar]
- DEINHARDT K, SALINAS S, VERASTEGUI C, WATSON R, WORTH D, HANRAHAN S, BUCCI C, SCHIAVO G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- DONG M, LIU H, TEPP WH, JOHNSON EA, JANZ R, CHAPMAN ER. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol Biol Cell. 2008;19:5226–37. doi: 10.1091/mbc.E08-07-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG M, RICHARDS DA, GOODNOUGH MC, TEPP WH, JOHNSON EA, CHAPMAN ER. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J Cell Biol. 2003;162:1293–303. doi: 10.1083/jcb.200305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG M, YEH F, TEPP WH, DEAN C, JOHNSON EA, JANZ R, CHAPMAN ER. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006;312:592–6. doi: 10.1126/science.1123654. [DOI] [PubMed] [Google Scholar]
- ERDMANN G, HANAUSKE A, WELLHONER HH. Intraspinal distribution and reaction in the grey matter with tetanus toxin of intracisternally injected anti-tetanus toxoid F(ab')2 fragments. Brain Res. 1981;211:367–77. doi: 10.1016/0006-8993(81)90708-3. [DOI] [PubMed] [Google Scholar]
- FISCHER A. Synchronized chaperone function of botulinum neurotoxin domains mediates light chain translocation into neurons. Curr Top Microbiol Immunol. 2013;364:115–37. doi: 10.1007/978-3-642-33570-9_6. [DOI] [PubMed] [Google Scholar]
- FOSTER L, CLAPP L, ERICKSON M, JABBARI B. Botulinum toxin A and chronic low back pain: a randomized, double-blind study. Neurology. 2001;56:1290–3. doi: 10.1212/wnl.56.10.1290. [DOI] [PubMed] [Google Scholar]
- FU FN, BUSATH DD, SINGH BR. Spectroscopic analysis of low pH and lipid-induced structural changes in type A botulinum neurotoxin relevant to membrane channel formation and translocation. Biophys Chem. 2002;99:17–29. doi: 10.1016/s0301-4622(02)00135-7. [DOI] [PubMed] [Google Scholar]
- FU ZJ, CHEN C, BARBIERI JT, KIM JJP, BALDWIN MR. Glycosylated SV2 and Gangliosides as Dual Receptors for Botulinum Neurotoxin Serotype F. Biochemistry. 2009;48:5631–5641. doi: 10.1021/bi9002138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALLOUX M, VITRAC H, MONTAGNER C, RAFFESTIN S, POPOFF MR, CHENAL A, FORGE V, GILLET D. Membrane Interaction of botulinum neurotoxin A translocation (T) domain. The belt region is a regulatory loop for membrane interaction. J Biol Chem. 2008;283:27668–76. doi: 10.1074/jbc.M802557200. [DOI] [PubMed] [Google Scholar]
- GILADI N. The mechanism of action of botulinum toxin type A in focal dystonia is most probably through its dual effect on efferent (motor) and afferent pathways at the injected site. Journal of the Neurological Sciences. 1997;152:132–5. doi: 10.1016/s0022-510x(97)00151-2. [DOI] [PubMed] [Google Scholar]
- GILIO F, CURRA A, LORENZANO C, MODUGNO N, MANFREDI M, BERARDELLI A. Effects of botulinum toxin type A on intracortical inhibition in patients with dystonia. Annals of Neurology. 2000;48:20–26. [PubMed] [Google Scholar]
- GILL DM. Bacterial Toxins - a Table of Lethal Amounts. Microbiol Rev. 1982;46:86–94. doi: 10.1128/mr.46.1.86-94.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JACKY BP, GARAY PE, DUPUY J, NELSON JB, CAI B, MOLINA Y, WANG J, STEWARD LE, BROIDE RS, FRANCIS J, AOKI KR, STEVENS RC, FERNANDEZ-SALAS E. Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A) PLoS Pathog. 2013;9:e1003369. doi: 10.1371/journal.ppat.1003369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAHN R, NIEMANN H. Molecular mechanisms of clostridial neurotoxins. Ann N Y Acad Sci. 1994;733:245–55. doi: 10.1111/j.1749-6632.1994.tb17274.x. [DOI] [PubMed] [Google Scholar]
- JANKOVIC J. Botulinum toxin in movement disorders. Curr Opin Neurol. 1994;7:358–66. doi: 10.1097/00019052-199408000-00014. [DOI] [PubMed] [Google Scholar]
- JANZ R, SUDHOF TC. SV2C is a synaptic vesicle protein with an unusually restricted localization: Anatomy of a synaptic vesicle protein family. Neuroscience. 1999;94:1279–1290. doi: 10.1016/s0306-4522(99)00370-x. [DOI] [PubMed] [Google Scholar]
- KALB SR, LOU J, GARCIA-RODRIGUEZ C, GEREN IN, SMITH TJ, MOURA H, MARKS JD, SMITH LA, PIRKLE JL, BARR JR. Extraction and inhibition of enzymatic activity of botulinum neurotoxins/A1, /A2, and /A3 by a panel of monoclonal anti-BoNT/A antibodies. PLoS One. 2009;4:e5355. doi: 10.1371/journal.pone.0005355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LALLI G, SCHIAVO G. Analysis of retrograde transport in motor neurons reveals common endocytic carriers for tetanus toxin and neurotrophin receptor p75NTR. J Cell Biol. 2002;156:233–9. doi: 10.1083/jcb.200106142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE GW, OVSEPIAN SV, WANG JF, AOKI KR, DOLLY JO. Extravesicular intraneuronal migration of internalized botulinum neurotoxins without detectable inhibition of distal neurotransmission. Biochemical Journal. 2012;441:443–452. doi: 10.1042/BJ20111117. [DOI] [PubMed] [Google Scholar]
- MAHRHOLD S, RUMMEL A, BIGALKE H, DAVLETOV B, BINZ T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006;580:2011–4. doi: 10.1016/j.febslet.2006.02.074. [DOI] [PubMed] [Google Scholar]
- MAHRHOLD S, STROTMEIER J, GARCIA-RODRIGUEZ C, LOU J, MARKS JD, RUMMEL A, BINZ T. Identification of the SV2 protein receptor-binding site of botulinum neurotoxin type E. Biochem J. 2013;453:37–47. doi: 10.1042/BJ20130391. [DOI] [PubMed] [Google Scholar]
- MARCHAND-PAUVERT V, AYMARD C, GIBOIN LS, DOMINICI F, ROSSI A, MAZZOCCHIO R. Beyond muscular effects: depression of spinal recurrent inhibition after botulinum neurotoxin A. Journal of Physiology-London. 2013;591:1017–1029. doi: 10.1113/jphysiol.2012.239178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTAL M. Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem. 2010;79:591–617. doi: 10.1146/annurev.biochem.051908.125345. [DOI] [PubMed] [Google Scholar]
- MONTECUCCO C, SCHIAVO G. Structure and function of tetanus and botulinum neurotoxins. Q Rev Biophys. 1995;28:423–72. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- NISHIKI T, KAMATA Y, NEMOTO Y, OMORI A, ITO T, TAKAHASHI M, KOZAKI S. Identification of protein receptor for Clostridium botulinum type B neurotoxin in rat brain synaptosomes. J Biol Chem. 1994;269:10498–503. [PubMed] [Google Scholar]
- PENG L, BERNTSSON RP, TEPP WH, PITKIN RM, JOHNSON EA, STENMARK P, DONG M. Botulinum neurotoxin D-C uses synaptotagmin I and II as receptors, and human synaptotagmin II is not an effective receptor for type B, D-C and G toxins. J Cell Sci. 2012;125:3233–42. doi: 10.1242/jcs.103564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENG L, TEPP WH, JOHNSON EA, DONG M. Botulinum neurotoxin D uses synaptic vesicle protein SV2 and gangliosides as receptors. PLoS Pathog. 2011;7:e1002008. doi: 10.1371/journal.ppat.1002008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIRAZZINI M, HENKE T, ROSSETTO O, MAHRHOLD S, KREZ N, RUMMEL A, MONTECUCCO C, BINZ T. Neutralisation of specific surface carboxylates speeds up translocation of botulinum neurotoxin type B enzymatic domain. FEBS Lett. 2013;587:3831–6. doi: 10.1016/j.febslet.2013.10.010. [DOI] [PubMed] [Google Scholar]
- POPOFF MR, BOUVET P. Genetic characteristics of toxigenic Clostridia and toxin gene evolution. Toxicon. 2013;75:63–89. doi: 10.1016/j.toxicon.2013.05.003. [DOI] [PubMed] [Google Scholar]
- PRIORI A, BERARDELLI A, MERCURI B, MANFREDI M. Physiological-Effects Produced by Botulinum Toxin Treatment of Upper-Limb Dystonia - Changes in Reciprocal Inhibition between Forearm Muscles. Brain. 1995;118:801–807. doi: 10.1093/brain/118.3.801. [DOI] [PubMed] [Google Scholar]
- PUHAR A, JOHNSON EA, ROSSETTO O, MONTECUCCO C. Comparison of the pH-induced conformational change of different clostridial neurotoxins. Biochem Biophys Res Commun. 2004;319:66–71. doi: 10.1016/j.bbrc.2004.04.140. [DOI] [PubMed] [Google Scholar]
- RESTANI L, ANTONUCCI F, GIANFRANCESCHI L, ROSSI C, ROSSETTO O, CALEO M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A) J Neurosci. 2011;31:15650–9. doi: 10.1523/JNEUROSCI.2618-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RESTANI L, GIRIBALDI F, MANICH M, BERCSENYI K, MENENDEZ G, ROSSETTO O, CALEO M, SCHIAVO G. Botulinum neurotoxins A and E undergo retrograde axonal transport in primary motor neurons. PLoS Pathog. 2012a;8:e1003087. doi: 10.1371/journal.ppat.1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RESTANI L, NOVELLI E, BOTTARI D, LEONE P, BARONE I, GALLI-RESTA L, STRETTOI E, CALEO M. Botulinum neurotoxin A impairs neurotransmission following retrograde transynaptic transport. Traffic. 2012b;13:1083–9. doi: 10.1111/j.1600-0854.2012.01369.x. [DOI] [PubMed] [Google Scholar]
- ROSSETTO O, PIRAZZINI M, MONTECUCCO C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol. 2014;12:535–49. doi: 10.1038/nrmicro3295. [DOI] [PubMed] [Google Scholar]
- ROTHMAN JE. Intracellular membrane fusion. Adv Second Messenger Phosphoprotein Res. 1994;29:81–96. doi: 10.1016/s1040-7952(06)80008-x. [DOI] [PubMed] [Google Scholar]
- RUMMEL A, EICHNER T, WEIL T, KARNATH T, GUTCAITS A, MAHRHOLD S, SANDHOFF K, PROIA RL, ACHARYA KR, BIGALKE H, BINZ T. Identification of the protein receptor binding site of botulinum neurotoxins B and G proves the double-receptor concept. Proc Natl Acad Sci U S A. 2007;104:359–64. doi: 10.1073/pnas.0609713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUMMEL A, HAFNER K, MAHRHOLD S, DARASHCHONAK N, HOLT M, JAHN R, BEERMANN S, KARNATH T, BIGALKE H, BINZ T. Botulinum neurotoxins C, E and F bind gangliosides via a conserved binding site prior to stimulation-dependent uptake with botulinum neurotoxin F utilising the three isoforms of SV2 as second receptor. J Neurochem. 2009a;110:1942–54. doi: 10.1111/j.1471-4159.2009.06298.x. [DOI] [PubMed] [Google Scholar]
- RUMMEL A, HAFNER K, MAHRHOLD S, DARASHCHONAK N, HOLT M, JAHN R, BEERMANN S, KARNATH T, BIGALKE H, BINZ T. Botulinum neurotoxins C, E and F bind gangliosides via a conserved binding site prior to stimulation-dependent uptake with botulinum neurotoxin F utilising the three isoforms of SV2 as second receptor. J Neurochem. 2009b;110:1942–1954. doi: 10.1111/j.1471-4159.2009.06298.x. [DOI] [PubMed] [Google Scholar]
- SALINAS S, BILSLAND LG, HENAFF D, WESTON AE, KERIEL A, SCHIAVO G, KREMER EJ. CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog. 2009;5:e1000442. doi: 10.1371/journal.ppat.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALINAS S, SCHIAVO G, KREMER EJ. A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol. 2010;8:645–55. doi: 10.1038/nrmicro2395. [DOI] [PubMed] [Google Scholar]
- SCHANTZ EJ, JOHNSON EA. Properties and use of botulinum toxin and other microbial neurotoxins in medicine. Microbiol Rev. 1992;56:80–99. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIAVO G, MATTEOLI M, MONTECUCCO C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–66. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- SCHMIEG N, MENENDEZ G, SCHIAVO G, TERENZIO M. Signalling endosomes in axonal transport: travel updates on the molecular highway. Semin Cell Dev Biol. 2014;27:32–43. doi: 10.1016/j.semcdb.2013.10.004. [DOI] [PubMed] [Google Scholar]
- SCOTT AB. Botulinum toxin injection into extraocular muscles as an alternative to strabismus surgery. J Pediatr Ophthalmol Strabismus. 1980;17:21–5. doi: 10.3928/0191-3913-19800101-06. [DOI] [PubMed] [Google Scholar]
- SILBERSTEIN S, MATHEW N, SAPER J, JENKINS S. Botulinum toxin type A as a migraine preventive treatment. For the BOTOX Migraine Clinical Research Group. Headache. 2000;40:445–50. doi: 10.1046/j.1526-4610.2000.00066.x. [DOI] [PubMed] [Google Scholar]
- SUN S, SURESH S, LIU H, TEPP WH, JOHNSON EA, EDWARDSON JM, CHAPMAN ER. Receptor binding enables botulinum neurotoxin B to sense low pH for translocation channel assembly. Cell Host Microbe. 2011;10:237–47. doi: 10.1016/j.chom.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SVERDLOV YS, ALEKSEEVA VI. Effect of tetanus toxin on presynaptic inhibition in the spinal cord. Fed Proc Transl Suppl. 1966;25:931–6. [PubMed] [Google Scholar]
- TAYLOR AM, BLURTON-JONES M, RHEE SW, CRIBBS DH, COTMAN CW, JEON NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURTON K, CHADDOCK JA, ACHARYA KR. Botulinum and tetanus neurotoxins: structure, function and therapeutic utility. Trends Biochem Sci. 2002;27:552–8. doi: 10.1016/s0968-0004(02)02177-1. [DOI] [PubMed] [Google Scholar]
- VON BARTHELD CS. Axonal transport and neuronal transcytosis of trophic factors, tracers, and pathogens. Journal of Neurobiology. 2004;58:295–314. doi: 10.1002/neu.10315. [DOI] [PubMed] [Google Scholar]
- WANG T, MARTIN S, PAPADOPULOS A, HARPER CB, MAVLYUTOV TA, NIRANJAN D, GLASS NR, COOPER-WHITE JJ, SIBARITA JB, CHOQUET D, DAVLETOV B, MEUNIER FA. Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. J Neurosci. 2015;35:6179–94. doi: 10.1523/JNEUROSCI.3757-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMSON LC, NEALE EA. Bafilomycin A1 inhibits the action of tetanus toxin in spinal cord neurons in cell culture. J Neurochem. 1994;63:2342–5. doi: 10.1046/j.1471-4159.1994.63062342.x. [DOI] [PubMed] [Google Scholar]
- YAO G, ZHANG S, MAHRHOLD S, LAM KH, STERN D, BAGRAMYAN K, PERRY K, KALKUM M, RUMMEL A, DONG M, JIN R. N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat Struct Mol Biol. 2016 doi: 10.1038/nsmb.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEH FL, DONG M, YAO J, TEPP WH, LIN G, JOHNSON EA, CHAPMAN ER. SV2 mediates entry of tetanus neurotoxin into central neurons. PLoS Pathog. 2010;6:e1001207. doi: 10.1371/journal.ppat.1001207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.