

Abstract
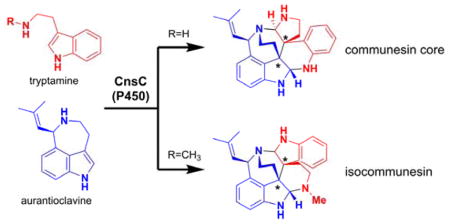
Dimeric indole alkaloids are structurally diverse natural products that have attracted significant attention from the synthetic and biosynthetic communities. Here we describe the characterization of a P450 monooxygenase CnsC from Penicillium that catalyzes the heterodimeric coupling between two different indole moieties, tryptamine and aurantioclavine, to construct vicinal quaternary stereocenters and yield the heptacyclic communesin scaffold. We show, using biochemical characterization, substrate analogs, and computational methods, that CnsC not only catalyzes the C3-C3 carbon-carbon bond formation, but also controls the regioselectivities of the pair of subsequent aminal bond formations to yield the communesin core. The use of ω-N-methyltryptamine and tryptophol in place to tryptamine led to the enzymatic synthesis of isocommunesin compounds, which have not been isolated to date.
Graphical Abstract
Dimeric indole alkaloids are a large subset of plant and fungal natural products that have a wide range of biological activities. Examples include the tryptamine-derived calycanthaceous alkaloids such as (+)-calycanthine, epipolythiodiketopiperazines such as chaetocin, and the perophoramidine and communesin alkaloids.1 A defining feature of these natural products is the presence of vicinal quaternary carbon stereocenters, which are particularly intriguing from both synthetic and biosynthetic perspectives.2 Synthetic approaches to the vicinal quaternary stereocenters of these natural products have been developed and include Heck cyclization methodology, bis(alkylation) chemistry, pericyclic reactions, and radical couplings.2 The biosynthetic pathways to these natural products, however, have been much less well-studied and the enzymes responsible for vicinal quaternary stereocenter formation have not been revealed. Identifying the biocatalysts that form the C3-C3′ linkage is an important objective towards better understanding of indole dimer biosynthesis and how Nature can efficiently construct complex scaffolds.
We have specifically targeted the biosynthesis of the communesin alkaloids, given their remarkable structures and the vast interest in these compounds from the synthetic community.3 The core structure of the communesins, as represented by the simplest isolated member, communesin K (3), consists of seven interconnected rings, two aminal linkages, and four contiguous stereocenters. Other family members include communesins A (1) and B (2), which present additional synthetic challenges (Figure 1 and S1). Several total syntheses have been reported.4 With regard to communesin biosynthesis, Stoltz et al proposed that the communesin core could be derived from the hetero-dimerization of tryptamine (5) and aurantioclavine (6), through either direct C3-C3′ indole coupling followed by aminal bond formation or via an exo inverse electron demand Diels-Alder reaction between 6 and the quinone methide imine derivative of 5.2b We recently identified the biosynthetic pathway of 1 and 2 in Penicillium expansum and verified that indeed, 5 and 6 are biosynthetic precursors to the core structure 4.5 We found that a cytochrome P450 monooxygenase, CnsC, may be involved in the oxidative coupling between 5 and 6, as deletion of Pe-cnsC in P. expansum abolished communesin production and instead accumulated 6. Sequence analysis showed that CnsC displays low sequence homology to known P450s in the database (Figure S2). While CnsC must play a key role in the selective dimerization and generation of the core structure, numerous mechanistic questions remain to be answered: 1) are there other enzymes involved in the biosynthesis of 4, which requires the formation of three bonds (C3-C3′, C2-N1′ and C2′-N10) between 5 and 6? 2) How are the sequences and regioselectivities of the bond-forming steps controlled to arrive at 4; and 3) what is the substrate specificity of CnsC towards two different indole building blocks in order to achieve the hetero-coupling?
Figure 1.

Formation of communesin core. A P450 (Pe-CnsC from P. expansum) has been implicated in the coupling of 5 and 6 into the core 4. Communesin K (3) is the simplest, stable communesin isolated, and is formed upon N-methylation of 4 by the methyltransferase Pe-CnsE.
To investigate the activity of CnsC, the intron-free cnsC and the P450 redox partner cytochrome P450 oxidoreductase (Pe-CPR) from P. expansum were cloned under the ADH2 promoter and transformed into Saccharomyces cerevisiae strain BJ5464-NpgA.6 However, western blotting analysis of the membrane fraction showed very weak expression of the P450, and no new product could be detected when 5 and 6 were supplemented to the yeast culture. To find a CnsC alternative that may be better expressed in yeast, we sequenced the genome of Penicillium rivulum, which is also a reported producer of communesin.3d The P. rivulum communesin cluster is highly similar to that of the cns cluster in P. expansum (Table S2), including the P450 of interest Pr-CnsC, which shows 84% sequence homology to Pe-CnsC and a different membrane anchoring region(Figures S3). Upon feeding 5 and 6 to the yeast strain expressing Pr-CnsC and Pe-CPR, we observed consumption of both substrates and production of trace amounts of 4 (m/z 385 [M+H]+). The low signal of 4 is due to its instability from the two labile aminal linkages, which was also observed in knockout studies with P. expansum.5 To further confirm formation of the core structure, we coexpressed the N-methyltransferase Pe-CnsE (Figure 1) with Pr-CnsC and Pe-CPR. Metabolite analysis of the yeast culture supplied with 5 and 6 showed clear accumulation of a new product that matches the retention time and m/z of the N1-methylated communesin K (3) (Figure S4A, ii). In contrast, untransformed yeast (Figure S4A, i) did not accumulate 3.
Microsomal fractions containing the overexpressed Pr-CnsC and Pe-CPR were purified from three-day cultures of the yeast expression strain. From the in vitro assay in which 0.5 mM 5, 0.5 mM 6 and 2 mM NADPH were incubated with microsomes (20 mg/mL total protein concentration), a clear product that corresponds to 4 emerged (Figure S4B, ii), albeit with low conversion. Introducing Pe-CnsE purified from Escherichia coli and 2 mM S-adenosyl-methionine (SAM) led to the formation of the expected 3, fully consistent with the yeast biotransformation results (Figure S4B, iii). Collectively, these results demonstrate that the P450 CnsC alone is sufficient to oxidatively couple the two different indole-containing substrates together to yield the heptacyclic core of communesins. In vitro assays using either 5 or 6 alone did not lead to the formation of homodimeric products, or any other detectable products. CnsC is therefore a rare example of a P450 that catalyzes C-C bond formation between two different substrates.7. It is particularly intriguing that CnsC strategically unifies 5 and 6 with controlled formation of four new stereocenters and three new bonds. The most likely order of bond formation is the C3-C3′ coupling between 5 and 6, followed by the two aminal bonds. It is also possible that the P450 active site orients the two substrates to promote aminal formation first, followed by the more difficult C3-C3′ bond formation.
To gain insight into the sequence of the bond forming steps and substrate specificities of CnsC, we tested enzyme activities towards different analogs of 5 and 6 using the in vitro reaction. When N1-methyl tryptamine or N1-methyl aurantioclavine was used in place of 5 or 6, respectively, no product formation was detected, indicating the N1 hydrogens of both substrates are essential for the coupling reaction (Figure S7). In contrast, when N10-methyl tryptamine 7 was used, we observed the formation of two oxidatively coupled products 8 and 9 with the same mass as 3 (m/z 399 [M+H]+) at a relative ratio of 5:1 (Figure 2A). To elucidate the structures of 8 and 9, a 4L culture of the yeast strain co-expressing Pr-CnsC and Pe-CPR was grown for three days then concentrated ten-fold and supplemented with 5.0 mg each of 6 and 7. Cultivation for one additional day afforded sufficient quantities of 8. The 1H and 13C NMR spectra of 8 showed characteristic signals of two aminals at δ 5.01 (H2′)/δ 84.8 (C2′) and δ 4.17 (H2)/δ 84.2 (C2) (Figure S8–S13 and Table S3), and with other key HMBC correlations, the skeleton of 8 was established. The NOESY spectrum showed the correlations of H2′ to H8′ (δ 2.25), H9 (δ 2.65) and H8 (δ 3.42); H2 to H8′ and H9 (δ 2.08); H11 (δ 5.26) to H7′ (δ 6.67); and no correlation of H9 to H11 confirming the relative stereochemistry at the corresponding carbon atoms. Based on the R configuration of C11 in 6,8 the absolute stereochemistry of 8 was confirmed to be 2′S, 3′S, 3R, 2S, 11R. We were not able to isolate sufficient amount of 9 for full structural characterization, however, the shift in retention time and mass increase are indicative of the N10′ methylated form of 4.
Figure 2.
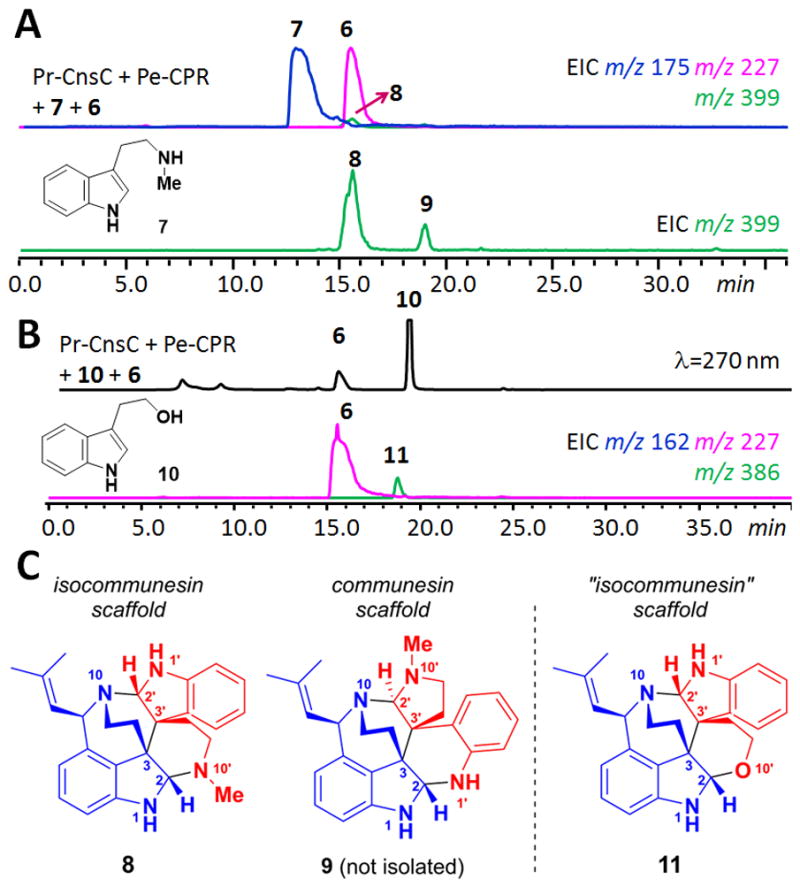
Formation of communesin and isocommunesin scaffolds in the presence of tryptamine analogs. (A) in vitro assay using 6 and 7 led to the synthesis of 8 and 9; (B) assay using 6 and 10 led to the hemiaminal ether containing 11; (C) structures of 8, 9 (putative) and 11.
The structure of 8 is unexpected and represents an alternative way in which the two indole-containing building blocks 6 and 7 can be coupled. Compound 8 remains a heptacyclic alkaloid with the C3-C3′ linkage and four contiguous stereocenters, but with both indoline moieties remaining intact. In the communesin configuration, the two indole nitrogen atoms and the two ω-nitrogen atoms are each paired to form the two aminal linkages (N1-C2-N1′, N10-C2′-N10′). However, in 8 two different C–N bonds (N1-C2-N10′ and N1′-C2′-N10) are formed to give the bis(indoline) “isocommunesin” scaffold of 8, which has not been isolated in any natural products to date. It is therefore intriguing that N-methylation in 7 can drastically alters the products formed by CnsC.
To learn more about these substituent effects, we coupled other tryptamine analogs with 6 and monitored formation of adducts using LC/MS and selective ion monitoring. Surprisingly, CnsC displayed significant substrate tolerance towards analogs of 5; product formation can be detected from substrates ranging in size between indole and ω-N-boc-tryptamine (Figures S5). Other C3-substituted indoles such as 3-methyl-indole, ω-N-dimethyl-tryptamine, 3-(2-azidoethyl)-indole all yielded products with retention time suggestive of the isocommunesin structures (Figures S6–S7). We were able to detect a single new product 11 (m/z 386 [M+H]+) when tryptophol 10 was used as a substrate together with 6 (Figure 2B). Subsequent feeding of 6 and 10 the yeast culture (6 mg each) led to the isolation of 11 (3 mg) for structural characterization (Figure S14–S18).
The 13C NMR spectrum of 11 showed characteristic signals of one aminal at δ 83.0 (C2′), and one hemiaminal at δ 92.5 (C2). The HMBC correlations showed key correlations that confirmed the skeleton of 11 to be also of the isocommunesin scaffold. The absolute stereochemistries of the four contiguous stereocenters of 11 were confirmed to be analogous to 8 (2′S 3′S, 3R, 2S, 11R) based on NOESY data and the known R configuration of precursor 6 at C7. Interestingly, when only 6 was supplemented to the yeast culture expressing Pr-CnsC and Pe-CPR, 11 was also detected in the organic extracts. This is due to 10 being a known metabolite in S. cerevisiae that is involved in quorum sensing.9 We also assayed the activities of CnsC towards N10-methyl analogs of 6, however, no coupled products were detected, suggesting the tolerance is low towards aurantioclavine analogs (Figure S7).
The reconstitution of CnsC activities and generation of either communesin or isocommunesin scaffolds provide significant insight into coupling between 5 and 6. The common C3-C3′ bond in 4, 8, and 11 suggests that this coupling step takes place first in the active site of CnsC and staples the heterodimers together. Because C3-C3′ indole coupling is widely observed among alkaloid natural products and only a few mechanistic proposals exist, we computationally examined four possible mechanisms of coupling that can afford 4 (Figure S23). These mechanisms include radical addition, radical cation addition, electrophilic aromatic addition, and radical combination. The radical addition mechanism involves the intermediacy of a tryptaminyl radical formed by CnsC-catalyzed N–H hemolysis, followed by addition to 6. Computations show that this mechanism has a 105-fold intrinsic preference for the formation of a C3-C2′ adduct, which was not observed experimentally (Figure S24). Similarly, reaction of a tryptaminyl radical cation (generated by CnsC-catalyzed single electron transfer) with 6 is predicted to also lead to facile formation of a C3-C2′ product (Figure S26). C–C bond formation via the capture a tryptaminyl cation, the product of the two-electron oxidation of 5, by 6 is predicted to be nonselective (Figure S25). None of these mechanisms are deemed likely given the exclusive C3-C3′ coupling observed here. Further discussion and details of this computational work are provided in the Supporting Information.
In contrast, the combination of two indole C3 radicals 12 and 13 to form the vicinal quaternary stereocenters and yield the product 14 must exclusively form the C3-C3 regioproduct (Figure 3). Both radicals can be generated by CnsC-catalyzed abstraction of the indole N1 hydrogen followed by facile migration of the radical to C3 positions. Such one-electron oxidation that initiate at the indole NH has been implicated in the P450-catalyzed dimerization of ditryptophenalanine and chaetocin.10–11 This mechanism is supported by the observation that CnsC is inactive towards either N1-methylated indole substrate.
Figure 3.

Proposed mechanism of CnsC catalyzed C3-C3′ coupling and formation of both communesin and isocommunesin scaffolds. 14 and 14′ are the immediate products of the enzymatic oxidative coupling between tryptamine and aurantioclavine. Assuming N-protonation of substrate, intermediates, and products, the relative Gibbs free energies (in kcal mol−1) of the aminal bond forming steps starting from 14 and 14′ are shown.
From 14 (or 14′), the two 3H-indoles in the two halves of the molecule can be subjected to intramolecular nucleophilic attack from the two amine groups and form the pair of aminal linkages. The regioselectivity of the aminal bond formation determines whether the communesin scaffold (as in 4) or the isocommunesin scaffold (as in 8) is formed. Our results indicate this regioselectivity is strongly influenced by the C3 substituent of 5. Energetics of intermediates and products of the reactions starting from 14 or 14′ were calculated to determine how the additional N-methyl substitution may influence the free energy of aminal C–N bond forming steps (Figure 3). Efforts to locate transition states for these reactions were unsuccessful. We calculated the energies with the N-protonated analogues of these structures to model the protonation states of these species in aqueous solution (Figure S28–30), but calculations with neutral species yielded similar results (Figure S27). All intermediates leading to the communesin (4 and 9) and isocommunesin scaffolds (21 and 8) are thermodynamically accessible at room temperature. The formation of either scaffold is irreversible as scaffold formation is highly exergonic (ca. −25 kcal mol−1 for the formation 4 and 21, and ca. −28 kcal mol−1 for the formation of 9 and 8). The only energetically uphill step in Figure 3 is the ring opening of pyrroloindole 15 to 16 required for formation of the communesin scaffold. Assuming the activation free energies of each step shown in Figure 3 are proportional to energies for the formation of the intermediates, we would expect that of the elementary step shown in Figure 3, the conversion of 15 to 16 would occur most slowly. Thus, formation of the isocommunesin scaffold would be more facile nonenzymatically than formation of the communesin scaffold. Given that the reaction of 5 and 6 with CnsC yields only the communesin product 4, both the initial oxidative coupling that forms 14 and the subsequent steps involving C-N bond formations and scission must be enzyme controlled. When the methylated 7 is coupled with 6 to form 14′, CnsC may no longer be able to exert complete control over the orientation of the intermediate of 14′, leading to the nonenzymatic formation of 8 and small amount of 9. Computational analysis also provided explanation to the exclusive formation of the isocommunesin scaffold 11 when 10 was used as a coupling partner to 6, which is due to a prohibitively high energy intermediate in the reaction path required for the formation of the communesin-like product (Figure S30).
In conclusion, we have identified a P450 monooxygenase CnsC that catalyzes the C3-C3′ coupling of two different indole substrates to introduce vicinal quaternary stereocenters. The enzyme also controls the regioselectivity of the subsequent aminal bond forming steps. The discovery of such a remarkable P450 further illustrates the biocatalytic power of fungal biosynthetic enzymes in the synthesis of complex molecular scaffolds.
Supplementary Material
Acknowledgments
This work was supported by the NIH (1DP1GM106413 to Y.T. and 1R56 AI101141 to Y.T. and N.K.G.) and the NSF (CHE-1361104 to K.N.H.) H-C. L. thanks the NSC of Taiwan (102-2917-I-564-008) and M.A.C. thanks the NSF for a graduate fellowship (DGE-1144087). Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575). These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631). We thank Prof. Frisvad for the P. rivulum strain (IBT 24420).
Footnotes
Experimental details, spectroscopic and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kim J, Movassaghi M. Acc Chem Res. 2015;48:1159. doi: 10.1021/ar500454v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Nat Chem Biol. 2013;9:136. doi: 10.1038/nchembio.1187. [DOI] [PubMed] [Google Scholar]
- 2.(a) Movassaghi M, Schmidt MA. Angew Chem Intl Ed. 2007;46:3725. doi: 10.1002/anie.200700705. [DOI] [PubMed] [Google Scholar]; (b) May JA, Stoltz BM. Tetrahedron. 2006;62:5262. [Google Scholar]; (c) Xu JB, Cheng KJ. Molecules. 2015;20:6715. doi: 10.3390/molecules20046715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Takayama H, Matsuda Y, Masubuchi K, Ishida A, Kitajima M, Aimi N. Tetrahedron. 2004;60:893. [Google Scholar]; (e) Kitajima M, Mori I, Arai K, Kogure N, Takayama H. Tetrahedron Lett. 2006;47:3199. [Google Scholar]; (f) Büschleb M, Dorich S, Hanessian S, Tao D, Schenthal KB, Overman LE. Angew Chem Int Ed. 2016;55:2. doi: 10.1002/anie.201507549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V, Steube K, Proksch P. J Nat Prod. 2004;67:78. doi: 10.1021/np030271y. [DOI] [PubMed] [Google Scholar]; (b) Hayashi H, Matsumoto H, Akiyama K. Biosci Biotechnol, Biochem. 2004;68:753. doi: 10.1271/bbb.68.753. [DOI] [PubMed] [Google Scholar]; (c) Andersen B, Smedsgaard J, Frisvad JC. J Agric Food Chem. 2004;52:2421. doi: 10.1021/jf035406k. [DOI] [PubMed] [Google Scholar]; (d) Dalsgaard PW, Blunt JW, Munro MHG, Frisvad JC, Christophersen C. J Nat Prod. 2005;68:258. doi: 10.1021/np049646l. [DOI] [PubMed] [Google Scholar]; (e) Wigley LJ, Mantle PG, Perry DA. Phytochemistry. 2006;67:561. doi: 10.1016/j.phytochem.2005.10.011. [DOI] [PubMed] [Google Scholar]; (f) Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Tetrahedron Lett. 1993;34:2355. [Google Scholar]
- 4.(a) Wu HX, Xue F, Xiao X, Qin Y. J Am Chem Soc. 2010;132:14052. doi: 10.1021/ja1070043. [DOI] [PubMed] [Google Scholar]; (b) Yang J, Wu HX, Shen LQ, Qin Y. J Am Chem Soc. 2007;129:13794. doi: 10.1021/ja075705g. [DOI] [PubMed] [Google Scholar]; (c) Zuo ZW, Xie WQ, Ma DW. J Am Chem Soc. 2010;132:13226. doi: 10.1021/ja106739g. [DOI] [PubMed] [Google Scholar]; (d) Zuo Z, Ma D. Angew Chem Intl Ed. 2011;50:12008. doi: 10.1002/anie.201106205. [DOI] [PubMed] [Google Scholar]; (e) Liu P, Seo JH, Weinreb SM. Angew Chem Intl Ed. 2010;49:2000. doi: 10.1002/anie.200906818. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Seo JH, Liu P, Weinreb SM. J Org Chem. 2010;75:2667. doi: 10.1021/jo100339k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Belmar J, Funk RL. J Am Chem Soc. 2012;134:16941. doi: 10.1021/ja307277w. [DOI] [PubMed] [Google Scholar]; (h) Crawley SL, Funk RL. Org Lett. 2003;5:3169. doi: 10.1021/ol034407v. [DOI] [PubMed] [Google Scholar]; (i) Han SJ, Vogt F, May JA, Krishnan S, Gatti M, Virgil SC, Stoltz BM. J Org Chem. 2015;80:528. doi: 10.1021/jo502534g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Han SJ, Vogt F, Krishnan S, May JA, Gatti M, Virgil SC, Stoltz BM. Org Lett. 2014;16:3316. doi: 10.1021/ol5013263. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) May JA, Zeidan RK, Stoltz BM. Tetrahedron Lett. 2003;44:1203. [Google Scholar]; (l) Siengalewicz P, Gaich T, Mulzer J. Angew Chem Intl Ed. 2008;47:8170. doi: 10.1002/anie.200801735. [DOI] [PubMed] [Google Scholar]
- 5.Lin HC, Chiou G, Chooi YH, McMahon TC, Xu W, Garg NK, Tang Y. Angew Chem Intl Ed. 2015;54:3004. doi: 10.1002/anie.201411297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma SM, Li JWH, Choi JW, Zhou H, Lee KKM, Moorthie VA, Xie XK, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, Moore BS. Nat Chem Biol. 2014;10:640. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Krishnan S, Bagdanoff JT, Ebner DC, Ramtohul YK, Tambar UK, Stoltz BM. J Am Chem Soc. 2008;130:13745. doi: 10.1021/ja804738b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Behenna DC, Krishnan S, Stoltz BM. Tetrahedron Lett. 2011;52:2152. [Google Scholar]
- 9.Albuquerque P, Casadevall A. Med Mycol. 2012;50:337. doi: 10.3109/13693786.2011.652201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saruwatari T, Yagishita F, Mino T, Noguchi H, Hotta K, Watanabe K. ChemBioChem. 2014;15:656. doi: 10.1002/cbic.201300751. [DOI] [PubMed] [Google Scholar]
- 11.(a) Walsh CT. ACS Chem Biol. 2014;9:2718. doi: 10.1021/cb500695k. [DOI] [PubMed] [Google Scholar]; (b) Gerken T, Walsh CT. ChemBioChem. 2013;14:2256. doi: 10.1002/cbic.201300513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.