

Abstract
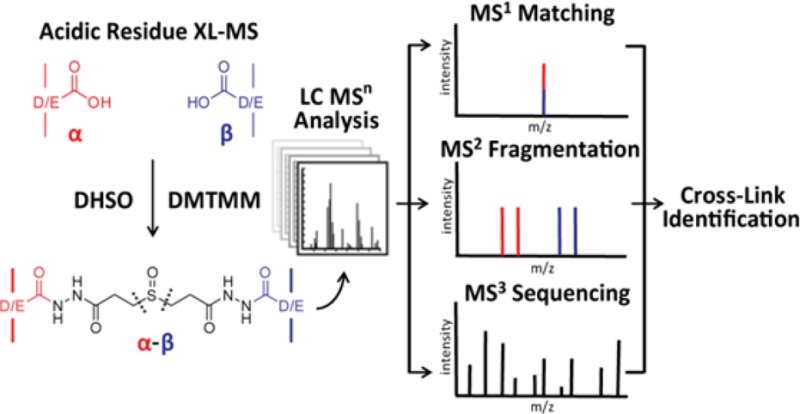
Cross-linking mass spectrometry (XL-MS) has become a powerful strategy for defining protein–protein interactions and elucidating architectures of large protein complexes. However, one of the inherent challenges in MS analysis of cross-linked peptides is their unambiguous identification. To facilitate this process, we have previously developed a series of amine-reactive sulfoxide-containing MS-cleavable cross-linkers. These MS-cleavable reagents have allowed us to establish a common robust XL-MS workflow that enables fast and accurate identification of cross-linked peptides using multistage tandem mass spectrometry (MSn). Although amine-reactive reagents targeting lysine residues have been successful, it remains difficult to characterize protein interaction interfaces with little or no lysine residues. To expand the coverage of protein interaction regions, we present here the development of a new acidic residue-targeting sulfoxide-containing MS-cleavable homobifunctional cross-linker, dihydrazide sulfoxide (DHSO). We demonstrate that DHSO cross-linked peptides display the same predictable and characteristic fragmentation pattern during collision induced dissociation as amine-reactive sulfoxide-containing MS-cleavable cross-linked peptides, thus permitting their simplified analysis and unambiguous identification by MSn. Additionally, we show that DHSO can provide complementary data to amine-reactive reagents. Collectively, this work not only enlarges the range of the application of XL-MS approaches but also further demonstrates the robustness and applicability of sulfoxide-based MS-cleavability in conjunction with various cross-linking chemistries.
The majority of proteins exert their functions in the form of protein complexes. These macromolecular assemblies and their protein–protein interactions play critical roles in regulating integral biological processes. As a result, perturbations of endogenous protein–protein interactions can result in deleterious effects on cellular activities. Structural analyses of these complexes by traditional biophysical structural techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) are frequently utilized to elucidate their topologies. Unfortunately, many large and heterogeneous complexes are refractory to such methods, ushering the development of new hybrid structural strategies. Cross-linking mass spectrometry (XL-MS) has emerged as a powerful and popular approach for delineating the protein interactions within large multisubunit protein complexes.1,2 Moreover, cross-linking can capture temporal protein interactions by forming covalent bonds between proximal amino acid residues, effectively freezing transient interactions and providing information on the identities and spatial orientations of interacting proteins simultaneously. These linkages are then utilized as distance constraints to facilitate three-dimensional modeling of protein complexes by refining existing high-resolution protein structures or complementing lower resolution biophysical structural techniques (e.g., cryo-electron microscopy) in order to position individual protein subunits or interacting regions.3−9
One of the major challenges in conventional XL-MS studies is the unambiguous identification of cross-linked peptides, due to difficulty in interpreting convoluted tandem mass spectra resulting from the fragmentation of covalently linked peptides. To this end, various types of cleavable cross-linkers have been developed to facilitate and simplify MS identification of cross-linked peptides, among which MS-cleavable cross-linkers appear to be the most attractive option due to their capability to improve MS identification of cross-linked peptides.10−16 In recent years, we have developed a new class of MS-cleavable cross-linking reagents containing sulfoxide group(s) within their spacer regions, i.e., disuccinimidyl sulfoxide (DSSO),14 dimethyl disuccinimidyl sulfoxide (DMDSSO),15 azide-tagged acid-cleavable disuccinimidyl bissulfoxide (Azide-A-DSBSO)16 (Figure 1A–C). These MS-cleavable reagents contain symmetric MS-labile C–S bonds (adjacent to the sulfoxide group) that can be selectively and preferentially fragmented prior to peptide backbone cleavage during collision induced dissociation (CID).14−16 Such fragmentation is robust and predictable, occurring independently of cross-linking types, peptide charges, and sequences. Ultimately this unique feature enables simplified and unambiguous identification of cross-linked peptides by MSn analysis and conventional database searching tools.14−16 Our newly developed sulfoxide-containing, MS-cleavable cross-linkers have been successfully applied not only to define protein–protein interactions and elucidate structures of protein complexes in vitro(5,9,14,17) and in vivo(16) but also to quantify structural dynamics of protein complexes.15,18
Figure 1.

Sulfoxide-containing MS-cleavable cross-linkers: (A) DSSO,14 (B) DMDSSO,15and (C) Azide-A-DSBSO.16 (D) Synthesis scheme of MS-cleavable cross-linker DHSO. (E) Characteristic MS2 fragmentation of DHSO interlinked heterodimer α-β.
In current XL-MS studies, amine-reactive reagents targeting lysine residues are the most widely used compounds for successful elucidation of protein structures. This is due to their effective and specific cross-linking chemistry as well as frequent occurrence of lysine residues in protein sequences, especially at surface-exposed regions of protein structures. However, it remains challenging to characterize protein interaction interfaces with little to no lysine residues. Therefore, there is a necessity for the development of additional cross-linking chemistries in order to increase the coverage of structural information obtainable from XL-MS experiments, particularly in systems where protein interacting regions are refractive to amine-specific cross-linking. Although several types of cross-linkers targeting other amino acids (e.g., sulfhydryl-reactive and nonspecific photoreactive) reagents are commercially available, their applications in studying protein–protein interactions thus far are very limited. For instance, sulfhydryl-reactive cross-linking reagents with specific chemistries targeting cysteine residues have not been widely adopted, most likely owing to the relatively low occurrence of cysteine residues and their participation in forming disulfide bonds in protein structures. In comparison, although photochemical cross-linking reagents can improve the coverage of protein interaction contacts by reacting with any amino acids nonspecifically,19 the resulting cross-linked products are often unpredictable, making their unambiguous MS identification even more difficult. In addition, nonspecific cross-linking has a higher chance of introducing more nonspecific interactions. Therefore, a specific cross-linking chemistry targeting other amino acid residues abundant at protein interaction sites would be ideal for complementing lysine targeting cross-linkers. While hydrophobic amino acid residues often constitute the cores, charged hydrophilic residues such as lysine, arginine, aspartic acid (Asp), and glutamic acid (Glu) often occupy surface-exposed regions of protein complexes, making them ideal targets for mapping protein interactions. According to a recent SwissProt database release,20 aspartic and glutamic acids comprise roughly 12.2% of all amino acid residues, compared to the 5.8% of lysines. Therefore, acidic residues (i.e., aspartic and glutamic acids) represent high potential targets for cross-linking studies due to their abundance and prevalence at interaction interfaces.
A recent study by Leitner et al. has demonstrated the feasibility of acidic residue-specific cross-linking chemistry to study protein interactions using noncleavable homobifunctional dihydrazide cross-linkers in conjunction with the coupling reagent 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM).21 This methodology is an improvement on the acidic residue cross-linking chemistry involving the coupling reagent 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide hydrochloride (EDC) that requires the cross-linking reaction to occur at a pH of 5.5.22 In comparison, DMTMM coupling with dihydrazide cross-linkers is compatible with proteins at neutral pH (7.0–7.5) and therefore better suited for studying the structures of proteins and protein complexes under physiological conditions. However, this cross-linking strategy remains susceptible to the challenges associated with traditional cross-linking reagents in unambiguously identifying cross-linked peptides and their linkage sites. Because of the increased prevalence of Asp and Glu in protein sequences, the accurate and unambiguous identification of peptides containing noncleavable dihydrazide cross-linked acidic residues would be intrinsically more complicated than the identification of lysine cross-linked peptides. To simplify MS analysis and facilitate the identification of acidic residue cross-linked peptides, we have developed a sulfoxide-containing MS-cleavable acidic residue-specific homobifunctional cross-linking reagent, dihydrazide sulfoxide (DHSO, a.k.a. 3,3′-sulfinyldi(propanehydrazide)). This reagent adopts the same MS-labile sulfoxide chemistry as our previously developed amine-reactive MS-cleavable cross-linkers (i.e., DSSO, DMDSSO, and Azide-A-DSBSO), thus enabling robust and unambiguous identification of cross-linked peptides via the same XL-MSn workflow.14−16 DHSO represents a novel class and the first generation of acidic residue-targeting cross-linking reagents with MS-cleavability. We expect that DHSO-based XL-MS strategies will become an invaluable tool in providing a complementary subset of cross-linking data toward a comprehensive structural elucidation of protein complexes by XL-MS.
Experimental Procedures
Materials and Reagents
General chemicals were purchased from Fisher Scientific or VWR International. Bovine serum albumin (≥96% purity), myoglobin from equine heart (≥90% purity), and DMTMM (≥96% purity) were purchased from Sigma-Aldrich. Ac-SR8 peptide (Ac-SAKAYEHR, 98.22% purity) was custom ordered from Biomatik (Wilmington, DE).
DHSO Cross-Linking of Synthetic Peptides
DHSO was synthesized as described (Figure 1D and Supplemental Methods). Synthetic peptide Ac-SR8 was dissolved in DMSO to 1 mM and cross-linked with DHSO in a 1:1 molar ratio of peptide to cross-linker in the presence of 1 equiv of diisopropylethylamine and DMTMM. The resulting samples were diluted to 10 pmol/μL in 3% ACN/2% formic acid prior to MSn analysis.
DHSO Cross-Linking of Equine Myoglobin and Bovine Serum Albumin
A volume of 50 μL of 50 μM BSA or 200 μM myoglobin in PBS buffer (pH 7.4) was reacted with DHSO in molar ratios of 1:5, 1:10, 1:20, and 1:30. The cross-linking reaction was initiated by adding equivalent concentrations of DHSO and DMTMM to protein solutions, reacted for 1 h at room temperature.
Digestion of DHSO Cross-Linked Proteins
Cross-linked protein samples were subjected to either SDS-PAGE followed by in-gel digestion or directly digested in solution prior to MS analysis23 (Supplemental Methods).
Liquid Chromatography–Multistage Tandem Mass Spectrometry (LC–MSn) Analysis
DHSO cross-linked peptides were analyzed by LC–MSn utilizing an Easy-nLC 1000 (Thermo Fisher, San Jose, CA) coupled online to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher, San Jose, CA).14,15 LC–MSn data extraction and database searching for the identification of DHSO cross-linked peptides were performed similarly as previously described14 (see the Supplemental Methods).
Results and Discussion
Design and Synthesis of a Novel Acidic Residue-Targeting Sulfoxide-Containing MS-Cleavable Cross-Linker
In order to facilitate accurate identification of acidic residue cross-linked peptides, we aimed to develop a novel MS-cleavable cross-linking reagent specific to Asp and Glu residues. This requires the incorporation of a functional group with robust MS-inducible cleavage sites located in the spacer region of the cross-linker. Previously, we successfully developed a novel class of amine-reactive, sulfoxide-containing MS-cleavable cross-linkers, i.e., DSSO,14 DMDSSO,15 and Azide-A-DSBSO16 (Figure 1A–C). The C–S bonds adjacent to the sulfoxide group(s) in these reagents have proven to be reliable labile bonds that fragment selectively and preferentially prior to the breakage of the peptide backbone during collision induced dissociation. Additionally, such fragmentation is predictable and occurs independently of peptide charge and sequence. These unique features facilitate the simplified analysis of sulfoxide-containing cross-linked peptides and their unambiguous identification by MSn.14−16 Following the success of our MS-cleavable, amine-reactive cross-linkers, we designed a novel acidic residue-reactive, MS-cleavable homobifunctional dihydrazide cross-linker incorporating a sulfoxide group in the spacer region, i.e., dihydrazide sulfoxide (DHSO). DHSO is synthesized from DSSO with two additional synthesis steps (Figure 1D). As shown, DHSO is composed of two hydrazide reactive groups and two symmetrical C–S cleavable bonds flanking a central sulfoxide. The spacer length of DHSO is 12.4 Å (calculated between the terminal nitrogen atoms). In comparison to existing cross-linkers for XL-MS studies,14−16,21 DHSO carries a linker length well suited for defining interaction interfaces between and within protein complexes.
CID Fragmentation Patterns of DHSO Cross-Linked Peptides
A previous study has shown that the reaction of hydrazide cross-linkers with acidic residues first requires activation of the terminal carboxyl groups of Asp (D) and Glu (E) side chains or protein C-termini.21 The coupling reagent DMTMM has been demonstrated to be effective in activating carboxylic acid groups to form a reactive intermediate that can be displaced by nucleophilic attack from hydrazides under physiological pH21 (Figure S-1A). Therefore, in this work, we have adopted DMTMM as the activating agent for DHSO cross-linking of acidic residues. Similar to lysine-reactive cross-linkers, DHSO cross-linking would result in the formation of three types of cross-linked peptides: dead-end (type 0), intralink (type 1), and interlink (type 2) modified peptides, among which interlinked peptides provide the most informative data on the relative spatial orientation of cross-linked acidic residues.24 Since all of the MS-cleavable, homobifunctional NHS esters we have previously developed display the same characteristic fragmentation patterns in MS2 due to the cleavage of either of the two symmetric CID-cleavable C–S bonds adjacent to the sulfoxide functional group,14−16 we expect that DHSO cross-linked peptides will behave similarly during MSn analysis even though their residue-targeting functional groups are different.
To elaborate this process, Figure 1E and Figure S-1B,C illustrate the predicted MS2 fragmentation patterns of DHSO interlinked, intralinked, and dead-end modified peptides, respectively. For a DHSO interlinked peptide α-β, the cleavage of one of the two symmetric C–S bonds would result in one of the two predicted peptide fragment pairs (i.e., αA/βS or αS/βA). The resulting α and β peptide fragments are modified by complementary cross-linker remnant moieties, i.e., alkene (A) or sulfenic acid (S). However, the sulfenic acid moiety can undergo dehydration to become a more stable unsaturated thiol moiety (i.e., T) (Figure S-1D). This conversion has been commonly observed in amine-reactive, sulfoxide-containing MS-cleavable cross-linked peptides, thus leading to the detection of αA/βT and αT/βA pairs instead as the four dominant MS2 fragment ions.14−16 Therefore, these two MS2 fragment pairs (i.e., αA/βT and αT/βA) are expected for a DHSO cross-linked heterodimer as well (Figure 1E), which can then be subjected to MS3 analysis for unambiguous identification of cross-linked peptide sequences and cross-linking sites. For a DHSO intralinked peptide αintra in which proximal D or E amino acid residues are cross-linked within the same peptide, one peptide fragment (i.e., αA+T) is expected in MS2 analysis (Figure S-1B). In reality, this particular ion would represent two populations of ion species that have identical peptide sequences and m/z values but transposed DHSO remnant-modified acidic residues. Lastly, a DHSO dead-end modified peptide αDN would potentially fragment into two ion species during MS2 analysis. Depending on the position of the cleaved C–S bond, αA or αT fragments would be observed, resulting in a pair of daughter ions detected during MS2 (Figure S-1C). The distinct MS2 fragmentation patterns of sulfoxide-containing MS-cleavable cross-linked peptides result in predictable mass relationships between parent ions and their respective fragments. These mass relationships are utilized as an additional verification of cross-linked peptide identification at the MS2 level. Along with mass fingerprinting by MS1 and peptide sequencing by MS3, three lines of evidence can be obtained and integrated to accurately identify DHSO cross-linked peptides using the identical MSn workflow that has been developed for the analysis of DSSO, DMDSSO, and DSBSO cross-linked peptides.14−16
Characterization of DHSO Cross-Linked Model Peptides by MSn Analysis
Despite the similarities in spacer arm structure to DSSO, it is necessary to verify whether DHSO cross-linked peptides indeed fragment as described above during MSn analysis (Figure 1E). Initial characterization of DHSO was performed on a synthetic peptide containing a single acidic residue, Ac-SR8 (Ac-SAKAYEHR). Interlinked Ac-SR8 homodimer was detected as quadruply charged (m/z 548.76234+) and quintuply charged (m/z 439.21175+) ion species, respectively. Since the two peptide sequences of interlinked homodimer are the same, only one pair of MS2 fragment ions (i.e., αA/αT) would be expected. Indeed, MS2 analysis of the quadruply charged parent ion produced a pair of dominant fragment ions αA/αT (m/z 536.272+/552.262+), demonstrating effective physical separation of the two cross-linked peptides as expected (Figure 2A). Similarly, MS2 analysis of the quintuply charged parent ion (m/z 439.21175+) yielded a single pair of dominant fragment ions αA/αT (m/z 357.853+/552.262+) as well (Figure 2B), demonstrating the characteristic fragmentation independent of peptide charges as expected. Subsequent MS3 analysis of the αA (m/z 536.272+) fragment ion (Figure 2C) resulted in a series of y and b ions that unambiguously confirmed the peptide sequence as Ac-SAKAYEAHR in which the glutamic acid was modified with a DHSO alkene (A) moiety. Similarly, MS3 analysis of the αT fragment (m/z 552.262+) determined its identity as Ac-SAKAYETHR, in which the glutamic acid was modified with a DHSO unsaturated thiol (T) moiety (Figure 2D). Therefore, the cross-linked peptide was identified as [Ac-SAKAYE6HR] interlinked to [Ac-SAKAYE6HR] through E6 in both peptides. This result indicates that DHSO interlinked peptides indeed display the same characteristic MSn fragmentation as sulfoxide-containing lysine interlinked peptides and can be analyzed using the same data analysis workflow as previously described.14−16
Figure 2.

MSn analysis of DHSO interlinked Ac-SR8 homodimer. MS2 spectra of DHSO interlinked Ac-SR8 at two different charge states: (A) [α-α]4+ (m/z 548.76234+) and (B) [α-α]5+ (m/z 439.21175+). MS3 spectra of MS2 fragment ions detected in part A: (C) αA (m/z 536.272+) and (D) βT (m/z 552.262+).
Characterization of DHSO Cross-Linked Model Proteins by MSn Analysis
To evaluate the capability of DHSO for protein cross-linking in vitro, we used equine myoglobin and bovine serum albumin (BSA) as our model proteins. These two proteins contain above-average acidic residue content (16.3% and 13.6%, respectively), making them well suited for evaluating DHSO cross-linking. In addition, BSA was employed previously for acidic residue cross-linking by noncleavable dihydrazides.21 To identify DHSO cross-linked peptides in myoglobin and BSA, we have performed in-gel digestion of gel-separated DHSO cross-linked proteins or in solution digestion of DHSO cross-linked proteins followed by peptide SEC as illustrated (Figure S-2). The resulting peptides were subjected to LC–MSn analysis. Figure 3A displays the MS1 spectrum of an exemplary interlinked peptide (α-β) (m/z 517.27035+) identified from myoglobin. Its MS2 analysis resulted in the detection of two peptide fragment pairs, i.e., αA/βT (m/z 429.742+/569.633+) and αT/βA (m/z 445.722+/559.643+) (Figure 3B), characteristic for DHSO interlinked heterodimers. MS3 analysis of αA (m/z 429.742+) (Figure 3C) determined its sequence as ASEADLKK, in which the glutamic acid residue at the third position from the N-terminus was modified with an alkene moiety. MS3 analysis of βT (m/z 569.633+) identified its sequence as VEADTIAGHGQEVLIR, with the aspartic acid residue at the fourth position from the N-terminus carrying an unsaturated thiol moiety (Figure 3D). Collectively, the interlinked peptide was unambiguously identified as [18VEADTIAGHGQEVLIR32 cross-linked to 58ASEADLKK64], describing an interlink formed between D21 and E60 of equine myoglobin.
Figure 3.

MSn analysis of a representative DHSO interlinked myoglobin peptide. (A) MS spectrum of the interlinked peptide α-β (m/z 517.27035+). (B) MS2 spectrum of the interlinked peptide detected in part A. MS3 spectra of MS2 fragment ions: (C) αA (m/z 429.742+) and (D) βT (m/z 569.633+).
Figure S-3 displays MSn analysis of a representative DHSO interlinked BSA peptide, which was measured as a quadruply charged ion (m/z 692.84754+) in MS1 (Figure S-3A). Its MS2 spectrum revealed two pairs of complementary MS2 fragment ions, i.e., αA/βT and αT/βA (Figure S-3B), further demonstrating the robust fragmentation expected of DHSO interlinked peptides. Together with MS3 sequencing of MS2 fragments αA (m/z 616.322+) and βT (m/z 760.362+) (Figure S-3C,D), this DHSO interlinked peptide was unambiguously identified as [66LVNEALTEFAK75 interlinked to 89SLHTLFGDETLCK100], in which residue E69 cross-linked to residue E97 in BSA.
In addition to interlinked peptides, intralinked peptides were also observed as a result of DHSO cross-linking of our model proteins. For example, MS2 fragmentation of an intralinked myoglobin peptide (Figure S-4) produced a single fragment ion peak αA+T (m/z 514.024+) 18 Da less than its parent ion, consistent with the expected fragmentation pattern described in Figure S-1B following dehydration of the sulfenic acid moiety to an unsaturated thiol moiety. Analysis of the αA+T ion in subsequent MS3 analysis (Figure S-4C) yielded a series of b and y ions permitting the unambiguous identification of two peptides sharing identical sequences but transposed alkene and unsaturated thiol moieties: 105YLEAFISDTAIIHVLHSK119 and 05YLETFISDAAIIHVLHSK119, indicating an intralink between residues E106 and D110.
MS2 fragmentation of a myoglobin dead-end modified peptide (m/z 604.30953+) resulted in the detection of a single pair of fragment ions αA/αT (m/z 559.303+/569.963+) (Figure S-5), consistent with the expected fragmentation pattern described in Figure S-1C. These fragment ions were then identified by MS3 analysis as 18VEAADIAGHGQEVLIR32 and 18VETADIAGHGQEVLIR32, respectively, representing a dead-end cross-link located on E19 of myoglobin (Figure S-5C,D).
In total, LC–MSn analysis of DHSO cross-linked myoglobin identified 33 unique interlinked peptides, representing 32 unique D|E-D|E linkages (Table S-1). Similarly, 62 unique DHSO interlinked BSA peptides were identified, describing 69 unique D|E-D|E linkages (Table S-2). Collectively, the results presented thus far indicate that DHSO can effectively cross-link acidic residue containing peptides and proteins in the presence of DMTMM at neutral pH. More importantly, our results have demonstrated that DHSO cross-linked peptides indeed exhibit the same characteristic MS2 fragmentation patterns as expected to allow their facile and accurate identification.
DHSO Cross-Linking Maps of Myoglobin and BSA
In order to assess the efficacy and sequence coverage of DHSO cross-linking on our model proteins, we generated cross-linking maps of myoglobin and BSA based on their identified DHSO interlinked peptides. The secondary structures of equine myoglobin comprise of eight α-helices and one short 310 helix (PDB 1DWR) (Figure 4A). The globular nature of myoglobin suggests that many of the helices are in close proximity to one another in three-dimensional space. The DHSO cross-link map of myoglobin based on the 33 unique D|E-D|E linkages is illustrated in Figure 4B, describing numerous intra- and inter-secondary structure interactions (i.e., α1-α5, α1-α8, α2-α4, α3-α4, α3-α8, α4-α5, α4-α8, α6-α8, α7-α8, and α8-α8). To further evaluate the identified cross-links, we mapped the cross-linked residues onto the crystal structure of myoglobin and calculated the distances between their alpha carbons (Cα-Cα distances) (Figure 4D,F). Considering the spacer length of DHSO (12.4 Å) and the distances contributed by D|E side chains (3.8 Å|4.9 Å, respectively), as well as backbone flexibility and structural dynamics, the upper limit for the Cα-Cα distances between DHSO cross-linked acidic residues is estimated to be ∼30 Å. Therefore, we have set the distance threshold for cross-linkable D|E residues as 30 Å. A total of 27 of the 32 myoglobin DHSO cross-links were mapped in the structure, with 26 having Cα-Cα distances <30 Å and one link slightly over the maximum distance at 31.1 Å. The remaining 5 linkages were not mapped on to the structure because they were identified as sites of oligomerization, in which identical residues or peptide sequences were cross-linked together.
Figure 4.

Myoglobin cross-link maps. (A) Myoglobin linear sequence showing locations of the 8 α-helices (blue) and 310 helix (yellow). (B) DHSO cross-link map on myoglobin linear sequence. (C) DSSO cross-link map on myoglobin linear sequence. (D) DHSO cross-link map on myoglobin crystal structure (PDB 1DWR). (E) DSSO cross-link map on myoglobin crystal structure (PDB 1DWR). (F) The distribution plot of identified linkages vs their spatial distances of D|E-D|E for DHSO (red) or K–K for DSSO (blue) in myoglobin structure.
Similarly, a DHSO cross-link map of BSA was generated based on the 69 unique D|E-D|E linkages (Figure 5A). When mapped to a previously published BSA crystal structure (PDB 4F5S), 64 out of 69 BSA linkages (93%) were calculated to have Cα-Cα distances below 30 Å (Figure S-6A,C). Structural flexibility and/or oligomerization of BSA likely contribute to the other five identified linkages found to be >30 Å. As shown in Figure 5A, DHSO interlinks were distributed throughout the primary sequence of BSA, with regions of dense cross-link clusters identified in regions with higher α-helix density. This even distribution is likely due to the dispersion of aspartic acid and glutamic acid residues throughout the protein. Collectively, our results suggest that DHSO cross-linking yields cross-links within expected distance constraints useful for structural elucidation for computational modeling in the same way as lysine cross-linked data.
Figure 5.
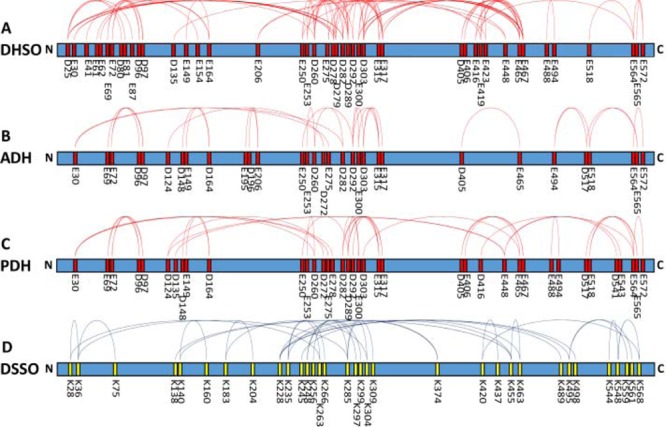
BSA cross-link maps on its linear sequence: (A) DHSO cross-link map, (B) ADH cross-link map, (C) PDH cross-link map, and (D) DSSO cross-link map. Note: ADH and PDH cross-link maps are generated based on data obtained by Leitner et al.21
Comparison of MS-Cleavable and Noncleavable Acidic Residue Cross-Linking
Previously, two noncleavable acidic residue cross-linkers, i.e., adipic acid dihydrazide (ADH) and pimetic acid dihydrazide (PDH), were used for probing the structure of bovine serum albumin,21 which resulted in the identification of 27 and 35 unique acidic residue linkages, respectively.21 A comparison of the linkage maps generated for DHSO, ADH, and PDH cross-linking of BSA (Figure 5A–C) revealed a high degree of similarity in proximally cross-linked regions. Apart from covering interaction regions cross-linked by ADH and PDH, DHSO cross-linking resulted in 34 additional unique D|E-D|E linkages. These unique DHSO cross-links are generally clustered in regions of particularly high acidic residue density, such as the regions between D25 and D97, E250 and E344, and D405 to E494 (Figure 5A–C). Limitations in bioinformatics software for analyzing noncleavable cross-linked peptides have been previously noted,21 which made the accurate identification of acidic residue cross-linked peptides considerably more challenging due to their higher frequency and corresponding increase in search space. In contrast, CID induced cleavage of DHSO cross-linked peptides during MS2 significantly simplified subsequent peptide sequencing in MS3. Given the same acidic residue reactive chemistry, the increase in identified cross-links using DHSO is mainly attributed to the simplified cross-link identification with improved accuracy afforded by MS-cleavability of DHSO cross-linked peptides. This ultimately facilitates unambiguous identification of individual linkages amidst peptides with multiple acidic residues in sequence. These results demonstrate the advantage of using DHSO, a MS-cleavable cross-linking reagent targeting acidic residues for probing protein–protein interactions over noncleavable reagents.
Comparison of DHSO and DSSO Cross-Linking
To assess the complementarity between acidic residue and primary amine cross-linking data, we examined the similarities and differences between DHSO and DSSO cross-linking of our selected model proteins. To this end, we also carried out LC–MSn analyses of DSSO cross-linked myoglobin and BSA, respectively. As summarized in Tables S-3 and S-4, 19 unique DSSO interlinked myoglobin peptides and 33 unique DSSO interlinked BSA peptides were identified. These linkages were then mapped onto their corresponding protein linear sequences (Figures 4C and 5D) and crystal structures (Figure 4E and Figure S-6B). As a result, all of myoglobin DSSO cross-links (Figure 4F) and 94% of BSA DSSO cross-links corresponded to Cα-Cα distances ≤30 Å (Figure S-6C). The two BSA cross-links that are outside the distance range may be a result of unexpected structural flexibility.
In the case of myoglobin, DSSO cross-linking identified several proximal helicase regions, such as α4-α5, α4-α7, α5-α8, α6-α8, and α5-310. In comparison, there is limited overlap between DHSO and DSSO cross-link maps except in regions containing α4-α5 and α6-α8 (Figure 4B,C), indicating that DHSO and DSSO cross-linking mapped different parts of interactions within myoglobin. The identified helicase interacting regions unique to DHSO or DSSO cross-linking correspond well with the number of cross-linkable residues and specific reactive chemistries. This is due to the fact that lysine and acidic residues are distributed unevenly across the myoglobin sequence. For example, the N-terminal region of myoglobin (residues 1–41) spanning helices α1 through α3 contains only one lysine but four glutamic acids and two aspartic acid residues. Therefore, profiling the interactions of the N-terminus within itself and with other parts of the protein will be difficult with amine-reactive cross-linking reagent such as DSSO. In contrast, acidic residue reactive cross-linker DHSO would be better suited for this purpose. Indeed, while DSSO was not able to cover this region as expected, DHSO cross-linking enabled the identification of 11 interlinked peptides describing multiple interactions between the N-terminus and other parts of the protein (i.e., α1-α5, α1-α8, α2-α4, α3-α4, and α3-α8). While DHSO provided exclusive data from the lysine scarce N-terminus, the lysine-rich 310 helix and many of the loop regions between the helical structures were better analyzed by DSSO due to the higher abundance of lysine residues in these regions. Together, these results demonstrate that acidic residue cross-linking can provide complementary structural information to that obtained using amine-reactive cross-linkers.
Interestingly, unlike myoglobin, DHSO and DSSO cross-linking of BSA have resulted in much more similar cross-linking profiles, meaning that similar interactions within BSA were identified (Figure 5A,D). This is most likely owing to the fact that BSA has more evenly dispersed distribution of lysine, aspartic acid, and glutamic acid residues throughout the protein sequence. Thus, combined usage of DHSO and DSSO can strengthen the validity of the cross-links identified by any of the two reagents individually. More importantly, this will generate complementary structural information to facilitate a more comprehensive understanding of protein structures.
Conclusion
Here we report the development and characterization of a new acidic residue-targeting, sulfoxide-containing MS-cleavable cross-linker, dihydrazide sulfoxide (DHSO), which is a new derivative of our previously developed amine-reactive MS-cleavable reagent, DSSO.14 Our analyses here have proven that DHSO cross-linked peptides possess the same characteristics distinctive to peptides cross-linked by sulfoxide-containing amine-reactive cross-linkers,14−16 thus permitting their fast and accurate identification by MSn analysis. The unique features of DHSO will significantly facilitate cross-linking studies targeting acidic residues, which has been difficult in the past due to the large number of D|E present in protein sequences and complexity of their resulting cross-linked peptides for MS analysis. Comparison of DHSO and DSSO cross-linking confirms the need of expanding the coverage of protein interactions using cross-linkers targeting different residues, especially when the distribution of specific amino acids is uneven. In summary, this work further demonstrates the robustness and potential of our XL-MS technology based on sulfoxide-containing MS-cleavable cross-linkers and provides a viable analytical platform for the development of new MS-cleavable cross-linker derivatives to further define protein–protein interactions. The development of these new tools will aid in the goal of understanding the structural dynamics of protein complexes at the global scale in the future.
Acknowledgments
We thank Drs. A. L. Burlingame and Robert Chalkley for the developmental version of Protein Prospector. This work was supported by National Institutes of Health Grants RO1GM074830 and RO1GM074830-10S2 to L.H. and Grant R01GM106003 to L.H. and S.D.R. E.J.N. was supported by an institutional Chemical and Structural Biology Training Grant predoctoral fellowship (Grant T32-GM10856).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b02240.
Supplemental methods, figures, and tables (PDF)
Author Contributions
§ C.B.G. and C.Y. contributed equally
The authors declare no competing financial interest.
Supplementary Material
References
- Sinz A. Mass Spectrom. Rev. 2006, 25, 663–682. 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- Walzthoeni T.; Leitner A.; Stengel F.; Aebersold R. Curr. Opin. Struct. Biol. 2013, 23, 252–260. 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Chen Z. A.; Jawhari A.; Fischer L.; Buchen C.; Tahir S.; Kamenski T.; Rasmussen M.; Lariviere L.; Bukowski-Wills J. C.; Nilges M.; Cramer P.; Rappsilber J. EMBO J. 2010, 29, 717–726. 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog F.; Kahraman A.; Boehringer D.; Mak R.; Bracher A.; Walzthoeni T.; Leitner A.; Beck M.; Hartl F. U.; Ban N.; Malmstrom L.; Aebersold R. Science 2012, 337, 1348–1352. 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- Kao A.; Randall A.; Yang Y.; Patel V. R.; Kandur W.; Guan S.; Rychnovsky S. D.; Baldi P.; Huang L. Mol. Cell. Proteomics 2012, 11, 1566–1577. 10.1074/mcp.M112.018374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger J. P.; Stengel F.; Pellarin R.; Zhang S.; Schaefer T.; Aylett C. H.; Cimermancic P.; Boehringer D.; Sali A.; Aebersold R.; Ban N. Cell 2014, 158, 1123–1135. 10.1016/j.cell.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Fernandez-Martinez J.; Tjioe E.; Pellarin R.; Kim S. J.; Williams R.; Schneidman D.; Sali A.; Rout M. P.; Chait B. T. Mol. Cell. Proteomics 2014, 13, 2927–2943. 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng-Elmore X.; Gao X. Z.; Pellarin R.; Schneidman-Duhovny D.; Zhang X. J.; Kozacka K. A.; Tang Y.; Sali A.; Chalkley R. J.; Cote R. H.; Chu F. J. Mol. Biol. 2014, 426, 3713. 10.1016/j.jmb.2014.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yu C.; Hu X.; Kim J. K.; Bierma J. C.; Jun H. I.; Rychnovsky S. D.; Huang L.; Qiao F. Cell Rep. 2015, 12, 2169–2180. 10.1016/j.celrep.2015.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Tanasova M.; Borhan B.; Reid G. E. Anal. Chem. 2008, 80, 9279–9287. 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Tang X.; Munske G. R.; Tolic N.; Anderson G. A.; Bruce J. E. Mol. Cell. Proteomics 2009, 8, 409–420. 10.1074/mcp.M800232-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M. Q.; Dreiocker F.; Ihling C. H.; Schafer M.; Sinz A. Anal. Chem. 2010, 82, 6958–6968. 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- Petrotchenko E. V.; Serpa J. J.; Borchers C. H. Mol. Cell. Proteomics 2011, 10, M110.001420. 10.1074/mcp.M110.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao A.; Chiu C. L.; Vellucci D.; Yang Y.; Patel V. R.; Guan S.; Randall A.; Baldi P.; Rychnovsky S. D.; Huang L. Mol. Cell. Proteomics 2011, 10, M110.002212. 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.; Kandur W.; Kao A.; Rychnovsky S.; Huang L. Anal. Chem. 2014, 86, 2099–2106. 10.1021/ac403636b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaake R. M.; Wang X.; Burke A.; Yu C.; Kandur W.; Yang Y.; Novtisky E. J.; Second T.; Duan J.; Kao A.; Guan S.; Vellucci D.; Rychnovsky S. D.; Huang L. Mol. Cell. Proteomics 2014, 13, 3533–3543. 10.1074/mcp.M114.042630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Rijkers D. T.; Post H.; Heck A. J. Nat. Methods 2015, 12, 1179–1184. 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Yu C.; Mao H.; Novitsky E. J.; Tang X.; Rychnovsky S. D.; Zheng N.; Huang L. Nat. Commun. 2015, 6, 10053. 10.1038/ncomms10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsom A.; Schneider M.; Fischer L.; Brock O.; Rappsilber J. Mol. Cell. Proteomics 2016, 15, 1105–1116. 10.1074/mcp.M115.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R.; et al. UniProt Consortium. Nucleic Acids Res. 2013, 41, D43–47. 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A.; Joachimiak L. A.; Unverdorben P.; Walzthoeni T.; Frydman J.; Forster F.; Aebersold R. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9455–9460. 10.1073/pnas.1320298111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak P.; Kruppa G. H. Eur. Mass Spectrom. 2008, 14, 355–365. 10.1255/ejms.963. [DOI] [PubMed] [Google Scholar]
- Wang X.; Chen C. F.; Baker P. R.; Chen P. L.; Kaiser P.; Huang L. Biochemistry 2007, 46, 3553–3565. 10.1021/bi061994u. [DOI] [PubMed] [Google Scholar]
- Schilling B.; Row R. H.; Gibson B. W.; Guo X.; Young M. M. J. Am. Soc. Mass Spectrom. 2003, 14, 834–850. 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.