

Summary
Over the past 20 years, there have been many advances in haemophilia treatment that have allowed patients to take greater control of their disease. However, the development of factor VIII (FVIII) inhibitors is the greatest complication of the disease and a challenge in the treatment of haemophilia making management of bleeding episodes difficult and surgical procedures very challenging. A meeting to discuss the unmet needs of haemophilia patients with inhibitors was held in Paris on 20 November 2014. Topics discussed were genetic and non-genetic risk factors for the development of inhibitors, immunological aspects of inhibitor development, FVIII products and inhibitor development, generation and functional properties of engineered antigen-specific T regulatory cells, suppression of immune responses to FVIII, prophylaxis in haemophilia patients with inhibitors, epitope mapping of FVIII inhibitors, current controversies in immune tolerance induction therapy, surgery in haemophilia patients with inhibitors and future perspectives for the treatment of haemophilia patients with inhibitors. A summary of the key points discussed is presented in this paper.
Keywords: FVIII products, genetics, haemophilia, inhibitors, prophylaxis, surgery
Introduction (Yesim Dargaud, France)
Over the past 20 years, advances in haemophilia treatment have allowed people with haemophilia to take greater control of their disease. However, the development of inhibitors complicates treatment, and inhibitor patients still have a range of unmet needs. Genotyping may provide individuals and their physicians with insight into bleeding severity, inhibitor risk and carrier status, which can help improve their knowledge of the disease and its management. Studies in molecular immunology have provided a greater understanding of the multifactorial causes of inhibitor development, and have examined the relevance of immunological danger signals. Recently, there has been increased interest in optimizing management of immune tolerance induction in patients with severe haemophilia A and inhibitors. Evidence is needed as to whether prophylaxis with bypassing agents can significantly delay/prevent the development of osteochondral changes in patients with inhibitors. Surgery in patients with haemophilia and inhibitors is still challenging even today. These and other issues concerning the unmet needs of haemophilia patients with inhibitors were discussed at a meeting in Paris on 20 November 2014 and a summary of the discussions is presented below.
Genetic and non-genetic risk factors for the development of inhibitory antibodies in patients with haemophilia (Anna Pavlova, Germany)
The development of FVIII inhibitors is the most severe and costly complication in the treatment of haemophilia A with an overall incidence of 20–30% [1]. The risk of development of inhibitors is associated with the severity of the disease. However, after seven decades, inhibitors are still a mystery. Back in the 1940s the first data emerged of FVIII inhibitors [2]; this discovery was followed in the 1980s–1990s with the cloning of the F8 gene and the detection and sequencing of mutations within the gene [3], as well as information on the link between human leukocyte antigen (HLA) type and inhibitor development [4]. In 2006 and 2009 reports were published on immune system polymorphisms and inhibitors [5,6]. With these discoveries, it is known that various factors influence inhibitor formation. These can be divided into modifiable and non-modifiable factors. The genetic factors are patient-related, non-modifiable and concern F8 gene mutation, family history, race or ethnicity, immune system, HLA type and polymorphisms in IL–10, TNFα, CTLA4 and HO–1. The environmental factors are non-patient-related and potentially modifiable, i.e. treatment regimen, intensity of treatment, whether treatment is prophylactic or on-demand, age at first treatment, type of concentrate used (recombinant or plasma-derived FVIII), and danger signals. The risk of developing FVIII antibodies is strongly related to the presence of stop mutations, large deletions and intrachromosomal recombinations [7]. A meta–analysis of 30 studies involving 5383 patients found that the inhibitor risk in patients with large deletions and nonsense mutations was higher than in those with intron 22 inversions (pooled odds ratio [OR] = 3.6, 95% confidence interval [95% CI], 2.3–5.7 and OR = 1.4, 95% CI, 1.1–1.8 respectively). The risk in those with intron 1 inversions and splice-site mutations was equal (pooled OR = 0.9; 95% CI, 0.6–1.5 and OR = 1.0; 95% CI, 0.6–1.5). In patients with small deletions/insertions and missense mutations the risk was lower (pooled OR = 0.5; 95% CI, 0.4–0.6 and OR = 0.3; 95% CI, 0.2–0.4 respectively). The relative risks (RR) for developing high-titre inhibitors were similar [8]. The general agreement is that null mutations such as nonsense mutations are associated with a high risk of developing inhibitors and non-null mutations such as missense mutations are associated with a low risk. In patients with non-severe haemophilia A, inhibitors are less frequently observed but when they do occur they can have a profound clinical impact as they may interact with endogenous FVIII to decrease the plasma FVIII level to under 1 IU dL−1.
The position and type of substitution of missense mutations may influence the risk of developing an inhibitor. The INSIGHT study analysed the association between F8 mutation and development of an inhibitor in 1112 patients with non-severe haemophilia A. Among a total of 214 different F8 missense mutations, 19 were associated with inhibitor development, of which the most prevalent were Arg593Cys (9.5%), Asn618Ser (5.2%), Arg2150His (5.1%) and Arg531Cys (3.2%) [9]. F8 genotyping could help to estimate an individual’s risk of inhibitor formation.
The Malmö International Brother Study (MIBS) highlighted the impact of family history in developing inhibitors. In this study, a 48% higher risk of developing an inhibitor was found in families with a history of inhibitors (95% CI 35–62%) [10]. Likewise the CANAL study found that the risk of developing inhibitors was 3-fold higher in these patients [11]. Race and ethnicity also affect inhibitor development: in the MIBS study a higher risk was found in patients of African origin compared to Caucasians (56 vs. 27% respectively) [10].
The presence of the H3 or H4 haplotype is also associated with a higher risk of inhibitor development compared to patients with the H1 and H2 haplotype (OR: 3.6) [12]. H1 and H2 are found in all racial groups but H3, H4 and H5 have only been found in black people.
Polymorphisms in IL–10 [13], TNFα [5] and CTLA4 [14] are associated with an increased risk of inhibitor development. The data describing the effect of HLA type on the development of inhibitors are contradictory: some studies show an increased risk while others show a decreased risk. Repessé et al. investigated the relationship between polymorphisms in the heme oxygenase–1-encoding gene (HMOX1). They found that inhibitor-positive patients had a higher frequency of alleles with large (GT) n repeats (L: n ≥ 30), meaning lesser HO–1 expression [15]. In the Hemophilia Inhibitor Genetics Study (HIGS), 53 single-nucleotide polymorphisms were found to be significant predictors of inhibitor status [16]. How these polymorphisms predispose to inhibitor development is still not clear. The evidence for an association between HLA, single nucleotide polymorphisms (SNPs) in the cytokine genes and the formation of FVIII inhibitors derives mainly from small case series and uncontrolled studies. The association also varies significantly because different ethnic groups from different geographic regions have been investigated. The presence of circulating inhibitors is the result of a complex interaction between many immune partners providing positive or negative signals affecting the production of inhibitors.
Environmental factors also affect inhibitor development. Inhibitors typically develop during the first 20–50 exposure days (EDs) and decrease to less than 1% after this time [11].
Early treatment increases the risk of inhibitor formation but this association might be explained by intensity of treatment [11]. Peak treatment moments may trigger inhibitor formation and have been considered the most significant determinant of inhibitor development [11]. Regular prophylaxis is associated with a 60% decreased risk of inhibitor development compared with on-demand treatment (RR: 0.4) [11]. However, the type of prophylaxis also affects inhibitor development. In a study, standard prophylaxis started at or after the first joint or other severe bleed, often using a Port-A-Cath, led to the formation of inhibitors in 47% of patients, compared to only 3.8% in patients given a low-dose prophylactic regimen started at manifested bleeding tendency, with no long or intensive treatment and without a Port-A-Cath[17].
The presence of danger signals (severe bleeds, trauma, surgery), associated with the use of high-dose FVIII and/or prolonged treatment leads to up-regulation of the cellular T and B cell lymphocyte response and an increased risk of inhibitor development. In contrast, the absence of danger signals is associated with a lower dose of antigen, regular prophylaxis and a decreased risk of inhibitor development [11].
With regard to the type of concentrate used, i.e. recombinant or plasma-derived, the data are conflicting and controversial. In the CANAL study, switching between FVIII products (recombinant or plasma-derived) did not increase the risk for inhibitors over the first 50 exposure days (RR: 1.1; CI: 0.6–1.8) [18] (Fig. 1). However, Chalmers et al. found that inhibitors developed more frequently in those initially treated with recombinant when compared with plasma-derived FVIII (P = 0.006) [19].
Fig. 1.
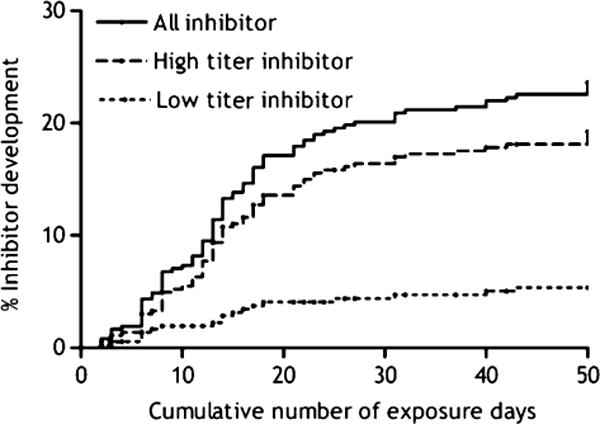
Cumulative incidence of inhibitor development: all inhibitors, and high- and low-titre inhibitors [11].
The aim of haemophilia treaters in minimizing inhibitor development is to identify a patient’s risk profile and use tailored treatment on an individual basis to reduce the risk of inhibitor development. It is known that inhibitor development is affected by different factors, but there may be other ones currently unknown that influence the risk.
Immunological aspects of inhibitor development (Sébastien Lacroix-Desmazes, France)
In patients with haemophilia A, the administration of therapeutic FVIII can lead to the development of anti-FVIII neutralizing (inhibitors) or non-neutralizing antibodies. Moreau et al. analysed the properties of anti-FVIII inhibitors from the IgG fraction of the plasma of healthy individuals and non-responder patients with haemophilia A. They found that the anti-FVIII antibodies neutralized FVIII activity and bound to FVIII with an affinity similar to that of anti-FVIII IgG affinity-purified from the plasma samples of haemophilia patients with inhibitor and individuals with anti-FVIII autoantibodies. The authors then further investigated the domain specificity of the antibodies by immunoprecipitation using radiolabelled FVIII, FVIII light chain, and the A1, A2 and C2 domains of FVIII. Results showed that anti-FVIII IgG which had been affinity-purifed from IVIg exhibited a higher titre of immunoprecipitating antibodies to the light chain of FVIII and the C2 domain than to intact FVIII and to the A2 domain. These observations suggest that FVIII inhibitors occurring in severe haemophilia and in patients with anti-FVIII autoimmune disease originate from the expansion of preexisting natural anti-FVIII clones with neutralizing properties [20]. Another study by Rossi et al. [21] found that anti-FVIII activity was inhibited by two different preparations of IVIg in the plasma of three of four patients with autoantibodies and two of three patients with alloantibodies, indicating that IVIg contains anti-idiotypes against anti-FVIII antibodies. FVIII-specific CD4+ T cells induce anti-FVIII antibody synthesis [22]. In a study, investigating the TCR (T cell receptor) repertoire of CD4+ T cells in patients with haemophilia and healthy controls, overlapping synthetic peptides, spanning the sequence of the FVIII A3 domain, were used to challenge blood CD4+ T cells in proliferation assays. The epitopes that were recognized in haemophilia A patients with or without inhibitors, acquired haemophilia patients, or healthy subjects overlapped, but had characteristic differences. Most members of one or more study groups recognized the sequence regions 1691–1710, 1801–1820, 1831–1850, and 1941–1960; however, haemophilia A patients with inhibitors recognized prominently only the sequence 1801–1820, which overlaps a known inhibitor binding site. It is possible that CD4+ T cells recognizing epitopes within residues 1801–1820 have a role in inducing inhibitor synthesis [23]. The depletion of natural CD4+/CD25high regulatory T cells may enhance or uncover FVIII-specific T cell responses in healthy individuals [24]. At the humoral level, tolerance to FVIII under physiological conditions relies on an equilibrium between the recognition of FVIII by naturally occurring potentially inhibitory anti-FVIII antibodies and their control by neutralizing anti-idiotypic antibodies. Neutralizing anti-idiotypic antibodies may also regulate the B cell clones that secrete the FVIII-specific antibodies [25].
There are several steps of immune system activation by FVIII. Initially, there is internalization of FVIII by antigen-presenting cells. This is followed by ‘immunogenic’ presentation of FVIII to naïve CD4+ T cells, and then by activation and differentiation of FVIII-specific naïve B cells. Finally, therapeutic FVIII is inhibited by anti-FVIII antibodies. Bleeding has been proposed as a risk factor for the development of FVIII inhibitors, with surgery, intensity of treatment and severe bleeds being rated highly as risk factors for inhibitor development [26].
Recurrent haemarthrosis leads to chronic synovitis and destructive arthropathy. In a murine model of haemarthrosis, the knee joint capsule of mice deficient in FVIII or IX and non-haemophilic wild type mice was punctured to induce a one-time, but massive haemorrhage. The puncture resulted in acute haemarthrosis in both types of haemophilic mice but not in wild type mice [27]. Further studies by Øvlisen et al. showed that the synovial fluid contained increased levels of the pro-inflammatory cytokines: IL–1 beta, IL–6, keratinocyte-derived chemokine (KC) and monocyte chemotactic protein–1 (MCP–1) [28]. It was suggested that these might in turn contribute to further progression of the inflammatory process. However, a recent study by Peyron et al. found that haemarthrosis and arthropathy do not lead to the development of FVIII inhibitors in severe haemophilia A mice [29]. In their studies, the levels of anti-FVIII IgG were similar in un-injured mice and those treated 1 or 14 days after injury (Fig. 2).
Fig. 2.
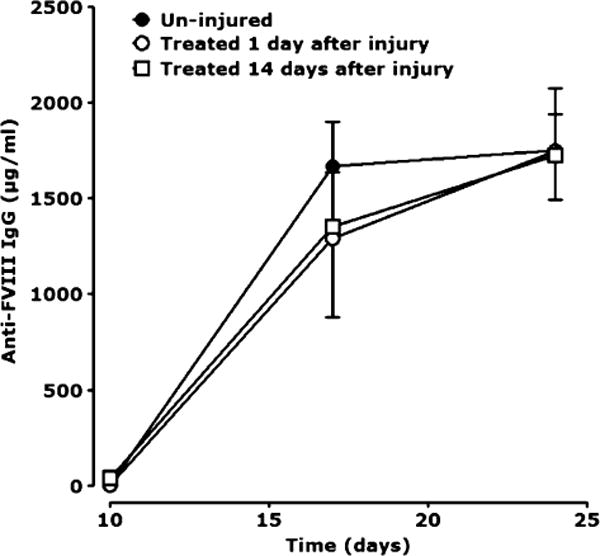
Levels of anti-FVIII IgG in un-injured mice and those treated 1 or 14 days after injury. From Peyron et al. Haemophilia 2015 [29].
Various receptors on scavenger cells play an important role in the endocytosis and degradation of FVIII. These are the low-density lipoprotein receptor-related protein (LRP, CD91) [30], the process of which is facilitated by cell surface heparan sulfate proteoglycans (HSPGs) [31], which acts in concert with low-density lipoprotein (LDL) receptor to regulate plasma levels of FVIII in vivo [32]. In addition, asparagine-linked oligosaccharide structures of the FVIII B domain recognize the carbohydrate recognition domains of asialoglycoprotein receptor (ASGPR) and an asialo–orosomucoid (ASOR)-sensitive mechanism, most likely ASGPR, contributes to the catabolism of coagulation FVIII in vivo [33]. In a study investigating the involvement of CD91 in FVIII endocytosis by human dendritic cells (DC), a model of professional antigen-presenting cells, it was found that CD91 and other members of the LDL receptor family are not strongly implicated in FVIII internalization by human monocyte-derived DC [34]. Results also showed that when DC from MHC-matched donors were incubated with a human FVIII-specific CD4+ T cell clone, D9E9 in the presence of increasing amounts of FVIII, D9E9 was activated in a dose-dependent manner. Another study showed that the entry of FVIII into human DCs leading to T cell activation is mediated by mannose-terminating glycans on FVIII. Mannose-terminating glycans have been identified as pathogen-associated molecular patterns that are essential for internalization of microbes by antigen-presenting cells, leading to presentation. The study also identified macrophage mannose receptor (CD206) as a candidate endocytic receptor for FVIII on DC [35].
Studies have been conducted in mice to investigate where therapeutic FVIII is distributed to in the body after administration. It has been found that FVIII preferentially accumulates in the spleen at the level of metallophilic macrophages in the marginal zone [36]. Surgical removal of the spleen resulted in a drastic reduction in anti-FVIII immune response.
FVIII products and inhibitor development in haemophilia A
European monitoring of inhibitor development in haemophilia A: 4-year results of EUHASS (Kathelijn Fischer, The Netherlands)
In patients treated with factor concentrates, alloantibodies to FVIII (inhibitors) can occur. Pharmacovigilance, i.e. the detection, monitoring and investigation of such adverse drug reactions, is performed either through voluntary reporting by health professionals and patients to the regulatory authorities, or by the product’s manufacturers. Inherited bleeding disorders such as haemophilia are rare, so a pharmacovigilance programme to monitor inhibitor development and other adverse events needs to be multicentric, probably multinational, simple and comprehensive. It was against this background that the European Haemophilia Safety Survey (EUHASS) was set up. EUHASS is a European, prospective multicentre surveillance system in the field of inherited bleeding disorders that began on 1 October 2008 [37]. Data include adverse events, inhibitors (reported at least quarterly), and patients at risk – previously untreated patients (PUPs) reaching 50 exposure days without inhibitors, and previously treated patients (PTPs) treated with concentrates in the last year. Between October 2008 and January 2013, 74 centres submitted data, with a mean of 75 patients with severe haemophilia per centre (IQR [interquartile range]: 34–131; range 7–428). In PUPs with severe haemophilia A, there were 108 patients with inhibitors out of a total of 417, representing an incidence of 26%. For haemophilia B, there were five cases of inhibitors out of a total of 72 patients (7%). There were no significant differences in developing inhibitors regarding the type of FVIII product used. This is in contrast to previous reports: Gouw et al. reported that although recombinant and plasma-derived FVIII products conferred similar risks of inhibitor development, second-generation full-length products were associated with an increased risk in patients with severe haemophilia A, as compared with third-generation products. The baseline risk was 32% [38]. A UK study by Collins et al. found that of 128 patients treated with Kogenate Bayer/Helixate NexGen, 45 (35.2%, 95% CI: 27.4–43.8) developed an inhibitor compared with 42/172 (24.4%, 95% CI: 18.6–31.4) with Advate (P = 0.04) [39]; overall baseline risk was 29%. Reasons for the differences found between these studies and EUHASS could include different data collection and analysis (although unlikely to explain 100% of the findings), and centre characteristics, such as different treatment regimens or concentrate selection (but also unlikely to be an explanation). Previously, mathematical calculus and simulation of data were applied to estimate whether the cumulative incidence of inhibitors is biased or flawed in EUHASS. It was found that for existing concentrates, both longitudinal cohort study methods and the EUHASS method yielded similar estimates of the cumulative incidence of inhibitor cases over a 5-year time period. For a newly introduced concentrate, however, a reliable estimate of inhibitor incidence with the EUHASS method could only be made after 3–4 years, even in large datasets [40]. The choice of concentrate may bias the assessment of inhibitor incidence. In a study by Kreuz et al., the safety and efficacy of a full-length sucrose-formulated recombinant FVIII product (rFVIII–FS; Kogenate FS; Kogenate Bayer) was evaluated in PUPs and minimally treated patients with severe haemophilia A. The incidence of FVIII inhibitor was 15% (9/60 patients) [41]. In view of these different results, should Kogenate/Helixate be prescribed to high-risk patients? There is no explanation for the results found in Kreuz et al.’s [39] study – all analyses in the RODIN [38], France Coag [42] and UKHCDO studies were adjusted for additional risk factors. In a comparison of inhibitor incidence associated with the use of concentrates used in non-RODIN vs. RODIN centres, the only significant difference was in the use of Advate, which was used in 30% of patients treated in non-RODIN centres compared with 50% of patients treated in RODIN centres [43]. In a comparison of rFVIII products in severe PTPs, no increase in inhibitor incidence was found for Kogenate/Helixate.
The next steps are to perform a pooled analysis of the results from the RODIN, FranceCoag and UKHCDO studies (this will be performed by the European Medicines Agency [EMA]), and combine these data with the EUHASS data (univariate analysis only). To definitively answer the question regarding the incidence of inhibitors according to rFVIII concentrate, it may be necessary to perform a randomized controlled trial, randomizing patients to Kogenate/Helixate or Advate.
Surveillance for inhibitors in the United States (Mike Soucie, USA)
The US Food and Drug Administration’s (FDA) Med-Watch is their safety information and adverse event reporting programme. It is a passive system that relies on voluntary reporting, does not collect denominator data, and inhibitors are an ‘expected’ adverse event in patients given FVIII concentrates. One of the major challenges facing scientists who work on rare disorders, such as haemophilia, is the lack of uniform health data. To address this issue and to advance health research, the Centers for Disease Control and Prevention (CDC) created a national public health surveillance project in 1998 called the Universal Data Collection (UDC) system. UDC was carried out with the help of federally funded haemophilia treatment centres (HTCs) in the USA and its territories. UDC collected data on the results of local inhibitor testing, however, local testing methods vary by site. Also, the UDC had limited ability to assess prevalence and no data were collected on new-onset inhibitors.
The Hemophilia Inhibitor Research Study (HIRS) was initiated in 2006 and was designed as a pilot project for national inhibitor surveillance to comply with FDA and European Medicines Agency (EMA) recommendations. The goals were to determine the feasibility of methods (test and exposure data), identify the population at risk and to characterize the risk factors for inhibitors (genetic, treatment-related and interactions). Prospective treatment data were collected on 1163 patients, including 129 inhibitor cases, from 17 sites. Patients were followed up for 3329 person-years, with infusion records for 113,205 exposure days. A total of 3048 inhibitor tests have been performed and 23 new FVIII inhibitors were identified (nine at enrollment and 14 during follow-up) [44]. Key findings of the study are that centralized laboratory testing is feasible, and the population at risk for inhibitors includes all patients. One-third of new inhibitors were in non-severe patients and one-half were in children aged over 5 years. In 61% of the patients who developed the 23 new FVIII inhibitors, there were no clinical effects at detection.
In a HIRS investigation of inhibitor frequency in USA patients with severe haemophilia A, the link between mutation type and inhibitor risk was confirmed in this population [45]. The study also confirmed the two-fold higher incidence of inhibitors in USA black and Hispanic patients – see Table 1.
Table 1.
Inhibitor frequency in USA haemophilia patients according to race [45].
| Race/ethnicity | Inhibitor | Frequency |
|---|---|---|
| White | 63/321 | 19.6% |
| Black | 13/35 | 37.1% |
| Hispanic | 15/32 | 46.9% |
| P = 0.0003 |
Lessons learned from HIRS are that national inhibitor surveillance should include:
Centralized inhibitor testing (modified Nijmegen– Bethesda assay [NBA] method, which allows testing of infused patients. Positive results should be confirmed by repeat specimens, and low-titre inhibitors should be tested with more specific assays)
Incident case surveillance (product exposure and environmental risk factor data collected retrospectively for 4 months prior to inhibitor detection)
Key findings from national surveillance would be: determination of prevalence and incidence of inhibitors in the USA, monitoring of trends in occurrence over time, and identification of clusters or ‘outbreaks’ of inhibitors. This is important as several new FVIII products will be launched in the next few years.
In December 2013, a new USA bleeding disorders surveillance system began, termed ‘Community Counts’, which builds on the work of the UDC project and collects information about common health issues, complications and causes of death among people with bleeding disorders receiving care at HTCs. It includes a standardized, centralized testing system with a standardized protocol specifying patients to be tested and the testing intervals. The protocol includes confirmatory testing and there is incident inhibitor case surveillance for newly identified cases.
Future plans are to disseminate CDC-developed screening methods to local and HTC laboratories. The data from screened patients will be used to assess genetic and environmental risk factors and to conduct epidemiological, laboratory and prevention research on risk factors for inhibitors. The aim is to support patient and provider education to promote best practises that prevent or eradicate inhibitors.
Recombinant and plasma-derived FVIII: what is the impact on the occurrence of inhibitors? (Ségolène Claeyssens, France)
In order to improve our knowledge of inhibitors large, international multicentre cohort series are needed, with well-defined age cohorts, well-defined inclusion criteria, data for all exposure days (EDs) (bleeding, product brand, etc. up to 50 EDs), data on gene mutations, intensive treatment and surgery, and a similar definition of outcome.
The Suivi Thérapeutique National des Hémophiles (SNH–National Therapeutic Survey of Haemophiliacs) was introduced in France in 1994. In 2003, it was replaced by the FranceCoag Network (Réseau France-Coag, RFC) project, a prospective cohort of patients suffering from inherited deficiencies of coagulating proteins. The main objective of the RFC is to obtain exhaustive information on the geographical distribution, the characteristics and the evolution of this patient population. This project also aims to create a pharmacosurveillance network allowing quick investigation in case of suspicion of an inhibitor or a new agent transmitted by FVIII treatments, and to monitor prophylaxis for feasibility, tolerance and impact on health. The RFC comprises a coordinating centre (CC) attached to the department of chronic diseases of the Institut de Veille Sanitaire (INvS). They confirm all new inhibitor cases and analyse data on risk factors of inhibitors. On 30 September 2014, the RFC contained data on 5165 patients with haemophilia A. The definition of the PUPs (previously untreated patients) cohort is a FVIII/IX level <2% and children born from 1 January 2000 for whom all the infusions are known. Follow-up is every 3 months until 150 EDs, then every year until the patient is 18 years old. Fixed data recorded include genetic mutation, family history of haemophilia and inhibitors, ethnicity and first treatment. Variable data include surgery, vaccinations and product switches. Of 741 boys with severe haemophilia A born between 1991 and 2013 screened for the RFC study, 303 were included in the analyses after exclusion of 50 patients who participated in the RODIN cohort. Inhibitor titres were measured locally using standard Bethesda or Nijmegen assays and an inhibitor was diagnosed if there had been a positive assay on any two dates. A clinically significant inhibitor was diagnosed in 114 boys (37.6%) after a median of 13 EDs (interquartile range [IQR], 8–19 EDs) [42]. A high-titre inhibitor was diagnosed in 63 boys (20.8%).
In contrast, the RODIN study of 574 patients [38] found that inhibitors developed in 177 children (cumulative incidence, 32.4%) of whom 116 patients had a high-titre inhibitory antibody (cumulative incidence, 22.4%). The median EDs were 15 (IQ: 10–20). Unexpectedly, the risk of inhibitor development was 60% higher among children receiving a second-generation full-length recombinant product than among those receiving a third-generation full-length product. This association may be a biased finding (through confounding, selection bias or information bias), a chance finding, or a causal effect.
In the UK PUP cohort study [39], the incidence of inhibitors in the 407 patients (including 88 patients in the RODIN cohort) in the main analysis was 118 (29%). Of the 118 inhibitors in the main analysis, 60 (14.7%) were high titre. Inhibitors developed after a median of 16 EDs (IQR: 9–30).
Thus, development of inhibitors in PUPs with severe haemophilia A is a multifactorial phenomenon. It is impossible to analyse the role of one factor without knowing the others. The risk of inhibitor development is perhaps not equal for the different brands of rFVIII. This may also be the case for plasma-derived FVIII and could explain part of the previous contradictory results published to date.
The results of the RFC PUPs cohort who received plasma-derived FVIII product are awaited. The size of this cohort—about 110, the use of a single plasmatic product, the analyses of almost every known risk factor except for immunological items are important endpoints to validate the quality of these results.
To summarize, the research into the differences between inhibitor development incidences in rFVIII and plasma-derived FVIII products is still relevant. The RFC PUPs cohort is a high quality tool to improve our knowledge of this major complication in the treatment of haemophilic patients.
Generation and functional properties of engineered antigen-specific human T regulatory cells: specific suppression of immune responses to FVIII (David Scott, USA)
Regulatory T cells (Tregs) have great potential in treating a range of diseases including haemophilia [46,47]. However, a problem with polyclonal Tregs is that they contain multiple specificities and may have non-specific immunosuppressive effects. Based on the pioneering work of June [48] and Eshhar [49] and their colleagues with chimeric antigen receptors (CARs), our group decided to create specific human regulatory cells using receptors constructed from specific clones. Thus, specific regulatory T cells have now been created using T cell receptors and single chain antibodies in our laboratory. The initial efforts involved the transduction of a recombinant T cell receptor obtained from a haemophilia A subject’s T cell clone into expanded human FoxP3+ Tregs [50,51]. These engineered FVIII-specific Tregs efficiently suppressed the proliferation and cytokine production of FVIII-specific T effector cells. Moreover, studies in mice demonstrated that antibody production from FVIII-primed spleen cells in vitro could be profoundly inhibited in the presence of these FVIII-specific Tregs [50,52]. When mock-transduced (irrelevant) human Tregs were added in excess, they increased the immune response to FVIII non-specifically but this effect could also be suppressed by the specific Tregs. These data suggest potential utility in the treatment of FVIII inhibitors in haemophilia A patients [50,52].
It is encouraging that an antibody response could be suppressed by targeting one epitope within FVIII. C2-specific Tregs can inhibit C2-specific helpers, but what about other epitopes in FVIII that recognize A2? The results provide indirect evidence that this bystander suppression can occur. Since FVIII has multiple epitopes presented by antigen-presenting cells, there could also be effector cells that recognize many different epitopes in FVIII [52]. To test whether bystander suppression to other unrelated epitopes could be achieved by C2-specific Tregs, T cell receptors from a patient with multiple sclerosis were cloned into a retroviral vector to create effectors recognizing a different antigen. Effector T cells, derived from these T cell receptors, specifically proliferated in the presence of myelin basic protein. In order to test bystander suppression for these unrelated epitopes, experiments were carried out with two different types of T cells: regulatory cells that were FVIII C2-specific and effector cells that were myelin basic protein specific. It was found that, in the presence of C2 peptide, the response to myelin basic protein was suppressed by C2-specific Tregs; however, the C2-specific Tregs did not suppress in the presence of ovalbumin (OVA). Thus, there is ‘specificity’ to bystander suppression.
To formally test this concept with the response to multiple FVIII epitopes, it is planned to use a novel CAR created from a single chain antibody prepared by Anja Naumann Schmidt and Christoph Königs in Frankfurt. They have isolated FVIII-specific single chain variable fragments (scFvs) from synthetic phage display libraries and characterized their FVIII domain specificity (e.g. A2) to develop a parallel approach [53]. Recent data in our laboratory suggests that CAR Tregs created from this scFv can suppress effector cells for FVIII (Schmidt AN, Kim YC, Heon JH, Königs C and Scott DW, in preparation).
When FVIII enters the spleen, for example, if Tregs were present they would suppress multiple antibodies targeting different epitopes. What if it were possible to take patients’ regulatory cells, expand them, put in an engineered T cell receptor or single chain antibody, expand these regulatory cells and then re-infuse them back into the patient? In the future this technique would hopefully suppress the antibody response to FVIII (see Fig. 3). These data suggest that the T-cell dependent antibody response in vitro can be suppressed, to prove this in vivo requires T cell receptor transduced Tregs from mice; these studies are in progress.
Fig. 3.
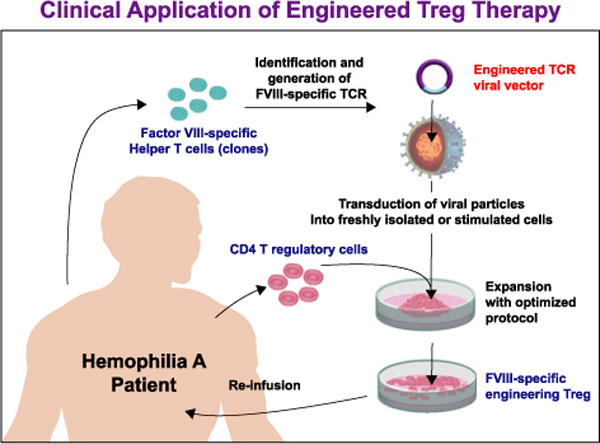
Clinical application of engineered Treg therapy.
In summary, antigen-specific human Tregs have been constructed that recognize FVIII epitopes and these cells specifically suppress FVIII effector cells. With the myelin basic protein cells, there is a possibility of not only looking at treatment of multiple sclerosis but proving that bystander suppression from multiple epitopes can be done using a T cell that is specific for even a single peptide in FVIII. This has great potential not only in haemophilia but in other monogenic diseases as well as autoimmunity.
Prophylaxis in haemophilia patients with inhibitors (Roseline d’Oiron, France)
In severe haemophilia patients without inhibitors, primary prophylaxis is the standard of care. Secondary prophylaxis may reduce and possibly arrest recurrent joint bleeding, avoid new target joint appearance, halt, or at least slow, the progression of joint destruction, improve quality of life (QoL) and reduce the risk of other serious haemorrhages, such as intracranial bleeding. The same goals should apply similarly to haemophilia patients with inhibitors who have an increased morbidity and impaired QoL compared to non-inhibitor patients. For years, lacks of data, the short half-life of bypassing agents and cost have limited the use of prophylaxis in inhibitor patients. However, increasing evidence of efficacy in terms of reduction of number of bleeds and improved QoL has helped the concept to be expanded. In a meta–analysis of six studies (34 inhibitor patients) reporting prophylactic treatment with activated prothrombin complex concentrates (aPCC), Valentino found a 64% reduction (49–100) of the bleeding rate in 31 of the 33 patients in parallel to improved QoL [54]. However, the treatment regimen was not effective in preventing or halting joint deterioration in patients with significant joint disease prior to initiation of prophylaxis, whereas it appeared useful to prevent joint damage in previously unaffected joints.
In the retrospective, observational PRO–PACT study [55] of 86 inhibitor patients with at least one bleeding episode or one bleeding episode per month during the 6 months prestudy period and receiving secondary prophylaxis with recombinant activated FVII (rFVIIa) there was a 46 or 52% overall reduction of bleeding rate respectively. No adverse events were recorded. In addition, there was a reduced burden of disease with decreased duration and number of hospitalizations.
Three prospective trials have investigated the use of secondary prophylaxis in haemophilia patients with inhibitors, one with rFVIIa and two with aPCC [56–58]. The studies found a 45–72.5% reduction in bleeding rate and a 43–75% reduction in target joint bleeding episodes in patients who underwent secondary prophylaxis. In the PROOF study, the occurrence of new target joints was substantially lower in the prophylaxis arm than in the on-demand arm: there were seven new target joints in 5 (29.4%) out of 17 prophylaxis subjects (range per subject: 1–2) vs. 23 new target joints in 11 (57.9%) out of 19 on-demand subjects (range per subject: 1–6), Fig. 4 [58].
Fig. 4.
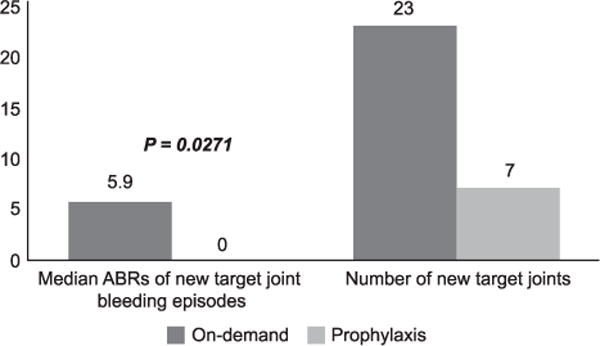
New target joints and associated bleeding episodes [58].
In the PROOF and Pro-FEIBA studies, a 61–73.8% reduction in joint bleeding rate was also found [57,58]. In the PROOF study, 12 of 16 (75%) subjects receiving prophylaxis were good responders (i. e. they had a ≥ 50% reduction in the number of bleeding episodes), compared to only two of 19 patients (10%) receiving on-demand therapy [58]. Two of 17 patients on prophylaxis had no bleed (one of the 17 patients later withdrew from the trial due to an adverse event). This compares with 16 of 26 patients (62%) who were good responders in the Pro-FEIBA study, six of whom had no bleeds [57]. QoL was also improved in patients on prophylaxis in all three studies, but only significantly for patients included in the Konkle et al. study and for good responders in the Pro-FEIBA study.
These three studies show a strong evidence of actual improvement of care for inhibitor patients given prophylaxis. However, prophylaxis in these patients is not as effective as prophylactic treatment in those without inhibitors. There were inferior outcomes compared to published retrospective cases, but the patients in the studies had a high incidence of bleeding. If secondary prophylaxis is given, prevention of progression of arthropathy is unlikely given the number of joint bleeds. No thrombotic events were reported in the studies, but only a few elderly patients were included. The question remains although, is a reduction of bleeding rate of 45–74% enough? An ‘acceptable’ annualized bleeding rate depends on the objective. For children, the major aim would be to avoid any joint bleeding and to preserve joint health, whereas in adults it would rather be to decrease the number of bleeds/target joints and to improve QoL and joint status. Efficacy and safety may be improved by tailoring prophylaxis, according to frequent and regular assessment of outcome measures such as physical, functional, radiological and QoL scores. The product, dose and frequency of bypassing therapy should be adjusted to find a regimen that is both effective and practical in parallel to other concurrent treatments such as analgesics and physiotherapy. Prophylaxis with bypassing agents is a major step forward in the care of haemophilia patients with inhibitors, but eradication of the inhibitor remains a priority. Finally, long-term cost–benefit analyses are needed.
Epitope mapping of anti-factor FVIII antibodies (Géraldine Lavigne-Lissaide, France)
FVIII is composed of a light chain containing the domains A3–C1–C2 and a heavy chain containing the domains A1–A2–B [59]. The acidic region of the light chain and the C2 domain are both directly involved in forming a high-affinity binding site for von Willebrand factor (VWF). Most inhibitory antibodies to human FVIII bind to epitopes in the A2, ap–A3, or C2 domains [60]. Various substances interact with FVIII at specific domains, such as FIXa, phospholipids and DCs. The activation of FVIII by thrombin requires cleavage of a peptide bond in the A2 domain after Arg372, and the removal of the B domain linked to the 41 residue N-terminal region of the A3 domain (the a3 acidic region) by thrombin cleavage after Arg1689 [61]. Since 1988 several epitope mapping studies of human FVIII inhibitor antibodies have been published identifying critical components of epitopes for the antibodies. These include FVIII Glu389, 390, 391 in the A2 domain [62], amino acid residues Gln1778–Met1823 in the A3 region [63], and Ile2098Ser, Ser2119Tyr, Asn2129Ser, Arg2150His, and Pro2153Gln in the C1 domain [64]. There have been many reports in the literature of the prevalence of inhibitors in patients with haemophilia, withrates varying between 12.2% [65] and 53.8% [66]. In such patients, the antibodies produced are capable of hydrolysing the therapeutic FVIII. In a study in 1999, Lacroix-Desmazes et al. demonstrated that FVIII is proteolysed by alloantibodies in patients with severe haemophilia A, demonstrating a previously unknown mechanism by which FVIII inhibitors may prevent the pro-coagulant function of FVIII [67].
Inhibitors may prevent the action of FVIII by inhibition by steric hindrance and other mechanisms, such as immune complex formation and FVIII clearance [68]. In a study to examine the contribution of Th1 and Th2 cells in the anti-FVIII antibody response, Reding et al. measured the concentration of Th1- and Th2-driven anti-FVIII IgG subclasses in 17 patients with severe haemophilia A and 18 patients with acquired haemophilia. They found that both patient groups had similar and comparable proportions of Th1- and Th2-induced anti-FVIII antibodies, suggesting a more important role of Th1 cells in the immune response to FVIII than previously appreciated [69]. More intense anti-FVIII antibody responses and higher inhibitor titres correlated with a predominance of Th2-driven subclasses. In contrast, Th1-driven anti-FVIII antibodies were predominant in patients who had low anti-FVIII antibody concentrations. FVIII inhibitors have been shown by immunoblotting or immunoprecipitation assays to bind predominantly to epitopes within the A2 and/or C2 domains of the FVIII protein [70].
It is important to study the anti-FVIII antibody response and profile epitopes of the C2 and A2 domains in order to understand more about inhibitors in patients with haemophilia. Various methods have been used to achieve this: these include the Bethesda method, phage display, ELISA and x-MAP/Luminex. The latter technique was used in a study to simultaneously detect and map the epitope specificity of inhibitors from haemophilia A patients by screening plasma against both heavy and light chains of human plasma-derived FVIII. The format used was a two-site sandwich assay, where one monoclonal antibody specific for the heavy or light chain was first immobilized on beads, and then incubated with the different forms of plasma-derived FVIII. Samples from haemophilia patients with autoantibodies preferentially recognized the light chain, whereas samples from patients with alloantibodies were directed against both the light and heavy chains [71]. An unmet need is fine epitope profiling in all fragments of FVIII. In a study in haemophilia mice, it was found that antihuman FVIII C2 domain antibodies recognize a functionally complex spectrum of epitopes dominated by inhibitors of FVIII activation [72]. The first challenge is to produce separate A2, C2 and C1 domains of FVIII. This was achieved by Lapalud et al. in 2012 [73]. Prior to that, in 2008 Greninger et al. found that the FVIII B cell epitope profile was associated with immune tolerance induction (ITI) outcome in a VWF/FVIII-treated cohort of patients with haemophilia A and high-titre inhibitors. The B cell epitope specificity in all four successful and in one of two partially successful ITI subjects included the C2 but excluded the A2 domains. Conversely, FVIII B cell epitopes in one partially successful ITI and in all three failed ITI subjects mapped to both the C2 and the A2 domains [74]. A study to monitor the distribution of IgG subclasses of anti-FVIII antibodies during ITI found that FVIII-specific antibodies of subclass IgG1 were detected in all inhibitor patients tested, anti-FVIII IgG4 in 16, IgG2 in 10 and IgG3 in one of 20 patients analysed. Levels of anti-FVIII IgG1 and IgG4 correlated well with inhibitor titres as measured by Bethesda assay [75]. In low-titre inhibitor patients, anti-FVIII antibodies consisted primarily of subclass IgG1, whereas anti-FVIII antibodies of subclass IgG4 were more prominent in patients with high-titre inhibitors who needed prolonged treatment or in whom ITI had failed.
Several Registries have tried to predict the response to ITI in patients with haemophilia and inhibitors. However, while records are not always in agreement, the recent concept of prognostic FVIII epitope needs to be taken into consideration.
In a national retrospective study [76], the principal objective was to identify predictive markers of ITI efficacy. The methods used, the isotypic and epitopic repertoires of inhibitory antibodies were analysed in plasma samples collected before ITI initiation from 15 children with severe haemophilia A and high-titre inhibitors, and their levels were compared in the two outcome groups (ITI success [n = 7] and ITI failure [n = 8]). The predictive value of these candidate biomarkers and of the currently used indicators (inhibitor titre and age at ITI initiation, highest inhibitor titre before ITI, and interval between inhibitor diagnosis and ITI initiation) were then compared by statistical analysis (Wilcoxon test and receiver operating characteristic [ROC] curve analysis). Thirty-four potential markers (five current indicators and 29 IgG biomarkers) were retrospectively compared in 15 children who had successful ITI (n = 7) or in whom ITI failed (n = 8). The values of the currently used indicators (inhibitor titre at ITI initiation, patient age at ITI initiation, highest inhibitor titre before ITI and during ITI, and time interval between inhibitor diagnosis and ITI initiation) were not significantly different (P > 0.05) between outcome groups (ITI success and failure). Then, the levels of the 29 IgG-associated biomarkers (five epitopic biomarkers [chains or domains recognized by anti-FVIII antibodies], four isotypic biomarkers [IgG1 to IgG4] and 20 combination biomarkers [combining information of both epitopic and isotypic repertoires of anti- FVIII antibodies]) were measured by the use of multiplexed assays in plasma samples collected before ITI. Among these biomarkers, only the values of three epitopic biomarkers (HC, P = 0.0240; A1, P = 0.0177; and A2, P = 0.0128) were significantly different between outcome groups and seemed to be particularly relevant for the prediction of ITI failure (Fig. 5a–f). The A2 and A1 markers could be directly considered as prognostic tests on their own, based on the established threshold values. Accordingly, a patient with an anti-A2 IgG or anti-A1 IgG level lower than 7.67 RAR (ratio antigenic reactivity) or 9.07 RAR, respectively, had a 99% chance of ITI success (negative predictive value [NPV] of 99%).
Fig. 5.
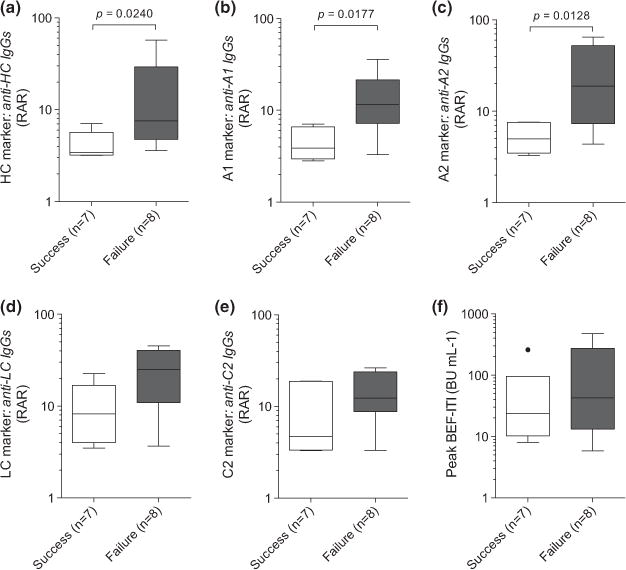
Predictive performance of the best markers. (a–e) The plasma levels, expressed as relative antigenic reactivity (RAR), of IgGs against the (a) heavy chain (HC) (b) A1 domain, (c) A2 domain, (d) light chain (LC) and (e) C2 domain of FVIII, measured before immune tolerance induction (ITI) initiation in 15 children with haemophilia A who had successful ITI (n = 7) (white boxplot) in whom ITI failed (n = 8) (grey boxplot), were compared by use of the Wilcoxon test. Similar analyses were carried out for currently used indicators. (f) An example of the highest inhibitor peak recorded before ITI (PEAK BEF–ITI), expressed in Bethesda Units (BU) per mL. The Tukey boxplots show the median value as a dash within the box, and outside dots mark the outliers. The significance level was set at 5% [76].
Although, these results need to be confirmed in a larger population, they bring new hope for the development of ITI outcome predictive tests for children with haemophilia A. Moreover, these two plasma biomarkers could be used in combination with other bioclinical markers. For example, the association of marker A1 with anti- FVIII IgG4 Abs, which constitute an isotypic biomarker that might be useful for the identification of patients at risk of ITI failure, could further improve ITI outcome prediction. However, because of the small size of the present cohort (n = 15), it was not possible to perform multivariate analyses.
Future goals are to develop an approach to design peptides mimicking discontinuous epitopes, synthesize epitopic peptides and screen for discontinuous peptides in a cohort of plasma anti-FVIII antibodies using x-MAP tools. The computer algorithm PEPOP uses the 3D coordinates of a protein both to predict clusters of surface accessible segments that might correspond to epitopes and to design peptides to be used to raise antibodies that target the cognate antigen at specific sites. More successful predictions of immunogenic peptides were obtained when a peptide was continuous as compared with peptides corresponding to discontinuous epitopes [77]. PEPOP was also used in another study, which found that C2 domain epitopes are organized as an epitopic mosaic distributed around the FVIII molecule, and confirmed the complexity and variability of the immune response against the C2 domain of FVIII [78]. The ability to finely map epitopes could be further used to follow the antibody specificity modifications over time.
Current difficulties in ITI therapy and daily practice
The burden of haemophilia care: inhibitors and ITI, a viewpoint from Israel (Gili Kenet, Israel)
Inhibitors are a major treatment challenge in patients with haemophilia. When they occur, treatment is often by ITI, typically involving the daily infusion of large doses of FVIII over many months. The dosage regimen depends on whether the patient has good or poor prognostic factors. The question of when to stop ITI is a matter of debate, along with when alternative management strategies should be considered. Finally, which patients should be given rituximab? In Israel, there is a single tertiary multidisciplinary treatment centre taking comprehensive care of 632 patients with haemophilia. The issues debated will be discussed by presentation of several demonstrative real-life cases.
Case 1: when to start ITI and which product should be used?
In the first case, a baby with familial haemophilia was born (in another hospital) by vacuum delivery. He developed a massive cephalohaematoma and was treated with 22 doses of recombinant FVIII therapy. At 9 months of age, he presented with a knee haemarthrosis and an inhibitor of 1 BU (Bethesda Unit) was detected. As this was clearly a low responding (LR) inhibitor and potentially maybe even transient, the decision had to be made as to whether or not to start ITI and which product should be used?
In the International Immune Tolerance Study, 115 ‘good risk’ severe high-titre inhibitor haemophilia A patients were treated with either a high-dose (200 IU kg−1 day−1) or a low-dose (50 IU kg−1 three times a week) ITI regimen. Successes did not differ between treatment arms (24 of 58 low-dose vs. 22/57 high-dose, P = 0.909). Peak historical (P = 0.026) and on-ITI (P = 0.002) titres were correlated inversely with success, but only peak titre on ITI predicted outcome in a multivariate analysis (P = 0.002) [79]. Product type (plasma-derived FVIII vs. recombinant FVIII) did not result in a significant difference (P = 0.58). Taking into account the fact that our patient is a young child with good prognostic factors, low-dose ITI was preferred, three times weekly, and central line placement was recommended. The decision was made not to wait for the anamnestic response for the initiation of ITI and to use prophylaxis if it is required.
Case 2: Tailoring ITI in patients with high-responding inhibitors
In our second case, a 3-year-old patient with sporadic haemophilia A presented with a high-responding (HR) inhibitor, diagnosed at 18 EDs. Repeated central venous line infections interrupted ITI. The inhibitor titre increased to 680 BU with poor response to bypassing therapy. He was given high doses of rFVIIa for bleeds but did not respond well. The question is can a potentially preferred therapeutic regimen for such patients be ‘tailored’ and monitored? In the International Immune Tolerance Study, there was a significant difference in the time to achieve treatment milestones by treatment arm. Time to achieve first normal recovery was 13.6 (IQR: 9.7–18.9) months for the low-dose regimen, vs. 6.9 (3.5–11.9) months for the high-dose regimen, P = 0.001 [79].
Treatment options for bleeding episodes of HR inhibitor patients (in whom complete FVIII inhibition occurs) are bypassing therapy with rFVIIa or aPCC (FEIBA). For LR patients with incomplete FVIII inhibition, ‘replacement’ therapy with FVIII or porcine FVIII is given. Unresponsiveness to bypassing agents may occur in patients with HR inhibitors. Sequential therapy, with plasma derived-aPCC and rFVIIa in patients with severe haemophilia and HR inhibitors, has been reported and even recommended for ‘resistant’ patients [80]. In patients refractory to rFVIIa and probably other haemophilia A patients with inhibitors, concomitant infusion of low-dose rFVIIa and low-dose aPCC seems to be safe, efficacious and economical [81]. Clinical observations of patients undergoing ITI showed decreased bleeding frequency during treatment with high-dose FVIII, possibly due to residual plasma trace FVIII activity. The low dose International Immune Tolerance Study was interrupted due to safety issues and the effectiveness of bypass prophylaxis could not be evaluated [79].
Notably, the combination of bypassing agents and FVIII showed a positive in vitro effect on thrombin generation in HR inhibitor patients and on the thrombin generation defect induced by anti-FVIII monoclonal antibodies compared to the use of FVIII alone [82]. In a previously published study by our group, the potential contribution of FVIII to rFVIIa-induced haemostasis in inhibitor plasma was defined by thrombin generation. Following a single combined FVIII– rFVIIa dose, 90% haemostasis was documented when patients with inhibitors were treated for various bleeding episodes (either within ITI or without it) and neither thrombosis nor any other complications were reported. During the study period, inhibitor levels and bleeding frequency declined significantly [83].
Thus, according to our experience, high-dose ITI should not be the only solution in patients with poor prognostic factors. Low-dose ITI may be used and a central venous line is mandatory. Regular prophylaxis should be given, starting with rFVIIa with the ITI dose, possibly switching to aPCC. In the ITI study, the dose used was 50 IU kg−1 three times a week [79].
If ITI has been successful or partially successful, prophylaxis should continue. In patients in whom ITI has failed, it is necessary to wait for perhaps 3 years before declaring complete or partial failure. In the ITI study, treatment failed in 17 of the 67 patients (25%) [79]. Is ITI failure a laboratory definition or a clinical definition? The following cases demonstrate this dilemma.
Cases 3 and 4: Is ITI failure a laboratory or a clinical definition?
A patient had been very resistant to ITI with HR inhibitors, 3 years after tolerance to low-dose FVIII plus rFVIIa prophylaxis, still had a borderline inhibitor and no good recovery. A plasma-derived FVIII with high VWF content was given. His response was much better than with the full-length recombinant product. In this case, despite laboratory definitions, he cannot be described as a treatment failure and he hardly ever bleeds.
In another case, an older patient was given various products, plus rituximab, yet the inhibitor could not be eradicated, so the ITI failed. However, in the case of surgery or a severe bleed he could be treated with high bolus FVIII followed immediately by continuous infusion. He underwent total hip replacement on FVIII alone despite the (currently LR) inhibitor. Therefore, the question of ITI failure is a clinical definition and not just a laboratory one.
Other treatment options for inhibitor patients are tranexamic acid (which may enhance the effectiveness of rFVIIa), prophylaxis with rFVIIa or FEIBA (may reduce the bleed frequency), rituximab (may reduce the inhibitor titre, at least temporarily) and radionuclide synovectomy (may help control recurrent bleeding in a target joint). In the future, patient-tailored therapy is the goal. Our centre suggests the use of thrombin generation-guided therapy, for ex vivo plasma spiking with either FVIII or bypassing agents and their combinations, in order to propose an individually tailored therapeutic approach [81,83].
Alternative management strategies should always be considered. Inhibitor titre does not express the kinetic behaviour of the inhibitor or an individual’s response to FVIII therapy. Ex vivo studies may identify inhibitor patients who could benefit from specific FVIII brand related/bypass tailored therapy (regardless of inhibitor titre). The wide range of variability in inhibitor patients’ response, combined with the development of various FVIII concentrates, emphasizes the need for personalized tailoring of treatment as well as FVIII-specific ITI regimens for haemophilia A patients with inhibitors.
A routinely used ITI protocol in Sweden (the Malmö protocol) combines FVIII administration with immunosuppression. Extracorporeal immunoadsorption is used as required to remove high-titre inhibitors and to achieve an inhibitor titre below 10 BU at the initiation of ITI. Most patients are then given oral corticosteroids, followed by cyclophosphamide and intravenous immunoglobulin with the start of high daily doses of FVIII. Whether the addition of such agents can reduce the time and intensity of ITI required, and whether this would aid individuals who did not succeed with traditional ITI with FVIII alone, remains unanswered [84]. There are limited data regarding immune modulation and FIX tolerance.
The use of rituximab
Rituximab is a chimeric monoclonal antibody against the CD20 antigen that blocks the proliferation of normal B cells, thus interfering with IgG antibody production. A national survey of all 23 comprehensive care haemophilia centres in the UK reported 15 consecutive severe (<1% FVIII) haemophilia A patients with loss of a detectable inhibitor in 40% of patients on rituximab with ITI, but a lasting response was seen in only two patients [85]. A Registry through the Hemophilia and Thrombosis Research Society was collecting data on congenital haemophilia patients who had been treated with rituximab; however, the Registry was closed last year and is no longer in existence. Currently, data interpretation is complicated by the variable FVIII regimens used, different definitions of response, various durations of follow-up and the potential for preferential reporting. Furthermore, the long-term adverse effects of B cell depletion, particularly in children, are unknown. Rituximab treatment should be considered in haemophilia patients with HR inhibitors (‘difficult to treat’), in those with a FVIII allergy impairing ITI, in those in whom a therapeutic window is required (e.g. at times of surgery) and in elderly patients with acquired haemophilia.
Current controversies in ITI therapy and daily practice, the German view (Carmen Escuriola-Ettingshausen, Germany)
ITI is the optimal treatment option in inhibitor patients after inhibitor development. It has a success rate of up to 96% [86]. The benefits to tolerize patients are regular FVIII administration for bleeds/surgery, regular FVIII prophylaxis to prevent bleeds/haemophilic arthropathy and an increase in QoL. Current controversies in ITI management are the therapeutic regimen (i.e. high-dose vs. low-dose vs. the Bonn protocol), when treatment should be started (≤10 BU and >10 BU), the type of concentrate to be used (plasma-derived vs. recombinant FVIII), the use of prophylaxis around the time of ITI, the use of ITI in adults and the use of immunosuppressive agents.
In the International ITI study [79], ITI success rates did not differ between the treatment arms (24/58 on the low-dose regimen vs. 22/57 on the high-dose regimen; P = 0.909). However, there was a significantly shorter time to achieve negative inhibitor titre (P = 0.017) and to reach normal recovery (P = 0.001) with the high-dose regimen. Furthermore, low-dose subjects bled more often (OR 2.2; P = 0.019). Early mean bleed rate per month was 0.623 for patients on the low-dose regimen and 0.282 for those on the high-dose (P = 0.00024). Significantly, more patients on the low-dose regimen required hospitalization for bleeding and significantly fewer experienced a bleed-free course on ITI compared with patients on the high-dose regimen. This could have potential effects on the incidence of haemophilic arthropathy in young children.
The rationale for peri-ITI prophylaxis with bypassing agents during the high-risk bleeding phase (>2 BU) is that intercurrent bleeds during ITI can cause or deteriorate haemophilic arthropathy and may interfere with the duration/outcome of ITI. The aim is to prevent bleeds in these patients. A substantial number of patients already respond to ITI but there are many patients in whom a bleeding tendency is observed. In these patients peri-ITI prophylaxis should be used in order to prevent the onset or deterioration of haemophilic arthropathy. In good prognosis patients in the International ITI study, bleeds during ITI were not found to have an influence on outcome [79]. However, in poor prognosis patients in the ObsITI study (an international open-label, uncontrolled, non-interventional, multicentre observational programme conducted by the Haemophilia Centre Rhine-Main [HZRM], Frankfurt-Mörfelden, Germany), the monthly bleeding rate correlated with ITI treatment outcome (P = 0.0009) [87,88].
Should ITI begin when the pre-ITI titre is ≤10 BU or >10 BU? Results from Registries have shown it makes sense to wait before starting ITI until the pre-ITI titre is ≤10 BU. In the ObsITI study, it was found that there was a higher rate of success of ITI and it was achieved earlier in patients with an inhibitor titre of ≤10 BU compared to those with a titre of >10 BU (Fig. 6) [87,88].
Fig. 6.
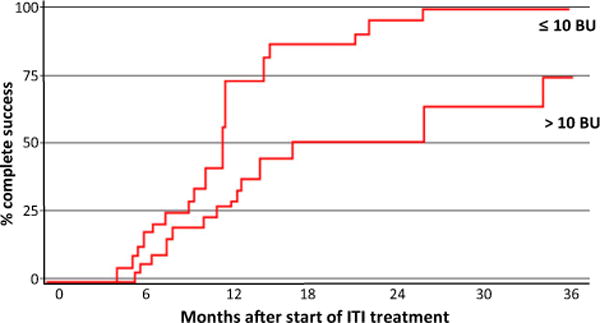
Similar results were found in the Grifols-ITI (G–ITI) study, an international, multicentre, retrospective study designed to assess the outcome of primary and rescue ITI in children and adults with haemophilia A using a single plasma-derived FVIII/VWF product (Fanhdi®; Grifols, Barcelona) [89]. Complete success rate was found to decrease continuously with increasing inhibitor titres at the start of ITI.
In Germany, the Bonn protocol has been used to treat haemophilia patients with inhibitors. It involves the regular administration of factor replacement over a prolonged period of time in conjunction with the use of aPCC. Kreuz et al. recommend early onset of a high-dosage ITI regimen in high responder patients as soon as possible after inhibitor detection, as this is associated with a greater success rate and shorter elimination time [90]. In a USA study of 58 patients with inhibitors undergoing ITI, Nakar et al. found a success rate of 79% (15/19 patients) in low responders when ITI was started within a month of detection. In high responders, success rate was 91% (21/23 patients) when ITI was started within a month of detection, compared to only 64% (7/11 patients) when begun after at least 6 months of detection regardless of the pre-ITI inhibitor titre [86]. In view of these results, it should be investigated if ITI should be started promptly as it is associated with a higher success rate, cost-effectiveness and prevents the ‘unprotected phase’ in small children with inhibitors. Several factors have been associated with a good prognosis for inhibitor patients. These are inhibitor titre at start of ITI <10 BU, maximum historical inhibitor titre <200 BU, age at start of ITI <7 years and inhibitor history <2 years. On the contrary, factors associated with a poor prognosis are inhibitor titre at start of ITI >10 BU, maximum historical inhibitor titre >200 BU, age at start of ITI >7 years and duration of inhibitor >2 years, as well as earlier ITI failure [89]. However, Oldenburg et al. [89] found no correlation between ITI outcome and patient age or time from diagnosis of inhibitor and start of ITI. These results have been confirmed by the ObsITI study as well as the PROFIT study (Italian ITI registry). ITI can be used in adults with poor prognosis, but according to current data the definition of poor prognosis should be revised. Also the episodic exposure to FVIII since the development of the inhibitor needs to be considered.
The type of concentrate used in ITI and its affect on treatment success has also been the topic of discussion. Data from Germany on ITI with FVIII/VWF concentrates indicate higher success rates with these than with pure recombinant FVIII (plasma-derived FVIII/VWF, pre-1993, success rate 87%; recombinant FVIII >1993, success rate 43%; plasma-derived FVIII/VWF >1993, success rate 82%) and higher success with the Bonn protocol [91]. Other studies have confirmed that the decrease in ITI success rate with recombinant FVIII was highly pronounced in high responders, and less so in the low responders [92]. Since then many patients who are poor responders have been treated with high concentrations of FVIII/VWF-containing products. However, there are no data from prospective, controlled studies yet which can clearly show which product to use. The RESIST study (REScue Immunetolerance STudy) is an international, randomized, prospective, controlled open-label study and a satellite study of the International Immune Tolerance Study [79]. The primary objective is to evaluate whether FVIII/VWF concentrates can induce more frequent or rapid immune tolerance to FVIII in haemophilia A patients with high-responding inhibitors, who are at high risk of failing ITI, in comparison with VWF-free FVIII concentrates. There are two patient groups: RESIST naïve (poor prognosis patients who have never had ITI; this sub-study is ongoing, but is no longer recruiting patients [93]) and RESIST experienced (earlier ITI failure [94]). FVIII/VWF concentrates are currently recommended for second-line and salvage ITI in patients who have failed previous attempts at tolerization using monoclonal or recombinant FVIII products. Higher success rates with plasma-derived FVIII/VWF products compared to recombinant FVIII have been found in the German experience [91,92].
Alternative therapeutic approaches in haemophilia patients with refractory inhibitors include the use of immunosuppressive agents. Rituximab, an anti-CD20 monoclonal antibody, has been used successfully alone or in combination with other immunosuppressive agents in patients with congenital haemophilia A or B and alloantibodies refractory to first-line ITI. However, well-designed, controlled studies are needed to clarify the use of immunosuppressive agents in the treatment of haemophilia patients with refractory inhibitors, particularly the long-term benefit of these agents and to collect data on adverse events, particularly in young children.
Prognostic factors of ITI, the French perspective (Annie Borel-Derlon, France)
The dosage regimen for ITI depends on the individual patient. In young PUPs (<5 years old) with FVIII inhibitor <5 BU and depending on the genotype (no large deletion), a low-dose regimen should be given: 50 IU every 2 days. There should be a short delay (<3 months) after diagnosis of the FVIII inhibitor before starting treatment and frequent biological FVIII inhibitor titre surveys need to be carried out. If there is a high anamnestic response then a high-dose ITI regimen should be given instead. In young PUPs with a high FVIII inhibitor titre >10 BU at diagnosis and high-risk genotype (e.g. large deletion or intron 22 inversion), a high-dose regimen should be administered at least ≥200 IU kg−1 everyday. Treatment (by central venous access for children) should begin early after diagnosis. The use of concurrent prophylaxis (with recombinant FVIIa or aPCC) depends on the bleeding tendency severity and the presence of localized bleeds. It should certainly be given if there is haemarthrosis or serious haematoma and particularly if the patient is undergoing physiotherapy.
ITI treatment should be stopped when the inhibitor has been eradicated or when ITI has failed (persistence of the FVIII inhibitor level and/or lack of compliance for treatment). Treatment should also be stopped after failure with different types of products: first the product that initiated the FVIII inhibitor, then plasma-derived FVIII/VWF. The laboratory definition of ITI failure is persistence of the same FVIII inhibitor titre for more than 6 months when the same or higher dosage regimen of ITI is given. In this situation, another FVIII should be tried, if possible. Clinical definition depends on compliance of treatment.
Good and bad prognosis factors in ITI (Thierry Lambert, France)
The management of haemophilia patients with inhibitors remains a major clinical challenge. ITI is usually given to eradicate inhibitors. Poor prognostic factors for treatment success are age >6 years, ITI started >1 year from inhibitor development, inhibitor peaks >200 BU, inhibitor titre >10 BU when ITI is started and previously failed ITI. In a patient aged 43 years with an inhibitor and poor prognostic factors, Factane (human FVIII) and CellCept (mycophenolate mofetil) were given in gradually reducing doses from 2003 to 2007 when the inhibitor was biologically eradicated, but the patient still required daily doses (30 IU kg−1) to avoid spontaneous bleedings. Another patient with poor prognostic factors and an inhibitor was given Advate from 2011 to 2014. The inhibitor level decreased, but was still present. In 2014, his treatment was switched to Octanate (high-purity human FVIII/VWF); the inhibitor then decreased slightly, but the patient still requires high daily doses of FVIII to reach clinical efficacy. A patient with mild-moderate haemophilia and an inhibitor was treated with rituximab, leading to a substantial drop in inhibitor level and a consequent rise in FVIII level.
In patients with either good or poor prognosis factors undergoing ITI, the use of implantable devices should be avoided. Adherence to treatment by the family/child is very important for treatment success. A high-dose regimen should be used if possible (according to international ITI recommendations), and prophylaxis given according to the bleeding pattern. If the ITI treatment is not successful, a rapid (3 months) change of regimen or dose should be considered. Deciding when to stop treatment is still a matter of debate. Treatment should be stopped if the patient gives up, if there is clear biological and clinical in efficacy after a number or months or even years, or in some cases, due to economic constraints.
The laboratory definition of complete success is an inhibitor titre <0.6 BU, FVIII recovery >66% and a half-life of >6 h. Partial success is a mixture of clinical and biological definitions: inhibitor titre to <5 BU with FVIII recovery <66% and/or FVIII half-life <6 h associated with clinical response to FVIII therapy, and not followed by a treatment-limiting anamnestic rise in inhibitors to >5 BU.
Alternative management strategies may be required if the patient/family refuses ITI or in the case of failure of ITI. In such circumstances, bypassing agent therapy should be commenced, and prophylaxis or on-demand therapy should be given according to the patient’s bleeding pattern. Rituximab therapy should be considered in patients with mild/moderate haemophilia in whom there has been no spontaneous return to their initial basal level of FVIII for months and who have a frequent bleeding pattern.
While good and bad prognosis factors are important, any individual patient might be considered for ITI. Before initiating therapy, patient/family motivation, willingness to start treatment, ability to accept and tolerate treatment should be carefully considered. Regular and scheduled follow-up is needed to assess laboratory and clinical data. Revisions of the treatment scheme may need to be made. Support of family and patient is crucial.
Surgery in patients with inhibitors
The surgeon’s view (Gianluigi Pasta, Italy)
There are two options for the treatment of haemophilic patients with inhibitors undergoing surgery: rFVIIa or aPCC. The dose, whether the treatment should be given by bolus or continuous infusion, and time of treatment are all factors that need to be considered. In 2003, Rodriguez-Merchan et al. evaluated 108 surgical procedures including 88 radiosynoviortheses and 20 major orthopaedic procedures (six total knee replacements, four osteosyntheses, two total hip replacements, two pseudotumours removed and six other major operations) in nine centres. This study concluded that rFVIIa allows haemophilic patients with high inhibitor titres to undergo elective orthopaedic surgery with a greater expectation of success [95]. However, of the 20 major orthopaedic procedures, postoperative bleeding complications occurred in three patients treated with insufficient doses of rFVIIa. In a paper published 4 years later, evaulating 91 procedures (80 minor and 11 major), Rodriguez-Merchan et al. concluded that the use of aPCC and rFVIIa allow for successful surgery in patients with inhibitors [96].
Radiosynoviorthesis is a very simple procedure with minor bleeding risk. After a synoviorthesis, there is no need for rehabilitation and after a 24-h period of moderate rest, the patient can gradually initiate regular physical activity [97]. A consensus statement on perspectives in surgery in haemophilia patients with inhibitors was developed following an international workshop [98]. The conclusion was to consider synoviorthesis and arthrocentesis as the only minor surgical procedures. Major procedures should include bone fixation for fractures, hip and knee replacements, arthrodeses, open synovectomies, pseudotumour removal and hardware removal. A study evaluated the efficacy of bypassing agents in providing haemostatic cover in a series of patients with haemophilia and inhibitors undergoing elective orthopaedic surgery between 1997 and 2008. Good control of haemostasis can be achieved with bypassing agents and rFVIIa was considered to be effective [99].
Various studies have investigated the use of bypassing agents in the management of haemophilia patients with inhibitors undergoing orthopaedic surgery. In a study of 12 patients with inhibitors (13 orthopaedic procedures), Ingerslev et al. used rFVIIa to promote haemostasis during surgery. In 12 of the cases, the result was considered to be excellent; in the other case a global evaluation of treatment was not reported [100]. There were no apparent adverse effects. Rodriguez-Merchan et al. used aPCC to provide haemostasis support in 26 patients undergoing surgery (20 radiosynoviortheses and seven major orthopaedic procedures) [101]. In the first group, there were 19 good results and one fair. In the second group, there were six good results and one fair. There was one case of arterial pseudoaneurysm. The authors concluded that orthopaedic surgery is now possible in haemophilia patients with inhibitors. However, a study by Goddard et al. on rFVIIa in haemophilia A patients with inhibitors undergoing orthopaedic surgery found significant bleeding at the third postoperative day. They concluded that optimal dosing regimens for rFVIIa as surgical cover are still to be determined [102].
In our centre in Italy, several major surgeries in haemophilia patients with inhibitors have been performed using rFVIIa. These have mainly been total knee replacement, knee arthroscopy, removal of total knee replacement and flat foot surgery as well as several other procedures. During 1997–2001, three infectious complications and one aseptic loosening were observed [103]. In one case, the infection was due to the presence of an infected central venous catheter. Red blood cells (RBC) transfusions are often required in these patients since a median haemoglobin drop of around 6 g dL−1 occurred.
For orthopaedic surgery in haemophilia patients, a multidisciplinary team is required comprising the orthopaedic surgeon, a haematologist, a physiotherapist and an infectivologist. Many interventions can reduce the need for allogeneic blood transfusion in elective orthopaedic surgery. These include hypotensive epidural anaesthesia, fibrin sealant and surgical technique [104]. The length of hospital stay required for haemophilia patients with inhibitors undergoing orthopaedic procedures has improved greatly in recent years: in general until 1997–2001 the average hospital stay was 14 days, compared to 7–9 days in more recent years.
Furthermore, fewer infectious complications and no aseptic loosening were observed (unpublished data). Finally, orthopaedic surgery in inhibitor patients is not for the faint-hearted and should only be carried out in centres with such experience [105].
The haematologist’s view (Yesim Dargaud, France)
The risks associated with major surgery in haemophilia patients with inhibitors are excessive/uncontrolled bleeding, death, poor wound healing and subsequent risk of infection, anamnestic response, thromboembolism/disseminated intravascular coagulation and increased cost of treatment. A key step for the success of a major elective surgery in inhibitor patients is excellent communication and collaboration between all relevant parties (patient, haematologist, experienced surgeon, anaesthesiologist, pharmacist, nurse, laboratory staff and physical therapist). Risk factors to consider are the bleeding risk of the surgical procedure itself, the patient’s inhibitor titre and anamnestic response, personal history of the patient to discriminate between ‘good responders’ and ‘bad responders’ to a particular by-passing agent, medical conditions affecting pharmacokinetic parameters and international guidelines. The latter state that if the inhibitor titre is <5 BU mL−1, human FVIII/FIX (or porcine FVIII) can be given because of the predictability of the clinical/biological efficacy (grade B or grade 1C). If the inhibitor titre is >5 BU mL−1, bypassing therapy is the treatment of choice (grade C or grade 1C) [106]. In a paper reporting 20 years of experience of surgery in haemophilia patients with inhibitors, the efficacy obtained with FVIII was good in 100% of the cases described; there were no haemorrhagic complications in the 18 procedures in which it was used (three major and 15 minor surgeries). With aPCC, excellent results were achieved in 96.87% of cases with only one haemorrhagic complication in a synoviorthesis out of 32 procedures. Good results were obtained with rFVIIa in 71.42% of cases (10/14) with few haemorrhagic complications [107], Table 2.
Table 2.
Efficacy of FVIII, prothrombin complex concentrates (aPCC) and recombinant activated FVII (rFVIIa) in haemophilia patients with inhibitors undergoing surgery [107].
| Type of study | Product | No. of episodes | Response | Adverse events |
|---|---|---|---|---|
| Retrospective | FVIII | 18 | Good 100% | – |
| Retrospective | aPCC | 32 | Good 96.87% (31/32) | Bleeding |
| Retrospective | rFVIIa | 14 | Good 71.42% (10/14) | Bleeding |
The FENOC (FEIBA NovoSeven Comparative) study was designed to test the equivalence of the bypassing agents, aPCC and rFVIIa, in the treatment of ankle, knee and elbow joint bleeding. The study reported that aPCC and rFVIIa exhibited similar effect on joint bleeds although the efficacy between products was rated differently by the majority of the patients [108]. Since the first surgery performed with rFVIIa, a synovectomy, the drug was evaluated in several surgical settings [109]. The only prospective, double-blind dose finding study was performed by Shapiro et al. and compared two doses of rFVIIa (35 μg kg−1 or 90 μg kg−1) during and after elective surgery. Results showed that rFVIIa 90 μg kg−1 was an effective first-line dose option in surgery [110]. Pruthi et al. carried out a comparative trial of bolus vs. continuous infusion of rFVIIa in haemophilia patients with inhibitors undergoing major surgery. Results showed that haemostatic efficacy of rFVIIa in each group were comparable: effective in 8/11 and 9/12 subjects in the bolus and continuous infusion arms, respectively, and ineffective in three subjects in each arm [111]. Two review papers reported results obtained with rFVIIa 90–150 μg kg−1 in haemophilia patients with inhibitors undergoing elective surgeries. The authors concluded that rFVIIa was effective in approximately 85% of cases and safe to use in patients undergoing orthopaedic surgery with very low thrombotic events (0.4%) [112,113]. Giangrande et al. published a consensus protocol for the use of rFVIIa in elective orthopaedic surgery in haemophilia patients with inhibitors. They recommended a bolus dose of rFVIIa of 120–180 μg kg−1 just prior to the first incision, with follow-up doses of 90 μg kg−1 every 2 h during the first 48 h. They suggested that a bolus dose of 90 μg kg−1 should be given 10 min before the removal of wound drains with a subsequent decrease of the frequency of rFVIIa infusions between days 2 and 5, 90 μg kg−1 every 3 h, and then between days 5–8, 90 μg kg−1 4 h and finally, after day 8, 90 μg kg−1 every 6 h [114].
Other studies reported data on the use of aPCC as a bypassing agent in haemophilia patients with inhibitors undergoing surgery. Rangarajan et al. reported the experience of four UK comprehensive care centres using aPCC for surgeries in patients with inhibitors. Peri-operative haemostatic outcome with aPCC was rated excellent or good in 78% of 18 major surgeries in 12 patients, including 11 major orthopaedic procedures. Haemostatic outcome was rated as excellent in all seven procedures in five patients with acquired FVIII inhibitors and in all eight minor surgical procedures in six patients. aPCC was well tolerated with no intra-operative haemostatic complications [115]. Négrier et al. published the results of the SURF study (SURgical interventions with FEIBA), an international Registry of surgery in haemophilia patients with inhibitors. Nineteen centres participated, involving 35 surgical procedures (37.1% high-risk surgery). Haemostasis was judged to be ‘good’ or ‘excellent’ in 91.2% (31 of 34) of surgical procedures and ‘fair’ in 8.8% (3 of 34). The authors concluded that aPCC can be safely and effectively used when performing surgical procedures in haemophilia patients with inhibitors [116]. Recently, Rangarajan et al. also produced consensus recommendations for the use of aPCC in haemophilia A patients with inhibitors undergoing elective surgery. For major procedures, they recommend 75–100 U kg−1 preoperatively, followed by 75–100 U kg−1 at 8-hourly intervals from days 1–7, 75–100 U kg−1 at 12-hourly intervals from days 8–21 and 75–100 U kg−1 once a day for a week, then every other day for weeks 5 and 6 [117].
Looking at risk, safety and efficacy assessments of bypassing agents, both aPCC and rFVIIa are associated with a low thrombotic risk [118]; however, aPCC may cause an anti-FVIII immune response, unlike rFVIIa [119]. Efficacy of aPCC ranges from 64–90% [118,120] and that of rFVIIa ranges from 80–95% [118,121]. In patients treated with rFVIIa, concomitant treatment with antifibrinolytics increases efficacy from 66% to 96% [122]. Combined treatment with aPCC and tranexamic acid was also reported with excellent haemostatic efficacy and safety with regard to thromboembolic complications and DIC in a limited number of patients [116,123].
In the event of a lack of haemostatic response to each of the bypassing agents used singly and at high dosage, it has been suggested that the agents might be used sequentially or in combination as a salvage treatment; however, the results reported with such strategies showed a high risk of thromboembolic complications [124]. A recent study using a hybrid regimen in four inhibitor patients undergoing eight major surgical procedures and given rFVIIa and tranexamic acid during surgery and in the initial two postoperative days followed by aPCC for the remaining period reported non-surgical bleeding in four of eight cases [125].
Individually tailored bypassing therapy may be the way forward in treating these patients. Global haemostasis assays such as thrombin generation test might be of interest for adapting the dose of bypassing therapy to the individual needs of each patient and for monitoring drug efficacy. A three-step protocol has been developed to individually tailor bypassing agent therapy for patients undergoing elective surgeries: first an in vitro spiking step to find the dose of each of aPCC and rFVIIa that normalizes thrombin generation capacity; second, ex vivo confirmation; and third, monitoring of efficacy of the chosen dose of the bypassing agent during surgery and the postoperative period [126]. The thrombin generation test has been used successfully to individually tailor by passing therapy in a prospective clinical study, in patients with severe haemophilia A and inhibitors undergoing major surgery using bypassing agents [127].
In conclusion, the choice of haemostatic treatment in inhibitor patients undergoing a surgery is dictated by a combination of several parameters. A multidisciplinary management approach to care is essential and bypassing therapy as recommended by guidelines and consensus protocols allows haemostatic efficacy in 80– 85% of cases. Thrombin generation assays may help physicians to individually tailor bypassing therapy and to early detect an abnormal activation of the coagulation system during treatment.
Future perspectives for the treatment of haemophilia with inhibitors (Claude Négrier, France)
Future approaches to the treatment of patients with haemophilia who have developed inhibitory antibodies consist of optimized treatment modalities with current products, modification of antigen recognition, new concepts for rebalancing the coagulation system, replacement of FVIII biological role with monoclonal antibodies and prevention of inhibitor formation. The International Immune Tolerance Study compared high-dose (200 IU kg−1 day−1) with low-dose (50 IU kg−1 three times a week) FVIII regimens in 115 ‘good risk’ haemophilia A patients with high-titre inhibitors. Sixty-six patients completed the trial. A Kaplan–Meier plot of 66 subjects achieving a success, partial success, or failure end point showed no significant difference between treatment arms in the subjects achieving tolerance (70%, P = 0.909). However, the time taken to achieve a negative titre (P = 0.027), a normal recovery (P = 0.002) and a normal half-life (P = 0.116, non-significant) were shorter with the high-dose ITI regimen [79].
In the ObsITI study, a satellite study is aiming to evaluate the haemostatic efficacy of FVIII during ITI in haemophilia A patients with inhibitors and the severity of the bleeding phenotype by measuring thrombin generation. The assay will also be used to evaluate the in vitro efficacy of two doses (low and high) of two different FVIII concentrates (recombinant and plasma-derived), and in vitro the combined procoagulant effect of FVIII and aPCC 85 ± 15 U kg−1 or rFVIIa at therapeutic concentrations. This sub-study will also evaluate the correlation between the individual response of patients to different ITI regimens and the functional targets against which anti-FVIII antibodies are directed (epitope mapping) [87].
Chronic joint disease following repeated bleeding into joints is a serious complication of haemophilia. In a study to evaluate the physical parameters in a large haemophilia population without and with inhibitors, Soucie et al. identified a statistically significant decrease in joint range of motion in patients who had developed inhibitors [128]. This study clearly demonstrates the clinical impact of these inhibitory antibodies in terms of physical morbidity.
New bypassing agents are being developed with the aim to minimize the physical consequences of recurrent bleedings in the inhibitor population. BAY 86–6150 is a rFVIIa protein characterized by a faster burst of thrombin generation and a more stable clot, as well as an increased circulating half-life. However, a phase II/III trial evaluating the efficacy and safety of BAY 86–6150 in haemophilia A or B patients with inhibitors has been discontinued due to the detection of neutralizing antibodies against FVIIa. Another product, NN1731, is structurally similar to rFVIIa with the exception of three amino acid substitutions (V158D/E296V/M298Q), which stabilize the active conformation of the molecule. The rationale for its development was that when FVIIa binds to tissue factor, the FVIIa molecule changes to a more active conformation. NN1731 is designed to mimic that conformation, thereby resulting in improved clotting. It has faster and more pronounced thrombin generation than rFVIIa, but similarly to the previous molecule, antibodies against FVIIa were raised and the clinical trial was discontinued.
It has been reported that thrombin generation test (TGT) may represent a useful tool to monitor bypassing therapy with rFVIIa or aPCC. However, TGT does not reflect the stability of fibrin clot and its resistance to fibrinolysis which are crucial. Dargaud et al. developed an in vitro model to assess fibrin clot stability using whole blood thromboelastography (TEG) and lysis with tissue plasminogen activator (tPA). After the addition of rFVIIa 90 μg kg−1 to haemophilia A plasma, the diameter of fibrin fibres was dramatically decreased (P = 0.006) and the density of the fibres was more pronounced. The TEG–tPA assay showed a dose-dependent improvement of clot stability in the presence of rFVIIa indicative of a specific link between thrombin generation, clot structure and stability [129].
In a prospective, randomized, crossover study comparing 6 months of anti-inhibitor coagulant complex (AICC) infused prophylactically at a dose of 85 U kg−1 three times per week with 6 months of on-demand therapy in 34 haemophilia patients with high-titre inhibitors (26 patients completed the study), 16 of 26 patients (62%) had a reduction of bleeding events and joint bleedings during the prophylaxis period (P < 0.001). In a sub-group of patients with a good response (reduction of at least 50% in bleeding events), the reduction in the bleeding rate was 84%, and six of these 16 patients had no bleeding events during the prophylaxis period [57]. The authors concluded that AICC prophylaxis significantly and safely decreased the frequency of joint and other bleeding events in patients with severe haemophilia A and FVIII inhibitors.
Turning to developments in antigen recognition, OBI–1, developed by Ipsen and Inspiration Biopharmaceuticals Inc, is a bioengineered porcine rFVIII that is highly purified. It has the procoagulant and biochemical properties of porcine pdFVIII, with improvements in risk of toxicity, infection and manufacture. It is formulated without animal-derived products and is 94% homologous with human FVIII. OBI–1 was effective in a phase II, open-label clinical trial in patients with haemophilia A and inhibitor against human FVIII, who experienced a non-life or -limb threatening bleed [130].
It is known that approximately 85% of FVIII inhibitors are directed against the A2 or C2 domains of the molecule, and most of the remainder at the amino-terminal region of the A3 domain. In approximately 60% of patients, there are two or three major inhibitor binding sites on the FVIII molecule. Anti-C2 and anti-A3 antibodies prevent FVIII from interacting normally with VWF and/or phospholipids, and the anti-A2 and anti-A3 antibodies interfere with the formation and activity of the tenase complex, blocking the pathway to generation of thrombin [131]. Mechanisms of inhibition prevent normal function of tenase complex, normal binding of phospholipids and normal binding to VWF.
New concepts for rebalancing the coagulation system include targeting tissue factor pathway inhibitor (TFPI) and antithrombin. In haemophilia A and B, the lack of FVIII and FIX, respectively, leads to a decrease in thrombin production, resulting in a bleeding phenotype. Inhibiting natural anticoagulants (e.g. antithrombin, protein C, protein S and TFPI) and therefore rebalancing the coagulation dynamics, has the theoretical potential to increase thrombin generation and correct the bleeding phenotype in haemophilia A or B. This molecule is currently under clinical investigation in a phase I trial. ARC19499 is an aptamer that inhibits TFPI, thereby enabling clot initiation and propagation via the extrinsic pathway. In a study by Waters et al., using a monkey model of haemophilia, FVIII neutralization resulted in prolonged clotting times as measured by thromboelastography and prolonged saphenous-vein bleeding times, consistent with FVIII deficiency. ARC19499 restored thromboelastography clotting times to baseline levels and corrected bleeding times. The authors concluded that ARC19499 inhibition of TFPI may be an effective alternative to current treatments of bleeding associated with haemophilia [132]. Unfortunately, these promising preclinical data were not confirmed by the phase I clinical trial.
Another approach to treating inhibitors is the replacement of FVIII procoagulant activity with a monoclonal antibody. Muto et al. recently identified an improved humanized bispecific antibody, ACE910, which mimics the action of FVIII. In a study in a nonhuman primate model of acquired haemophilia, they administered ACE910 or recombinant porcine FVIII (rpoFVIII). Results showed that a single bolus of 1 or 3 mg kg−1 ACE910 exhibited haemostatic activity comparable to that of 10 U kg−1 (twice daily) of rpoFVIII against bleedings [133]. An initial phase I study in 64 Japanese and Caucasian healthy adults indicated that ACE910 at doses up to 1 mg kg−1 had medically acceptable safety and tolerability profiles. These results suggest that ACE910 has therapeutic potential to prevent bleeding in haemophilia A patients. Recently, a new phase I study has been initiated to assess prophylactic efficacy as well as safety and pharmacokinetics in patients with/without inhibitors [134].
The final approach is prevention of inhibitor formation. Environmental risk factors for the development of high-titre inhibitors in haemophilia patients are duration of treatment at first FVIII exposure (RR = 4.1 [2.4–7.0] if FVIII is used for more than 5 consecutive days), reason for first FVIII treatment such as surgical procedure (RR = 3.2 [1.6–6.7]), and treatment intensity >50 IU kg−1 during the first 50 days of exposure (RR = 3.0 [1.3–6.9]) [11]. Prophylaxis was associated with a 42% lower risk for the development of high-titre inhibitors compared to on-demand treatment (crude HR, 0.58; 95% CI, 0.36–0.92). However, the inhibitor incidences in patients receiving prophylaxis and those receiving on-demand treatment did not differ during the first 20 EDs, and only after 20 EDs a markedly lower inhibitor incidence among patients receiving prophylaxis was observed [135], Fig. 7.
Fig. 7.
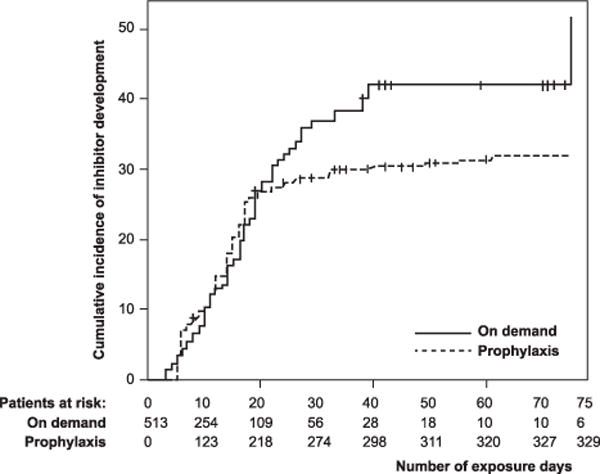
Regular prophylaxis and the cumulative incidence of inhibitor development. The RR for regular prophylaxis was 1.0 in the period from 1 to 10 exposure days, 0.95 from 11 to 20 exposure days, 0.22 from 21 to 30 exposure days, 0.27 from 31 to 40 exposure days and 0.32 from 41 to 75 exposure days [135].
In a study of 574 PUPs with severe haemophilia A treated with various FVIII concentrates, inhibitory antibodies developed in 177 of the 574 children (cumulative incidence, 32.4%). Recombinant and plasma-derived FVIII products conferred similar risks of inhibitor development, and the content of VWF in the products and switching among products were not associated with the risk of inhibitor development [18,38]. Second-generation full-length recombinant products were associated with an increased risk of inhibitor development, as compared with third-generation products [38]. Another study also confirmed the higher risk of inhibitor development with a second-generation full-length rFVIII product compared to other rFVIII products in PUPs with severe haemophilia A [42].
In conclusion, there are still many challenges to overcome to optimize haemophilia care using bypassing agents in patients with haemophilia who have developed inhibitors. Innovative possibilities to choose the product and tailor the dose for achieving effective haemostatic response should be promoted, in a new era of personalized medicine (associated with optimization of cost), particularly with regard to prophylaxis in inhibitor patients. Rebalancing the coagulation system by blocking the inhibitors of coagulation could represent a valuable treatment option. Replacement of FVIII with a bi-functional monoclonal antibody could result in efficient prophylaxis in haemophilia A patients with inhibitors. The future of the therapeutic modalities that could be used in patients with inhibitors might reduce the burden of the recurrent bleeding events, but it could also be more complicated due to the diverse modes of action of the drug candidates in an era where individualization of therapy is likely to play a major role.
Acknowledgments
Writing support was provided by Ros Kenn, freelance medical editor/writer, and funded by Octapharma.
Footnotes
Disclosures
Y. Dargaud has received speaker’s fees from Bayer, Baxter, CSL Behring, NovoNordisk, Octapharma, Pfizer and Stago; honoraria for attending advisory boards for NovoNordisk and Baxter, and has received research support from Alnylam, Bayer, CSL Behring, LFB, Novo Nordisk, Octapharma and Pfizer. A. Pavlova has received a fee from Octapharma and NovoNordisk for presentations. S. Lacroix-Desmazes has no conflicts of interest to declare. K. Fischer has received speaker’s fees from Bayer, Baxter, CSL Behring, Octapharma, Pfizer and NovoNordisk; performed consultancy for Bayer, Baxter, Biogen, NovoNordisk and Pfizer; and has received research support from Bayer, Wyeth/Pfizer, Baxter and Novo Nordisk. M. Soucie has no conflicts of interest to declare. S. Claeyssens has received a fee from Octapharma. D. Scott has no disclosures except for patent filed with Henry Jackson. Foundation for Engineered Tregs with funding from NIH. R. d’Oiron has received fees or honoraria for attending advisory boards, consultancy or speaking at symposia from Baxter, Bayer, NovoNordisk, Sobi, Pfizer and CSL Behring. G. Lavigne has no conflicts of interest to declare. G. Kenet has acted as a consultant for Prolor Biotech, Bayer, Pfizer, NovoNordisk and received research grants from BPL and Baxter. C. Escuriola Ettingshausen has received research support from Baxter, Biotest, CSL Behring, Octapharma and BPL, travel support from Baxter, Bayer, Biotest, CSL Behring, Grifols and Octapharma, consultancy fees from Octapharma and CSL Behring and honoraria from Baxter, Biotest, CSL Behring, Grifols, Octapharma and NovoNordisk. A. Borel-Derlon has received financial support for haemophilia A research grant from Bayer, SCL Behring and LFB. She has not received personal payment from industry adapted to haemophilia treatments. T. Lambert has received fees for consulting and/or reimbursement for attending symposia from Baxter, Bayer, Biogen, IPSPM, LFB, NovoNordisk, Octapharma, Pfizer and Roche. G. Pasta has been a paid consultant to NovoNordisk, Pfizer, Bayer-Schering and Octapharma. C. Négrier has received consultancy fees, travel support and research support from Alnylam, Baxter, Bayer, Biogen Idec, CSL Behring, Inspiration, LFB, Novo Nordisk, Octapharma, Pfizer and has been on scientific advisory boards.
References
- 1.Saint-Remy JM, Lacroix-Desmazes S, Oldenburg J. Inhibitors in haemophilia: pathophysiology. Haemophilia. 2004;10(Suppl 4):146–51. doi: 10.1111/j.1365-2516.2004.01009.x. [DOI] [PubMed] [Google Scholar]
- 2.Lozner EL, Joliffe LS, Taylor FHL. Hemorrhagic diathesis with prolonged coagulation time, associated with a circulating anticoagulant. Am J Med Sci. 1940;199:318. [Google Scholar]
- 3.Gitschier J, Wood WI, Tuddenham EG, et al. Detection and sequence of mutations in the factor VIII gene of haemophiliacs. Nature. 1985;315:427–30. doi: 10.1038/315427a0. [DOI] [PubMed] [Google Scholar]
- 4.Oldenburg J, Picard JK, Schwaab R, Brackmann HH, Tuddenham EG, Simpson E. HLA genotype of patients with severe haemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost. 1997;77:238–42. [PubMed] [Google Scholar]
- 5.Astermark J, Oldenburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in patients with hemophilia A. Blood. 2006;108:3739–45. doi: 10.1182/blood-2006-05-024711. [DOI] [PubMed] [Google Scholar]
- 6.Pavlova A, Delev D, Lacroix-Desmazes S, et al. Impact of polymorphisms of the major histocompatibility complex class II, interleukin-10, tumor necrosis factor-alpha and cytotoxic T-lymphocyte antigen-4 genes on inhibitor development in severe hemophilia A. J Thromb Haemost. 2009;7:2006–15. doi: 10.1111/j.1538-7836.2009.03636.x. [DOI] [PubMed] [Google Scholar]
- 7.Schwaab R, Brackmann HH, Meyer C, et al. Haemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost. 1995;74:1402–8. [PubMed] [Google Scholar]
- 8.Gouw SC, van den Berg HM, Oldenburg J, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012;119:2922–34. doi: 10.1182/blood-2011-09-379453. [DOI] [PubMed] [Google Scholar]
- 9.Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122:1954–62. doi: 10.1182/blood-2013-02-483263. [DOI] [PubMed] [Google Scholar]
- 10.Astermark J, Berntorp E, White GC, Kroner BL, MIBS Study Group The Malmö International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia. 2001;7:267–72. doi: 10.1046/j.1365-2516.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- 11.Gouw SC, van der Bom JG, Marijke van den Berg H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109:4648–54. doi: 10.1182/blood-2006-11-056291. [DOI] [PubMed] [Google Scholar]
- 12.Viel KR, Ameri A, Abshire TC, et al. Inhibitors of factor VIII in black patients with haemophilia. N Engl J Med. 2009;360:1618–27. doi: 10.1056/NEJMoa075760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozier JN, Rosenberg PS, Goedert JJ, Menashe I. A case-control study reveals immunoregulatory gene haplotypes that influence inhibitor risk in severe haemophilia A. Haemophilia. 2011;17:641–9. doi: 10.1111/j.1365-2516.2010.02473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Astermark J, Wang X, Oldenburg J, Berntorp E, Lefvert AK, MIBS Study Group Polymorphisms in the CTLA-4 gene and inhibitor development in patients with severe hemophilia A. J Thromb Haemost. 2007;5:263–5. doi: 10.1111/j.1538-7836.2007.02290.x. [DOI] [PubMed] [Google Scholar]
- 15.Repessé Y, Peyron I, Dimitrov JD, et al. ABIRISK consortium Development of inhibitory antibodies to therapeutic factor VIII in severe hemophilia A is associated with microsatellite polymorphisms in the HMOX1 promoter. Haematologica. 2013;98:1650–5. doi: 10.3324/haematol.2013.084665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Astermark J, Donfield SM, Gomperts ED, et al. Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort The polygenic nature of inhibitors in hemophilia A: results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Blood. 2013;121:1446–64. doi: 10.1182/blood-2012-06-434803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurnik K, Bidlingmaier C, Engl W, Chehadeh H, Reipert B, Auerswald G. New early prophylaxis regimen that avoids immunological danger signals can reduce FVIII inhibitor development. Haemophilia. 2010;16:256–62. doi: 10.1111/j.1365-2516.2009.02122.x. [DOI] [PubMed] [Google Scholar]
- 18.Gouw SC, van der Bom JG, Auerswald G, Ettinghausen CE, Tedgård U, van den Berg HM. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007;109:4693–7. doi: 10.1182/blood-2006-11-056317. [DOI] [PubMed] [Google Scholar]
- 19.Chalmers EA, Brown SA, Keeling D, et al. Paediatric Working Party of UKHCDO Early factor VIII exposure and subsequent inhibitor development in children with severe haemophilia A. Haemophilia. 2007;13:149–55. doi: 10.1111/j.1365-2516.2006.01418.x. [DOI] [PubMed] [Google Scholar]
- 20.Moreau A, Lacroix-Desmazes S, Stieltjes N, et al. Antibodies to the FVIII light chain that neutralize FVIII procoagulant activity are present in plasma of nonresponder patients with severe hemophilia A and in normal polyclonal human IgG. Blood. 2000;95:3435–41. [PubMed] [Google Scholar]
- 21.Rossi F, Sultan Y, Kazatchkine MD. Anti-idiotypes against autoantibodies and alloantibodies to VIII: C (antihaemophilic factor) are present in therapeutic polyspecific normal immunoglobulins. Clin Exp Immunol. 1988;74:311–16. [PMC free article] [PubMed] [Google Scholar]
- 22.Reding MT, Wu HY, Krampf M, et al. CD4+ T cell response to factor VIII in hemophilia A, acquired hemophilia, and healthy subjects. Thromb Haemost. 1999;82:509–15. [PubMed] [Google Scholar]
- 23.Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope repertoire of human CD4(+) T cells on the A3 domain of coagulation factor VIII. J Thromb Haemost. 2004;2:1385–94. doi: 10.1111/j.1538-7836.2004.00850.x. [DOI] [PubMed] [Google Scholar]
- 24.Kamaté C, Lenting PJ, van den Berg HM, Mutis T. Depletion of CD4+/CD25high regulatory T cells may enhance or uncover factor VIII-specific T-cell responses in healthy individuals. J Thromb Haemost. 2007;5:611–13. doi: 10.1111/j.1538-7836.2007.02336.x. [DOI] [PubMed] [Google Scholar]
- 25.Lacroix-Desmazes S, Navarrete AM, André S, Bayry J, Kaveri SV, Dasgupta S. Dynamics of factor VIII interactions determine its immunologic fate in hemophilia A. Blood. 2008;112:240–9. doi: 10.1182/blood-2008-02-124941. [DOI] [PubMed] [Google Scholar]
- 26.Astermark J, Altisent C, Batorova A, et al. Non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16:747–66. doi: 10.1111/j.1365-2516.2010.02231.x. [DOI] [PubMed] [Google Scholar]
- 27.Hakobyan N, Enockson C, Cole AA, Sumner DR, Valentino LA. Experimental haemophilic arthropathy in a mouse model of a massive haemarthrosis: gross, radiological and histological changes. Haemophilia. 2008;14:804–9. doi: 10.1111/j.1365-2516.2008.01689.x. [DOI] [PubMed] [Google Scholar]
- 28.Øvlisen K, Kristensen AT, Jensen AL, Tranholm M. IL-1 beta, IL-6, KC and MCP-1 are elevated in synovial fluid from haemophilic mice with experimentally induced haemarthrosis. Haemophilia. 2009;15:802–10. doi: 10.1111/j.1365-2516.2008.01973.x. [DOI] [PubMed] [Google Scholar]
- 29.Peyron I, Dimitrov JD, Delignat S, et al. Haemarthrosis and arthropathy do not favour the development of factor VIII inhibitors in severe haemophilia A mice. Haemophilia. 2015;21:e94–8. doi: 10.1111/hae.12579. [DOI] [PubMed] [Google Scholar]
- 30.Saenko EL, Yakhyaev AV, Mikhailenko I, Strickland DK, Sarafanov AG. Role of the low density lipoprotein-related protein receptor in mediation of factor VIII catabolism. J Biol Chem. 1999;274:37685–92. doi: 10.1074/jbc.274.53.37685. [DOI] [PubMed] [Google Scholar]
- 31.Sarafanov AG, Ananyeva NM, Shima M, Saenko EL. Cell surface heparan sulfate proteoglycans participate in factor VIII catabolism mediated by low density lipoprotein receptor-related protein. J Biol Chem. 2001;276:11970–9. doi: 10.1074/jbc.M008046200. [DOI] [PubMed] [Google Scholar]
- 32.Bovenschen N, Mertens K, Hu L, Havekes LM, van Vlijmen BJ. LDL receptor cooperates with LDL receptor-related protein in regulating plasma levels of coagulation factor VIII in vivo. Blood. 2005;106:906–12. doi: 10.1182/blood-2004-11-4230. [DOI] [PubMed] [Google Scholar]
- 33.Bovenschen N, Rijken DC, Havekes LM, van Vlijmen BJ, Mertens K. The B domain of coagulation factor VIII interacts with the asialoglycoprotein receptor. J Thromb Haemost. 2005;3:1257–65. doi: 10.1111/j.1538-7836.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 34.Dasgupta S, Navarrete AM, André S, et al. Factor VIII bypasses CD91/LRP for endocytosis by dendritic cells leading to T-cell activation. Haematologica. 2008;93:83–9. doi: 10.3324/haematol.11535. [DOI] [PubMed] [Google Scholar]
- 35.Dasgupta S, Navarrete AM, Bayry J, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci USA. 2007;104:8965–70. doi: 10.1073/pnas.0702120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navarrete A, Dasgupta S, Delignat S, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. 2009;7:1816–23. doi: 10.1111/j.1538-7836.2009.03571.x. [DOI] [PubMed] [Google Scholar]
- 37.Makris M, Calizzani G, Fischer K, et al. EUHASS: the European haemophilia safety surveillance system. Thromb Res. 2011;127(Suppl 2):S22–5. doi: 10.1016/S0049-3848(10)70150-X. [DOI] [PubMed] [Google Scholar]
- 38.Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368:231–9. doi: 10.1056/NEJMoa1208024. [DOI] [PubMed] [Google Scholar]
- 39.Collins PW, Palmer BP, Chalmers EA, et al. UK Haemophilia Centre Doctors’ Organization Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000–2011. Blood. 2014;124:3389–97. doi: 10.1182/blood-2014-07-580498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer K, Lewandowski D, Marijke van den Berg H, Janssen MP. Validity of assessing inhibitor development in haemophilia PUPs using registry data: the EUHASS project. Haemophilia. 2012;18:e241–6. doi: 10.1111/j.1365-2516.2011.02687.x. [DOI] [PubMed] [Google Scholar]
- 41.Kreuz W, Gill JC, Rothschild C, et al. International Kogenate-FS Study Group Full-length sucrose-formulated recombinant factor VIII for treatment of previously untreated or minimally treated young children with severe haemophilia A: results of an international clinical investigation. Thromb Haemost. 2005;93:457–67. doi: 10.1160/TH03-10-0643. [DOI] [PubMed] [Google Scholar]
- 42.Calvez T, Chambost H, Claeyssens-Donadel S, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124:3398–408. doi: 10.1182/blood-2014-07-586347. [DOI] [PubMed] [Google Scholar]
- 43.Fischer K, Lassila R, Peyvandi F, et al. Inhibitor development in haemophilia according to concentrate. Four-year results from the European HAemophilia Safety Surveillance (EUHASS) project. Thromb Haemost. 2015;114:670–5. doi: 10.1160/TH14-10-0826. [DOI] [PubMed] [Google Scholar]
- 44.Soucie JM, Miller CH, Kelly FM, et al. Haemophilia Inhibitor Research Study Investigators A study of prospective surveillance for inhibitors among persons with haemophilia in the United States. Haemophilia. 2014;20:230–7. doi: 10.1111/hae.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller CH, Benson J, Ellingsen D, et al. HemophiliaInhibitor Research Study Investigators F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18:375–82. doi: 10.1111/j.1365-2516.2011.02700.x. [DOI] [PubMed] [Google Scholar]
- 46.Tang Q, Bluestone JA, Kang SM. CD4(+) Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol. 2012;4:11–21. doi: 10.1093/jmcb/mjr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Putnam AL, Safinia N, Medvec A, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. 2013;13:3010–20. doi: 10.1111/ajt.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–35. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional expression of chimeric receptor genes in human T cells. J Immunol Methods. 2001;248:67–76. doi: 10.1016/s0022-1759(00)00343-4. [DOI] [PubMed] [Google Scholar]
- 50.Kim YC, Zhang AH, Su Y, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. 2015;125:1107–15. doi: 10.1182/blood-2014-04-566786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YC, Bhairavabhotla R, Yoon J, et al. Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood. 2012;119:2810–18. doi: 10.1182/blood-2011-09-377895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moorehead PC. Engineering regulatory T cells against factor VIII inhibitors. Blood. 2015;125:1053–4. doi: 10.1182/blood-2014-12-617530. [DOI] [PubMed] [Google Scholar]
- 53.Naumann A, Scherger AK, Neuwirth J, et al. Selection and characterisation of FVIII-specific single chain variable fragments. Hamostaseologie. 2013;33(Suppl 1):S39–45. [PubMed] [Google Scholar]
- 54.Valentino LA. Assessing the benefits of FEIBA prophylaxis in haemophilia patients with inhibitors. Haemophilia. 2010;16:263–71. doi: 10.1111/j.1365-2516.2009.02126.x. [DOI] [PubMed] [Google Scholar]
- 55.Young G, Auerswald G, Jimenez-Yuste V, et al. PRO-PACT: retrospective observational study on the prophylactic use of recombinant factor VIIa in hemophilia patients with inhibitors. Thromb Res. 2012;130:864–70. doi: 10.1016/j.thromres.2012.08.305. [DOI] [PubMed] [Google Scholar]
- 56.Konkle BA, Ebbesen LS, Erhardtsen E, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5:1904–13. doi: 10.1111/j.1538-7836.2007.02663.x. [DOI] [PubMed] [Google Scholar]
- 57.Leissinger C, Gringeri A, Antmen B, et al. Anti-inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N Engl J Med. 2011;365:1684–92. doi: 10.1056/NEJMoa1104435. [DOI] [PubMed] [Google Scholar]
- 58.Antunes SV, Tangada S, Stasyshyn O, et al. Randomized comparison of prophylaxis and on-demand regimens with FEIBA NF in the treatment of haemophilia A and B with inhibitors. Haemophilia. 2014;20:65–72. doi: 10.1111/hae.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saenko EL, Scandella D. The acidic region of the factor VIII light chain and the C2 domain together form the high affinity binding site for von Willebrand factor. J Biol Chem. 1997;272:18007–14. doi: 10.1074/jbc.272.29.18007. [DOI] [PubMed] [Google Scholar]
- 60.Barrow RT, Healey JF, Jacquemin MG, Saint-Remy JM, Lollar P. Antigenicity of putative phospholipid membrane-binding residues in factor VIII. Blood. 2001;97:169–74. doi: 10.1182/blood.v97.1.169. [DOI] [PubMed] [Google Scholar]
- 61.Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16:597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 62.Ware J, MacDonald MJ, Lo M, de Graaf S, Fulcher CA. Epitope mapping of human factor VIII inhibitor antibodies by site-directed mutagenesis of a factor VIII polypeptide. Blood Coagul Fibrinolysis. 1992;3:703–16. doi: 10.1097/00001721-199212000-00002. [DOI] [PubMed] [Google Scholar]
- 63.Fijnvandraat K, Celie PH, Turenhout EA, et al. A human alloantibody interferes with binding of factor IXa to the factor VIII light chain. Blood. 1998;91:2347–52. [PubMed] [Google Scholar]
- 64.Jacquemin M, Lavend’homme R, Benhida A, et al. A novel cause of mild/moderate hemophilia A: mutations scattered in the factor VIII C1 domain reduce factor VIII binding to von Willebrand factor. Blood. 2000;96:958–65. [PubMed] [Google Scholar]
- 65.Vincent AM, Lillicrap D, Boulanger A, et al. Non-neutralizing anti-FVIII antibodies: different binding specificity to different recombinant FVIII concentrates. Haemophilia. 2009;15:374–6. doi: 10.1111/j.1365-2516.2008.01909.x. [DOI] [PubMed] [Google Scholar]
- 66.Vianello F, Radossi P, Tison T, et al. Prevalence of anti-FVIII antibodies in severe haemophilia A patients with inversion of intron 22. Br J Haematol. 1997;97:807–9. doi: 10.1046/j.1365-2141.1997.1082922.x. [DOI] [PubMed] [Google Scholar]
- 67.Lacroix-Desmazes S, Moreau A, SooryaNarayana, et al. Catalytic activity of antibodies against factor VIII in patients with hemophilia A. Nat Med. 1999;5:1044–7. doi: 10.1038/12483. [DOI] [PubMed] [Google Scholar]
- 68.Lavigne-Lissalde G, Rothschild C, Pouplard C, et al. Characteristics, mechanisms of action, and epitope mapping of anti-factor VIII antibodies. Clin Rev Allergy Immunol. 2009;37:67–79. doi: 10.1007/s12016-009-8119-0. [DOI] [PubMed] [Google Scholar]
- 69.Reding MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1-and Th2-induced anti-factor VIII IgG subclasses in congenital and acquired hemophilia patients. Thromb Haemost. 2002;88:568–75. [PubMed] [Google Scholar]
- 70.Scandella D, Mattingly M, Prescott R. A recombinant factor VIII A2 domain polypeptide quantitatively neutralizes human inhibitor antibodies that bind to A2. Blood. 1993;82:1767–75. [PubMed] [Google Scholar]
- 71.Lavigne-Lissalde G, Tarrade C, Lapalud P, et al. Simultaneous detection and epitope mapping of anti-factor VIII antibodies. Thromb Haemost. 2008;99:1090–6. doi: 10.1160/TH07-08-0497. [DOI] [PubMed] [Google Scholar]
- 72.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110:4234–42. doi: 10.1182/blood-2007-06-096842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lapalud P, Ali T, Cayzac C, et al. The IgG autoimmune response in postpartum acquired hemophilia A targets mainly the A1a1 domain of FVIII. J Thromb Haemost. 2012;10:1814–22. doi: 10.1111/j.1538-7836.2012.04850.x. [DOI] [PubMed] [Google Scholar]
- 74.Greninger DA, Saint-Remy JM, Jacquemin M, Benhida A, DiMichele DM. The use of factor VIII/von Willebrand factor concentrate for immune tolerance induction in haemophilia A patients with high-titre inhibitors: association of clinical outcome with inhibitor epitope profile. Haemophilia. 2008;14:295–302. doi: 10.1111/j.1365-2516.2007.01620.x. [DOI] [PubMed] [Google Scholar]
- 75.van Helden PM, van den Berg HM, Gouw SC, et al. IgG subclasses of anti-FVIII antibodies during immune tolerance induction in patients with hemophilia A. Br J Haematol. 2008;142:644–52. doi: 10.1111/j.1365-2141.2008.07232.x. [DOI] [PubMed] [Google Scholar]
- 76.Lapalud P, Rothschild C, Mathieu-Dupas E, et al. Anti-A2 and anti-A1 domain antibodies are potential predictors of immune tolerance induction outcome in children with hemophilia A. Thromb Haemost. 2015;13:540–7. doi: 10.1111/jth.12846. [DOI] [PubMed] [Google Scholar]
- 77.Moreau V, Fleury C, Piquer D, et al. PEPOP: computational design of immunogenic peptides. BMC Bioinformatics. 2008;9:71. doi: 10.1186/1471-2105-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lebreton A, Moreau V, Lapalud P, et al. Discontinuous epitopes on the C2 domain of coagulation factor VIII mapped by computer-designed synthetic peptides. Br J Haematol. 2011;155:487–97. doi: 10.1111/j.1365-2141.2011.08878.x. [DOI] [PubMed] [Google Scholar]
- 79.Hay CR, DiMichele DM, International Immune Tolerance Study The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119:1335–44. doi: 10.1182/blood-2011-08-369132. [DOI] [PubMed] [Google Scholar]
- 80.Schneiderman J, Rubin E, Nugent DJ, Young G. Sequential therapy with activated prothrombin complex concentrates and recombinant FVIIa in patients with severe haemophilia and inhibitors: update of our previous experience. Haemophilia. 2007;13:244–8. doi: 10.1111/j.1365-2516.2007.01451.x. [DOI] [PubMed] [Google Scholar]
- 81.Martinowitz U, Livnat T, Zivelin A, Kenet G. Concomitant infusion of low doses of rFVIIa and FEIBA in haemophilia patients with inhibitors. Haemophilia. 2009;15:904–10. doi: 10.1111/j.1365-2516.2009.02028.x. [DOI] [PubMed] [Google Scholar]
- 82.Doshi BS, Gangadharan B, Doering CB, Meeks SL. Potentiation of thrombin generation in haemophilia A plasma by coagulation factor VIII and characterization of antibody-specific inhibition. PLoS ONE. 2012;7:e48172. doi: 10.1371/journal.pone.0048172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Livnat T, Martinowitz U, Azar-Avivi S, et al. Combined administration of FVIII and rFVIIa improves haemostasis in haemophilia A patients with high-responding inhibitors–a thrombin generation-guided pilot study. Haemophilia. 2013;19:782–9. doi: 10.1111/hae.12181. [DOI] [PubMed] [Google Scholar]
- 84.Benson G, Auerswald G, Elezović I, et al. Immune tolerance induction in patients with severe hemophilia with inhibitors: expert panel views and recommendations for clinical practice. Eur J Haematol. 2012;88:371–9. doi: 10.1111/j.1600-0609.2012.01754.x. [DOI] [PubMed] [Google Scholar]
- 85.Collins PW, Mathias M, Hanley J, et al. UK Haemophilia Centre Doctors’ Organisation Rituximab and immune tolerance in severe hemophilia A: a consecutive national cohort. J Thromb Haemost. 2009;7:787–94. doi: 10.1111/j.1538-7836.2009.03332.x. [DOI] [PubMed] [Google Scholar]
- 86.Nakar C, Manco-Johnson MJ, Lail A, et al. Prompt immune tolerance induction at inhibitor diagnosis regardless of titre may increase overall success in haemophilia A complicated by inhibitors: experience of two US centres. Haemophilia. 2015;21:365–73. doi: 10.1111/hae.12608. [DOI] [PubMed] [Google Scholar]
- 87.Observational Immune Tolerance Induction research program. Available at: http://www.obsiti.com/at-a-glance.php (accessed 16 January 2015) and Clini-calTrials.gov Identifier: NCT02207894.
- 88.Kreuz W, Escuriola Ettingshausen C, Vdovin V, Zozulya N, Plyushch O, Svirin P, et al. First prospective report on immune tolerance in poor risk haemophilia A inhibitor patients with a single factor VIII/von Willebrand factor concentrate in an observational immune tolerance induction study. Haemophilia. 2015 Jul 23; doi: 10.1111/hae.12774. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 89.Oldenburg J, Austin SK, Kessler CM. ITI choice for the optimal management of inhibitor patients - from a clinical and pharmacoeconomic perspective. Haemophilia. 2014;20(Suppl 6):17–26. doi: 10.1111/hae.12466. [DOI] [PubMed] [Google Scholar]
- 90.Kreuz W, Becker S, Lenz E, et al. Factor VIII inhibitors in patients with hemophilia A: epidemiology of inhibitor development and induction of immune tolerance for factor VIII. Semin Thromb Haemost. 1995;21:382–9. doi: 10.1055/s-2007-1000659. [DOI] [PubMed] [Google Scholar]
- 91.Auerswald G, Spranger T, Brackmann HH. The role of plasma-derived factor VIII/von Willebrand factor concentrates in the treatment of hemophilia A patients. Haematologica. 2003(88):EREPO5. [PubMed] [Google Scholar]
- 92.Kreuz W, Escuriola Ettingshausen C, Auerswald G, et al. Immune tolerance induction (ITI) in haemophilia A patients with inhibitors – the choice of concentrate affecting success. Haematologica. 2001;86(Suppl 4):16–22. [Google Scholar]
- 93.Study of First TIME Immunotolerance Induction in Severe Hemophilia A Patients With Inhibitor at High Risk of Failure: Comparison With FVIII Concentrates With or Without Von Willebrand Factor - RES.I.S.T. Naive (RESIST NAIVE). ClinicalTrials.gov Identifier: NCT01051544.
- 94.Rescue Immunotolerance Study in Induction of Immune Tolerance (ITI)-Experienced Patients (RES.I.S.T. Experienced) (RESIST EXP). ClinicalTrials.gov Identifier: NCT01051076.
- 95.Rodriguez-Merchan EC, Wiedel JD, Wallny T, et al. Elective orthopaedic surgery for inhibitor patients. Haemophilia. 2003;9:625–31. doi: 10.1046/j.1365-2516.2003.00803.x. [DOI] [PubMed] [Google Scholar]
- 96.Rodriguez-Merchan EC, Hedner U, Heijnen L, et al. Prevention of haemophilic arthropathy during childhood. May common orthopaedic management be extrapolated from patients without inhibitors to patients with inhibitors? Haemophilia. 2008;14(Suppl 6):68–81. doi: 10.1111/j.1365-2516.2008.01892.x. [DOI] [PubMed] [Google Scholar]
- 97.Rodriguez-Merchan EC, Quntana M, Jimenez-Yuste V. Orthopaedic surgery in haemophilia patients with inhibitors as the last resort. Haemophilia. 2008;14(Suppl 6):57–8. doi: 10.1111/j.1365-2516.2008.01891.x. [DOI] [PubMed] [Google Scholar]
- 98.Rodriguez-Merchan EC, Rocino A, Ewenstein B, et al. Consensus perspectives on surgery in haemophilia patients with inhibitors: summary statement. Haemophilia. 2004;10(Suppl 2):50–2. doi: 10.1111/j.1365-2516.2004.00941.x. [DOI] [PubMed] [Google Scholar]
- 99.Caviglia H, Candela M, Galatro G, Neme D, Moretti N, Bianco RP. Elective orthopaedic surgery for haemophilia patients with inhibitors: single centre experience of 40 procedures and review of the literature. Haemophilia. 2011;17:910–19. doi: 10.1111/j.1365-2516.2011.02504.x. [DOI] [PubMed] [Google Scholar]
- 100.Ingerslev J, Freidman D, Gastineau D, et al. Major surgery in haemophilic patients with inhibitors using recombinant factor VIIa. Haemostasis. 1996;26(Suppl 1):118–23. doi: 10.1159/000217252. [DOI] [PubMed] [Google Scholar]
- 101.Rodriguez-Merchan EC, Quintana M, Jimenez-Yuste V, Hernández-Navarro F. Orthopaedic surgery for inhibitor patients: a series of 27 procedures (25 patients) Haemophilia. 2007;13:613–19. doi: 10.1111/j.1365-2516.2007.01520.x. [DOI] [PubMed] [Google Scholar]
- 102.Goddard N. Case studies: orthopaedic surgery in adult patients with haemophilia A with inhibitors. Haemophilia. 2005;11(Suppl 1):32–7. doi: 10.1111/j.1365-2516.2005.01155.x. [DOI] [PubMed] [Google Scholar]
- 103.Solimeno LP, Mancuso ME, Pasta G, Santagostino E, Perfetto S, Mannucci PM. Factors influencing the long-term outcome of primary total knee replacement in haemophiliacs: a review of 116 procedures at a single institution. Br J Haematol. 2009;145:227–34. doi: 10.1111/j.1365-2141.2009.07613.x. [DOI] [PubMed] [Google Scholar]
- 104.Moonen AF, Neal TD, Pilot P. Peri-operative blood management in elective orthopaedic surgery. A critical review of the literature Injury. 2006;37(Suppl 5):11–16. doi: 10.1016/S0020-1383(07)70006-2. [DOI] [PubMed] [Google Scholar]
- 105.Ludham C. Identifying and managing inhibitor patients requiring orthopaedic surgery - the multidisciplinary team approach. Haemophilia. 2005;11(Suppl 1):7–10. doi: 10.1111/j.1365-2516.2005.01152.x. [DOI] [PubMed] [Google Scholar]
- 106.Teitel JM, Carcao M, Lillicrap D, et al. Orthopaedic surgery in haemophilia patients with inhibitors: a practical guide to haemostatic, surgical and rehabilitative care. Haemophilia. 2009;15:227–39. doi: 10.1111/j.1365-2516.2008.01840.x. [DOI] [PubMed] [Google Scholar]
- 107.Quintana-Molina M, Martínez-Bahamonde F, González-García E, et al. Surgery in haemophilic patients with inhibitor: 20 years of experience. Haemophilia. 2004;10(Suppl 2):30–40. doi: 10.1111/j.1365-2516.2004.00938.x. [DOI] [PubMed] [Google Scholar]
- 108.Astermark J, Donfield SM, DiMichele DM, et al. FENOC Study Group A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) Study. Blood. 2007;109:546–51. doi: 10.1182/blood-2006-04-017988. [DOI] [PubMed] [Google Scholar]
- 109.Hedner U, Glazer S, Pingel K, et al. Successful use of recombinant factor VIIa in patient with severe haemophilia A during synovectomy. Lancet. 1988;2:1193. doi: 10.1016/s0140-6736(88)90259-0. [DOI] [PubMed] [Google Scholar]
- 110.Shapiro AD, Gilchrist GS, Hoots WK, Cooper HA, Gastineau DA. Prospective, randomised trial of two doses of rFVIIa (NovoSeven) in haemophilia patients with inhibitors undergoing surgery. Thromb Haemost. 1998;80:773–8. [PubMed] [Google Scholar]
- 111.Pruthi RK, Mathew P, Valentino LA, Sumner MJ, Seremetis S, Hoots WK, NovoSeven in Surgery Study Investigators Haemostatic efficacy and safety of bolus and continuous infusion of recombinant factor VIIa are comparable in haemophilia patients with inhibitors undergoing major surgery. Results from an open-label, randomized, multicenter trial. Thromb Haemost. 2007;98:726–32. [PubMed] [Google Scholar]
- 112.Obergfell A, Auvinen MK, Mathew P. Recombinant activated factor VII for haemophilia patients with inhibitors undergoing orthopaedic surgery: a review of the literature. Haemophilia. 2008;14:233–41. doi: 10.1111/j.1365-2516.2007.01617.x. [DOI] [PubMed] [Google Scholar]
- 113.Valentino LA, Cooper DL, Goldstein B. Surgical experience with rFVIIa (NovoSeven) in congenital haemophilia A and B patients with inhibitors to factors VIII or IX. Haemophilia. 2011;17:579–89. doi: 10.1111/j.1365-2516.2010.02460.x. [DOI] [PubMed] [Google Scholar]
- 114.Giangrande PL, Wilde JT, Madan B, et al. Consensus protocol for the use of recombinant activated factor VII [eptacog alfa (activated); NovoSeven] in elective orthopaedic surgery in haemophilic patients with inhibitors. Haemophilia. 2009;15:501–8. doi: 10.1111/j.1365-2516.2008.01952.x. [DOI] [PubMed] [Google Scholar]
- 115.Rangarajan S, Yee TT, Wilde J. Experience of four UK comprehensive care centres using FEIBA® for surgeries in patients with inhibitors. Haemophilia. 2011;17:28–34. doi: 10.1111/j.1365-2516.2010.02360.x. [DOI] [PubMed] [Google Scholar]
- 116.Négrier C, Lienhart A, Numerof R, et al. SURgical interventions with FEIBA (SURF): international registry of surgery in haemophilia patients with inhibitory antibodies. Haemophilia. 2013;19:e143–50. doi: 10.1111/hae.12080. [DOI] [PubMed] [Google Scholar]
- 117.Rangarajan S, Austin S, Goddard NJ, et al. Consensus recommendations for the use of FEIBA(®) in haemophilia A patients with inhibitors undergoing elective orthopaedic and non-orthopaedic surgery. Haemophilia. 2013;19:294–303. doi: 10.1111/hae.12028. [DOI] [PubMed] [Google Scholar]
- 118.Tjønnfjord GE, Holme PA. Factor eight inhibitor bypass activity (FEIBA) in the management of bleeds in hemophilia patients with high-titer inhibitors. Vasc Health Risk Manag. 2007;3:527–31. [PMC free article] [PubMed] [Google Scholar]
- 119.Négrier C, Goudemand J, Sultan Y, Bertrand M, Rothschild C, Lauroua P. Multicenter retrospective study on the utilization of FEIBA in France in patients with factor VIII and factor IX inhibitors. French FEIBA Study Group. Factor Eight Bypassing Activity. Thromb Haemost. 1997;77:1113–19. [PubMed] [Google Scholar]
- 120.Freydin M. A review of treatment options for hemophilia. J Young Invest. 2007 [serial online] http://www.jyi.org/issue/a-review-of-treatment-options-for-hemophilia/
- 121.Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost. 1998;80:912–18. [PubMed] [Google Scholar]
- 122.Schulman S. Safety, efficacy and lessons from continuous infusion with rFVIIa. rFVIIa-CI Group. Haemophilia. 1998;4:564–7. doi: 10.1046/j.1365-2516.1998.440564.x. [DOI] [PubMed] [Google Scholar]
- 123.Holmström M, Tran HT, Holme PA. Combined treatment with APCC (FEIBA®) and tranexamic acid in patients with haemophilia A with inhibitors and in patients with acquired haemophilia A– a two-centre experience. Haemophilia. 2012;18:544–9. doi: 10.1111/j.1365-2516.2012.02748.x. [DOI] [PubMed] [Google Scholar]
- 124.Ingerslev J, Sørensen B. Parallel use of by-passing agents in haemophilia with inhibitors: a critical review. Br J Haematol. 2011;155:256–62. doi: 10.1111/j.1365-2141.2011.08854.x. [DOI] [PubMed] [Google Scholar]
- 125.van Veen JJ, Maclean RM, Hampton KK, Hamer A, Makris M. Major surgery in severe haemophilia A with inhibitors using a recombinant factor VIIa and activated prothrombin complex concentrate hybrid regimen. Haemophilia. 2014;20:587–92. doi: 10.1111/hae.12365. [DOI] [PubMed] [Google Scholar]
- 126.Dargaud Y, Lienhart A, Negrier C. Prospective assessment of thrombin generation test for dose monitoring of bypassing therapy in hemophilia patients with inhibitors undergoing elective surgery. Blood. 2010;116:5734–7. doi: 10.1182/blood-2010-06-291906. [DOI] [PubMed] [Google Scholar]
- 127.Dargaud Y, Lienhart A, Meunier S, et al. Major surgery in a severe haemophilia A patient with high titre inhibitor: use of the thrombin generation test in the therapeutic decision. Haemophilia. 2005;11:552–8. doi: 10.1111/j.1365-2516.2005.01141.x. [DOI] [PubMed] [Google Scholar]
- 128.Soucie JM, Cianfrini C, Janco RL, et al. Joint range-of-motion limitations among young males with hemophilia: prevalence and risk factors. Blood. 2004;103:2467–73. doi: 10.1182/blood-2003-05-1457. [DOI] [PubMed] [Google Scholar]
- 129.Dargaud Y, Prevost C, Lienhart A, Claude Bordet J, Negrier C. Evaluation of the overall haemostatic effect of recombinant factor VIIa by measuring thrombin generation and stability of fibrin clots. Haemophilia. 2011;17:957–61. doi: 10.1111/j.1365-2516.2011.02526.x. [DOI] [PubMed] [Google Scholar]
- 130.Toschi V. OBI–1, porcine recombinant Factor VIII for the potential treatment of patients with congenital hemophilia A and alloantibodies against human Factor VIII. Curr Opin Mol Ther. 2010;12:617–25. [PubMed] [Google Scholar]
- 131.Oldenburg J, El-Maarri O, Schwaab R. Inhibitor development in correlation to factor VIII genotypes. Haemophilia. 2002;8(Suppl 2):23–9. doi: 10.1046/j.1351-8216.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- 132.Waters EK, Genga RM, Schwartz MC, et al. Aptamer ARC19499 mediates a procoagulant hemostatic effect by inhibiting tissue factor pathway inhibitor. Blood. 2011;117:5514–22. doi: 10.1182/blood-2010-10-311936. [DOI] [PubMed] [Google Scholar]
- 133.Muto A, Yoshihashi K, Takeda M, et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12:206–13. [PubMed] [Google Scholar]
- 134.Shima M, Hermans C, de Moerloose P. Novel products for haemostasis. Haemophilia. 2014;20(Suppl 4):29–35. doi: 10.1111/hae.12413. [DOI] [PubMed] [Google Scholar]
- 135.Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121:4046–55. doi: 10.1182/blood-2012-09-457036. [DOI] [PubMed] [Google Scholar]