

Introduction
The total synthesis of complex organic architectures is often complicated by the presence of several different functional groups in the target molecule. These groups often present significant synthetic challenges, particularly if the reactions in a preparative sequence may affect one or more of these functional groups. Typically, protecting groups are employed to temporarily render the reactive functionalities inert while transformations are performed at other sites in the molecule. The protecting groups are then unmasked at strategic points later in the synthetic route. The choice of protecting group is often a key decision, as problems with their introduction or removal may affect the viability of the synthetic sequence. Alternatively, protecting groups can become a liability should they decompose at undesired times or contribute to the instability of intermediates. Therefore, efforts have been focused on developing reliable systems that have sufficient stability to withstand many common transformations but that can still be removed under mild conditions when desired. These efforts have resulted in the development of many different blocking groups for a variety of functionalities.1–3 In the search for the optimal protecting group, predicting which one will perform best in a complex system can be difficult. One approach to address this challenge is to use protecting groups that can be installed and removed under a variety of conditions, ensuring that options will be available if the planned installation or removal leads to unforeseen substrate decomposition.
To protect the carboxylate group, the 4-methoxybenzyl (PMB) ester has become known as an inexpensive “workhorse” protecting group. This ester may be readily introduced in high yield under a number of mild reaction conditions. The PMB ester possesses excellent stability under many reaction conditions; therefore, it can be used in a variety of settings. In addition, when the removal of a PMB ester is desired, many orthogonal methods may be employed to induce cleavage. This review discusses conditions for installing and removing PMB esters and presents many complex examples in which the PMB ester is featured. Although this survey is relatively comprehensive, covering the years 1959 to the end of 2014, it is not intended to be exhaustive. Rather, this manuscript will focus on the most interesting and useful examples that may prove beneficial for planning multi-step syntheses of complex molecules that require protection of the carboxylate function. However, this review will provide a more thorough discussion of the PMB ester protecting group than other sources that provide general overviews of protecting groups.1,2
I. Formation of PMB Esters
1. From Carboxylic Acids
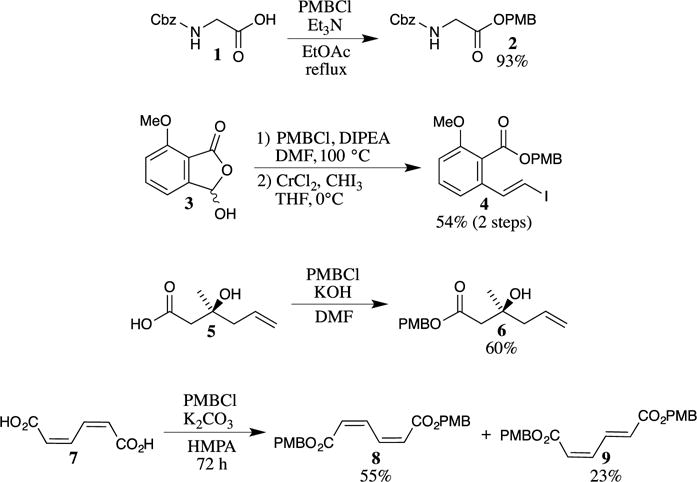
Similar to many protecting groups, the PMB ester was first explored in the context of protecting amino acids for peptide synthesis. Maclaren, Hiskey and Adams reported some of the first examples in their preparation of the PMB esters of N-protected amino acids, such as N-Cbz glycine (1). The protection of the carboxy group was accomplished by reacting the carboxylic acid with 4-methoxybenzyl chloride (PMBCl) in the presence of triethylamine (Scheme 1).4,5 This method was very successful with glycine derivatives, which could subsequently be used in the synthesis of more complex peptides. In cases where the carboxylic acid group is hindered, direct alkylation can be slow and may require heating. One excellent example of this problem is illustrated in the studies conducted by Georg and her group with the hindered carboxy aldehyde 3 (shown in Scheme 1 as its hemiacylal).6 Heating of the reaction mixture to 100 °C in dimethylformamide (DMF) was required to obtain a satisfactory yield of the PMB ester so that the next step in the sequence (a Takai olefination7) could be performed to access the vinyl iodide 4. Bases other than trialkylamines may also be used. For example, Fujino and Sugai showed that deprotonation of the carboxylic acid with potassium hydroxide followed by trapping with PMBCl is effective for obtaining PMB ester 6, which was used in an approach to obtain the enantiomerically pure (R)-3-hydroxy-3-methyl-4-pentenoic acid PMB ester.8 The silver salts of carboxylates have also been alkylated with PMBCl to synthesize the PMB esters of amino acids.9 This method reportedly provided higher yields of the esters with several amino acid substrates and avoided racemization, providing materials that are useful for custom peptide synthesis. Some isomerization of Z-unsaturated acids often occurs under basic conditions, as was observed during the protection of (Z,Z)-muconic acid (7) with PMBCl.10 The reaction conditions [K2CO3, hexamethylphosphoramide (HMPA), 72 h] resulted in a 7:3 mixture of the Z,Z isomer 8 and the Z,E isomer 9 in a 78% combined yield.
Scheme 1.
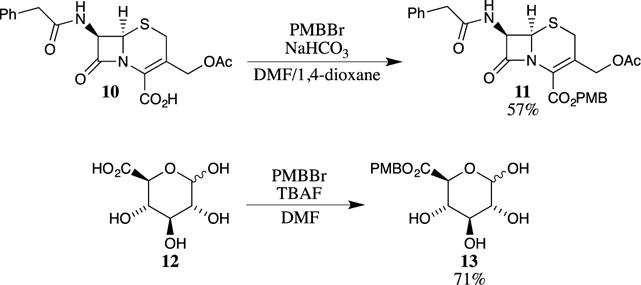
Other PMB halides have also been used to synthesize PMB esters. These reactions are less common because 4-methoxybenzyl bromide (PMBBr) is considerably less stable than PMBCl and polymerizes rapidly.11,12 However, PMBBr may be utilized to prepare PMB esters, as exemplified by the protection of cephalosporin 10 with PMBBr in 57% yield (Scheme 2).13 Typically, the ester can be obtained in the presence of unprotected polyols using conditions similar to those shown in Scheme 2.14 For example, glucuronic acid (12) is protected as its PMB ester by deprotonation with tetrabutylammonium fluoride (TBAF), which acts as a base in this case, and alkylation with PMBBr to provide ester 13 in 71% yield.15
Scheme 2.
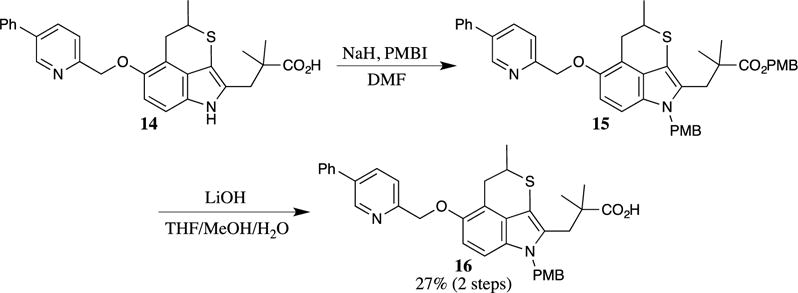
The even more reactive 4-methoxybenzyl iodide (PMBI) has rarely been utilized for the introduction of PMB esters. One report describes the alkylation of the hindered carboxylic acid 14 with PMBI (Scheme 3).16 During the formation of the PMB ester, the indole nitrogen is also alkylated, leading to the formation of ester 15. No yield was provided from compound 15; however, a 27% yield of indole 16 was reported over two steps following hydrolysis of the ester with aqueous lithium hydroxide.
Scheme 3.
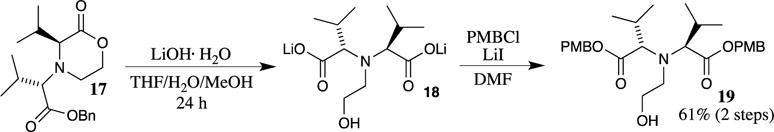
Difficult esterifications requiring a large amount of the unstable PMBI may be performed with PMBI generated in situ from another PMB halide, thereby avoiding the difficulties associated with the isolation and handling of the highly reactive (and unstable) iodide. Walker demonstrated this procedure in the synthesis of sterically congested iminodiacid 19 (Scheme 4).17 In this example, lactone 17 was first converted to the lithium dicarboxylate salt 18 with lithium hydroxide. Alkylation of the lithium carboxylates of 18 proved to be sluggish with PMBCl, which is understandable due to the steric constraints in this hindered system. The formation of the more reactive PMBI in situ resulted in a more rapid formation of PMB ester 19 in 61% overall yield from lactone 17.
Scheme 4.
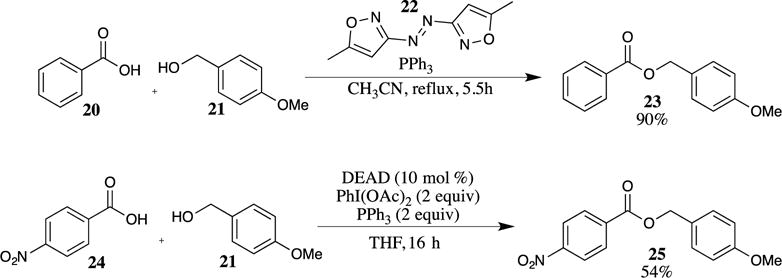
Mitsunobu conditions have also been used to prepare PMB esters.18 An obvious disadvantage of the Mitsunobu protocol is the formation of stoichiometric by-products, particularly the hydrazodicarboxylate and triphenylphosphine oxide. The development of recyclable azo reagents is one approach that reduces the waste generated by these processes. For example, employing 5,5′-dimethyl-3,3′-azoisoxazole (22) and triphenylphosphine with a variety of carboxylic acids and benzyl alcohols (including 4-methoxybenzyl alcohol (21), as shown in Scheme 5) led to the formation of the corresponding esters.19 The azo reagent 22 can be easily synthesized from 3-amino-5-methyl-isoxazole and sodium hypochlorite, and the hydrazodicarboxylate formed as the by-product is insoluble in organic solvents, a feature that facilitates its recovery and recycling. Other investigators have focused on developing reaction conditions that utilize only a catalytic amount of azo reagent.20 Typically, an oxidizing agent (such as PhI(OAc)2) is utilized to regenerate the azo compound in situ from the hydrazodicarboxylate, as shown for the formation of ester 25 (Scheme 5).21 The PMB ester 25 was shown to be stable under these oxidative conditions, providing a catalytic version of the Mitsunobu reaction that is capable of introducing the PMB ester.
Scheme 5.
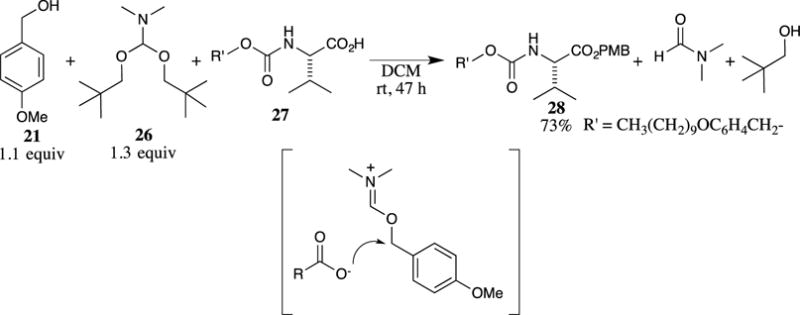
N,N-Dimethylformamide dineopentyl acetal (26) has also been described as a useful esterification reagent by Eschenmoser and co-workers.22,23 This reagent effectively acted as an activating reagent for 4-methoxybenzyl alcohol (21), producing an imidate-type intermediate that alkylates the carboxylic acid (as shown in Scheme 6). Several N-protected peptides (such as 27) were esterified using this method in excellent yields. Although this procedure does generate some waste in the formation of 2,2-dimethyl-1-propanol and DMF, these side-products can easily be removed by washing with water. Control experiments performed without the addition of the 4-methoxybenzyl alcohol led to the nearly quantitative recovery of the carboxylic acid, demonstrating that the more sterically hindered neopentyl alcohol does not compete for the carboxylate nucleophile.
Scheme 6.
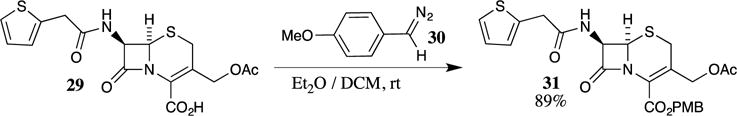
PMB esters may also be synthesized by reacting 4-methoxyphenyldiazomethane (30) with carboxylic acids.24 This method has frequently been employed with structurally complex or sensitive substrates. For example, the sensitive β-lactam 29 was converted to the corresponding PMB ester 31 in 89% yield (Scheme 7). The use of diethyl ether with dichloromethane as a co-solvent was shown to be the optimal medium for the transformation and led to the rapid formation of the PMB ester. One advantage of this method lies in the facile work-up of the reaction mixture because the only by-product is nitrogen gas, which often allows for the isolation of PMB esters without chromatography.
Scheme 7.
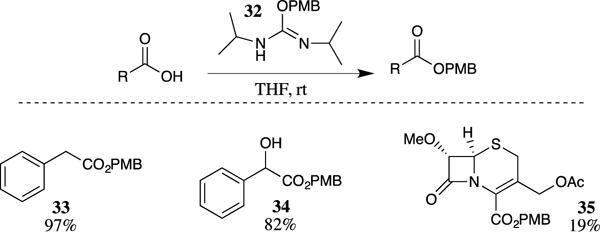
Protection of carboxylic acids as PMB esters may also be accomplished using N,N-diisopropyl-O-(4-methoxybenzyl)isourea (32) (Scheme 8).25 PMB isourea 32 is readily prepared through the copper-catalyzed addition of 4-methoxybenzyl alcohol to N,N-di(isopropyl)carbodiimide.26 Simple mixing of isourea 32 with carboxylic acids provides the corresponding PMB esters in good yields. The formation of PMB esters using this reagent proceeded well on a diverse range of carboxylic acids at room temperature in THF, acetone or acetonitrile. Methanol was found to be unsuitable as a solvent because of the competitive formation of 4-methoxybenzyl methyl ether due to methanol acting as a competing nucleophile when present at high concentrations. Alcohols were tolerated in the carboxylic acid substrates, however, as evidently at competitive alcohol concentrations the esterification was favored. This is demonstrated by the formation of PMB ester 34 in 82% yield. Finke and colleagues explored the formation of PMB esters with isourea 32 in their studies on cephalosporin esters for the inhibition of human leukocyte elastase.27 PMB ester 35 was formed from 7α-methoxycephalosporanic acid and isourea 32, although only in 19% yield. A similar reagent, 4-methoxybenzyl-1-piperidyl carbonate, has also been reported to be useful in the formation of PMB esters from carboxylic acids, but this method has not been widely utilized.28
Scheme 8.
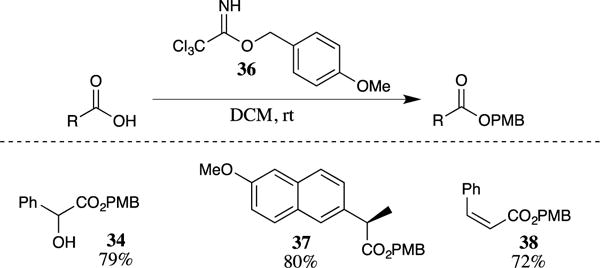
The generation of PMB esters has also been reported from the reaction of carboxylic acids with commercially available 4-methoxybenzyl-2,2,2-trichloro-acetimidate (36) (Scheme 9).29–31 The use of the trichloroacetimidate for esterification does not require an acid catalyst because the carboxylic acid itself promotes the transformation. This method was successful over a wide range of substrates, including electron-rich and electron-poor benzoic acids and carboxylic acids bearing halogens, alkenes, and alkynes.31 Esters such as 34, which have a free alcohol group, were also formed in excellent yields. The mild nature of these conditions is demonstrated by the formation of the PMB ester of naproxen 37 with no racemization of the nearby chiral center. Additionally, PMB ester 38 was formed with no detectable isomerization of the Z-alkene to the more stable E-isomer. Preliminary mechanistic studies30 showed that the addition of strong amine bases hampered this reaction. This implies that the carboxylic acid first protonates the imidate (which has a pKa of approximately 11); the imidate group is then displaced by the carboxylate anion, leaving trichloroacetamide as the only by-product of the transformation. The presence of an amine was found to be detrimental to the transformation because the basic amine protects the imidate from protonation, thus suppressing the esterification in these cases.
Scheme 9.
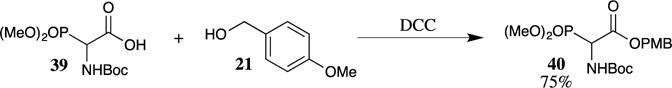
Carbodiimide coupling reagents are also effective in forming PMB esters. Early studies on the synthesis of PMB esters used dicyclohexylcarbodiimide (DCC) as a coupling agent between carboxylic acids and 4-methoxybenzyl alcohol (21).32,33 These reactions are greatly accelerated by the addition of the acylation catalyst 4-(dimethylamino)pyridine (DMAP) and additives such as hydroxybenzotriazole (HOBt).34 Carbodiimide couplings are still occasionally used for sensitive substrates. For example, Feldman and co-workers formed PMB ester 40 utilizing DCC to couple carboxylic acid 39 with 4-methoxybenzyl alcohol (21) (Scheme 10).35 These conditions were chosen to avoid disrupting or altering the sensitive phosphonate group in the reaction product.
Scheme 10.
2. From Activated Esters
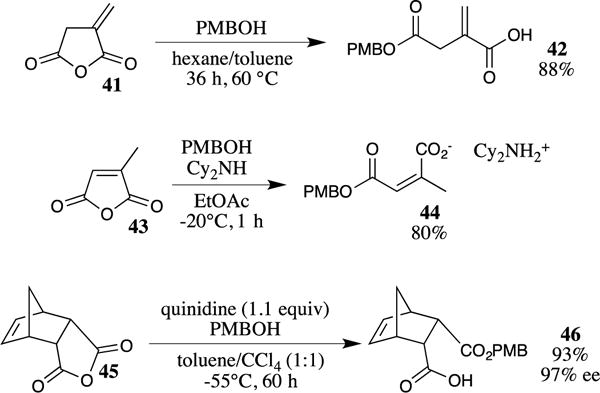
The addition of 4-methoxybenzyl alcohol to acid chlorides in the presence of a base to scavenge the HCl by-product is a common method for synthesizing PMB esters. These conditions are eminently suitable to a variety of substrates and are typically used when the required acid chloride is inexpensive and readily available.36 The acid chloride may also be generated in situ and then treated with 4-methoxybenzyl alcohol in a single flask procedure.37 Similarly, the addition of 4-methoxybenzyl alcohol to an anhydride may also be used to generate PMB esters.38,39 Many cyclic anhydrides (such as 4140) have been reported to react with 4-methoxybenzyl alcohol to provide the corresponding ester by simple heating (Scheme 11).41,42 The addition of an amine (e.g., dicyclohexylamine) accelerated these reactions, allowing them to proceed below room temperature, as illustrated for anhydride 43.43 The desymmetrization of meso-anhydrides using a chiral amine base has also been achieved by Bolm and co-workers.44 The utilization of a stoichiometric amount of quinidine as the chiral promoter led to the opening of meso-anhydride 45 in excellent yield with high enantioselectivity.
Scheme 11.
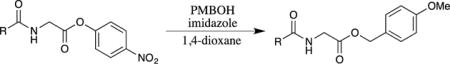
PMB esters may also be obtained through the addition of 4-methoxybenzyl alcohol to 4-nitrophenyl esters promoted by an amine base, such as imidazole (Table 1).45 For these reactions, glycine 4-nitrophenyl esters were prepared from the addition of 4-nitrophenol to a glycine-mixed anhydride46 or by the reaction of the free carboxylic acid with tris-(4-nitrophenoxy)-phosphine.47 The 4-nitrophenyl esters were highly crystalline, which facilitated the purification of these intermediates. Utilizing these methods, dipeptides with C-terminal glycine residues were initially converted to the 4-nitrophenyl ester and then transformed into the corresponding PMB ester. The displacement of 4-nitrophenyl esters in peptides was only applicable to substrates with an unprotected C-terminal glycine, as significant racemization was observed with other amino acid derivatives.
Table 1.
Formation of PMB Esters from 4-Nitrophenyl Esters
| ||
|---|---|---|
|
| ||
| Entry | Compounda | % Yield |
| 1 | Cbz-Gly-OPMB | 77 |
| 2 | Cbz-Ala-Gly-OPMB | 86 |
| 3 | NPS-Ala-Gly-OPMB | 86 |
| 4 | Boc-Ser(OBz)-Gly-PMB | 44 |
| 5 | NPS-Cys(SBz)-Gly-OPMB | 84 |
| 6 | NPS-Phe-Gly-OPMB | 61 |
Cbz = benzyloxycarbonyl, Boc = tert-butyloxycarbonyl, NPS = 2-nitrophenylsulfonyl, Bz = Benzoate
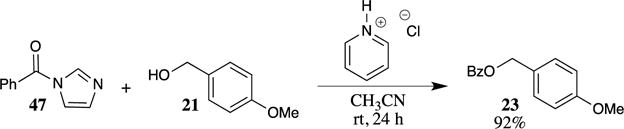
Sarpong and his group recently reported that PMB esters can be formed through the nucleophilic addition of 4-methoxybenzyl alcohol to N-acylimidazoles in the presence of pyridinium salts.48 The use of pyridinium salts to promote the process was key for the success of this transformation, as other acids and bases were less effective. For example, N-benzoylimidazole (47) was converted to 4-methoxybenzyl benzoate (23) in 92% yield in the presence of pyridinium chloride (Scheme 12). This method appears to hold great promise for facilitating difficult acylation reactions, with N-acylimidazole reagents being inexpensive and stable at room temperature.49,50
Scheme 12.
3. From Other Esters and Imides
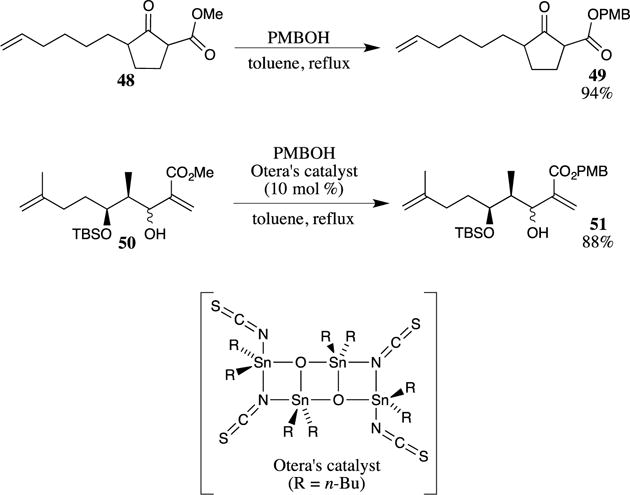
PMB esters may also be formed through the exchange reaction of another ester with 4-methoxybenzyl alcohol in the presence of an acid catalyst. These reactions are most often performed with methyl and ethyl esters as the starting material because the methanol or ethanol side product can be readily removed by distillation from the reaction mixture, which drives the equilibrium of the reaction toward the desired PMB ester product. In some cases, an exogenous acid catalyst is not required, as shown for ester 48 (Scheme 13).51 In this case, a methyl ester is refluxed in toluene with 4-methoxybenzyl alcohol to afford PMB ester in 94% yield. This uncatalyzed reaction may be limited to β-ketoesters that can enolize (such as 48, Scheme 13), forming an intramolecular hydrogen bond that activates the ester of 48 and facilitates the ester exchange reaction. Supporting this hypothesis, other β-ketoester-containing substrates have also been shown to undergo similar uncatalyzed trans-esterification reactions.52 In more complex systems, direct ester exchange reactions that are accelerated by protic acids are often problematic due to side reactions. In these cases, Otera’s catalyst (shown in Scheme 13 below)53,54 may be employed to transform methyl esters into PMB esters. One representative example is the conversion of methyl ester 50 to PMB ester 51.55 Ester 50 is quite sensitive because elimination of the allylic alcohol to form the diene or isomerization of the 1,1-disubstituted alkene to the more stable trisubstituted olefin are both quite facile and may be problematic side reactions under acidic conditions.
Scheme 13.
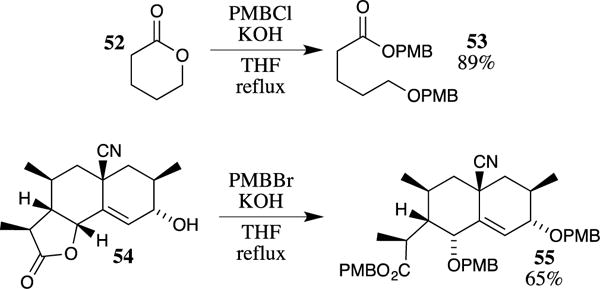
PMB esters may also be accessed in good yields by treating lactones with sodium or potassium hydroxide under anhydrous conditions in the presence of PMBCl. For example, heating lactone 52 with potassium hydroxide followed by the addition of an excess of PMBCl protects the newly formed alcohol and the carboxylate to provide the bis-protected substrate 53 in good yield (Scheme 14).56 Evidently opening of the lactone is significantly more rapid than hydrolysis of the PMBCl under these conditions. This method has also shown utility in more complex examples, such as with lactone 54, in which hydrolysis of the lactone followed by in situ trapping with PMBBr provides the completely protected system 55.57 A stepwise lactone hydrolysis followed by protection is also effective.58
Scheme 14.
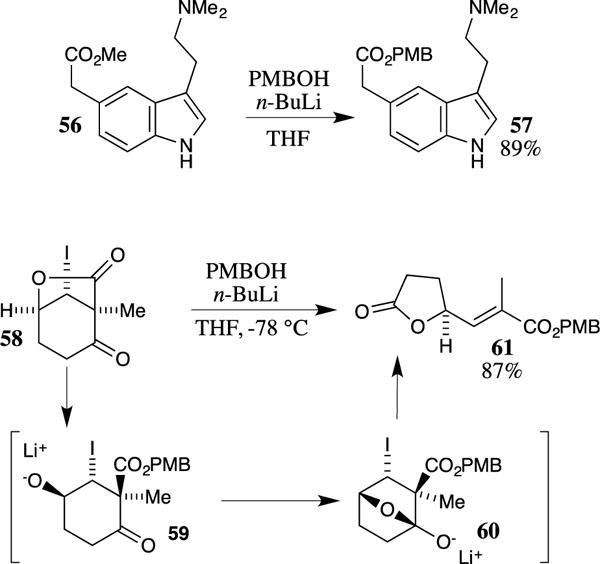
The addition of the lithium salt of 4-methoxybenzyl alcohol to a methyl ester or lactone is also a viable method for forming the corresponding PMB ester. For example, the methyl ester of indole 56 was transformed into the corresponding PMB ester 57 by treatment with PMBOLi,59 which was formed in situ from the deprotonation of PMBOH (21) with n-butyllithium (Scheme 15). This method provided the PMB ester in 89% yield. Occasionally, these reactions may lead to base-promoted rearrangements. For example, Schultz and co-workers attempted to prepare a PMB ester with lactone 58; however, this procedure led to the unsaturated ester 61 as the major product.60,61 This result was rationalized via the transannular addition of the alkoxide to the ketone in structure 59, leading to the bicyclic system 60. Fragmentation of 60 followed by elimination of the resulting iodide provides the observed product 61.
Scheme 15.
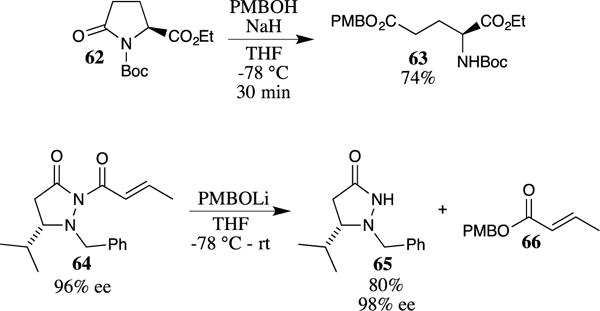
Unsymmetrical imides are also readily opened by the addition of the sodium or lithium alkoxide of 4-methoxybenzyl alcohol to afford the corresponding PMB ester. For example, N-Boc-protected pyroglutamate ethyl esters such as 62 are opened with the sodium salt of 4-methoxybenzyl alcohol to provide the ring-opened PMB ester 63 in good yield (Scheme 16).62 Alternatively, a catalytic amount of potassium cyanide facilitates analogous transformations, although longer reaction times are required.63 Similar methods have been used in the removal of chiral auxiliaries to provide PMB esters.64 For example, Sibi and his co-workers utilized the lithium salt of 4-methoxybenzyl alcohol to remove the acyl group from pyrazolidinone 6465 to obtain the highly enantioenriched heterocycle 65, which was then used in the synthesis of several chiral catalysts.66–68
Scheme 16.
4. From Aldehydes
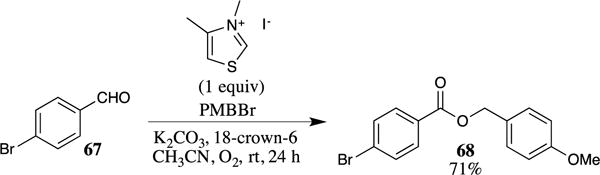
Aldehydes may be directly converted to their corresponding PMB esters utilizing Stetter-type chemistry.69 For example, the reaction of 4-bromobenzaldehyde with PMBBr in the presence of a thiazolium salt afforded the corresponding PMB ester 68 in 71% yield (Scheme 17). This process provided high yields for a number of electron-deficient aromatic aldehyde substrates. This procedure is notable as it provides a single-step transformation of an aldehyde (such as 67) to the corresponding ester (such as 68), which typically requires a multistep sequence.
Scheme 17.
II. Deprotection of PMB Esters
1. Deprotection with Brønsted Acids
PMB esters were developed explicitly such that the protected carboxylate may be unmasked selectively under acidic conditions. Weygard and Hunger31,32 were the first to evaluate the stability of the PMB ester in this area and found that PMB esters were readily cleaved from small peptides by exposure to neat trifluoroacetic acid (TFA) at 0 °C or in refluxing acetic acid; no cleavage occurred with acetic acid at room temperature. No racemization of the amino acid products was observed after cleavage with TFA, even in the case of N-protected S-benzylcysteine derivatives5 that are highly prone to racemization.4,45 In some cases, only 1 to 5 equiv. of TFA were required to cleave the ester. The use of an electron-rich aromatic system as an additive to minimize the formation of side products is common when near stoichiometric amounts of TFA are utilized, with anisole being typically employed for this purpose.70 Substoichiometric amounts of TFA have also proven to be effective when phenol was used as the solvent at elevated temperatures (45 °C).71 The nucleophilic additives and/or solvents are presumed to function as cation scavengers, reacting with the 4-methoxybenzyl cation generated in the transformation which protects the substrate from side reactions.
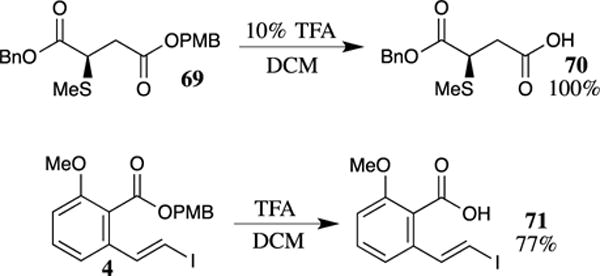
The use of TFA to deprotect PMB esters does not cleave benzyl esters. For example, Sutton and Clardy employed a PMB ester in their synthesis of the antibiotic pantocin B.72 The PMB ester is selectively and quantitatively cleaved from intermediate 69 using 10% TFA in dichloromethane (Scheme 18). No cleavage of the benzyl ester occurred under these conditions, and no epimerization of the stereogenic carbon adjacent to the ester was observed. Steric factors appear to play only a small role in the TFA mediated cleavage of PMB esters, as expected given the proposed mechanism in which protonation of the carbonyl group is followed by loss of the 4-methoxybenzyl carbocation. For example, the cleavage of hindered PMB ester 4 proceeded in excellent yield, despite its hindered nature.6
Scheme 18.
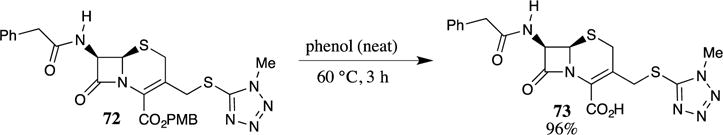
Several other Brønsted acids have been shown to be effective in cleaving PMB esters. For example, neat formic acid was effective for the removal of several PMB esters from N-protected amino acids.9 The use of HCl gas dissolved in an organic solvent has also been reported to cleave PMB esters from small peptides, with the rate of hydrolysis being dependent on the dielectric constant of the solvent (more polar solvents provide faster reaction rates, e.g., ethyl acetate is less effective than nitromethane).9 These studies led to a series of publications detailing the use of PMB esters in peptide synthesis,9,32,45 and these results have been reviewed.4 Subsequent investigations on β-lactam antibiotics demonstrated that in some cases, PMB esters could be cleaved by heating the substrate to 60 °C in neat phenol without the addition of a stronger acid.70 This method was particularly useful for the deprotection of acid-sensitive substrates, such as β-lactam 72 (Scheme 19). The deprotection of PMB ester 72 failed with many other methods (anisole/TFA, neat formic acid, or anisole/AlCl3) due to the sensitivity of the β-lactam ring.
Scheme 19.
The reactivity of PMB esters with Brønsted acids compared with other acid-labile protecting groups has been investigated by many researchers. Attempts to selectively cleave t-butyl esters or diphenylmethyl (DPM) esters without cleaving a PMB ester proved unsuccessful (N-benzyloxycarbonyl-L-proline esters were used in this study, with mixtures of the two esters placed in the same flask and treated with TFA, conc. aq. HCl or HCl gas dissolved in an organic solvent).9 Subsequent efforts determined that Boc carbamates could be cleaved in the presence of PMB esters by using a stoichiometric amount of p-toluenesulfonic acid (TsOH).72 t-Butyl esters were found to be stable under the stoichiometric TsOH conditions, providing a method for the selective deprotection of PMB esters in the presence of t-butyl esters.
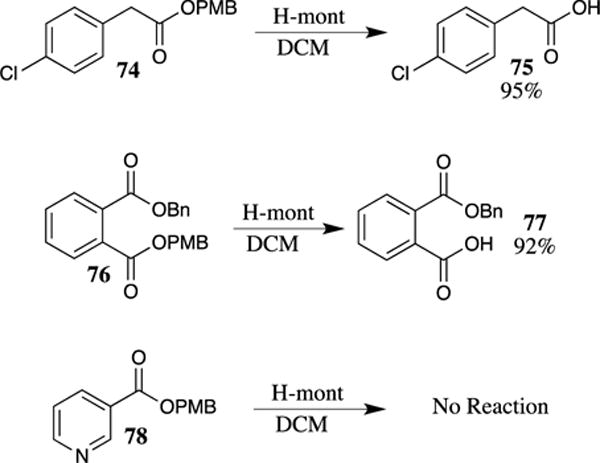
More recently, heterogeneous acids based on montmorillonite clays have been explored for the cleavage of PMB esters.73 These reagents have the advantages of low cost and facile removal from the reaction medium by simple filtration. A number of montmorillonite clays were found to be effective in the deprotection of PMB esters, including H-Mont, Mont K10 and Mont KSF. The fastest reactions occurred in dichloromethane (compared with toluene in which the reaction was slower, and acetone, which afforded only trace amount of product), although all of the montmorillonite catalyzed deprotections had reaction rates that were significantly slower compared to deprotections employing stoichiometric quantities of TFA. Utilizing H-mont in dichloromethane (DCM), a number of PMB esters were deprotected in excellent yields (Scheme 20). Benzyl esters were shown to be unaffected under these conditions, as demonstrated in substrate 76. The presence of a basic group, such as in picolinate 78, was detrimental to the reaction, which was expected as the pyridine nitrogen in 78 will act as a buffer and deter cleavage of the PMB ester.
Scheme 20.
2. Deprotection with Lewis Acids
Lewis acids have also been used to cleave PMB esters. Initially, strong Lewis acids, like AlCl3 and B(OTf)3, were found to be effective in cleaving PMB esters. At first the Lewis acid was added to the Brønsted acid (which was used as the solvent for the reaction) to accelerate the reaction. These examples utilized neat TFA with added stoichiometric amounts of B(OTf)3 to cleave the PMB ester from an α-hydroxyglycine-containing peptide.74 Subsequent processes based solely on Lewis acids were developed for the deprotection of the PMB esters in carbapenem antibiotics. Deprotection of these substrates with aqueous acid- or base-mediated hydrolysis conditions led to the opening of the β-lactam ring in some cases. Additionally, transition metal-catalyzed hydrogenolysis is problematic due to catalyst poisoning by the sulfide present in the substrate. Alternatively, treatment of the PMB ester 79 with 2.5 equiv. of AlCl3 in anhydrous dichloromethane at −50 °C in the presence of anisole as a cation scavenger afforded the corresponding carboxylic acid in 60% yield (Scheme 21).75 Similar conditions that cleave benzyl esters have been described, although they require substantially higher temperatures and longer reaction times (23 °C, 5 h).76
Scheme 21.

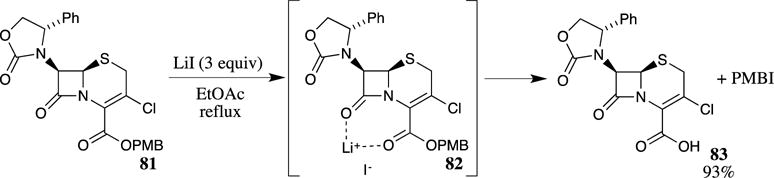
Weaker Lewis acids have also been used to cleave PMB esters. For example, metal salts such as lithium iodide in refluxing ethyl acetate have been shown to be effective in deprotecting β-amido PMB esters.77 The mechanism of the reaction is proposed to proceed through coordination of the lithium ion to the ester and amide carbonyl groups (as shown in 82, Scheme 22), activating the PMB group for nucleophilic attack from the iodide. Other solvents, such as THF or dichloromethane, could be employed, but the reaction rates were slower. The use of solvents with a dielectric constant above 10 (i.e., pyridine) stopped the reaction because the lithium ion is chelated by polar solvents; therefore, it is ineffective in activating the carbonyl group of the ester. This method was also shown to be effective for a number of other esters (Bn, PNB, and t-Bu) in similar β-lactam systems. With other reactive β-lactams, opening of the β-lactam ring was also observed due to the lithium ion not only activating the ester but also the β-lactam carbonyl through the same interaction.
Scheme 22.
Iodotrimethylsilane (TMSI) in the presence of triphenylphosphine has also been used to cleave PMB esters.78 This reagent does not form the highly reactive alkylating agent PMBI as a side product; rather, the less reactive 4-methyoxybenzyltriphenylphosphonium iodide salt is obtained. Because the reactive PMBI may lead to undesired side reactions, which lower the yields and complicate purifications, these new conditions appear to offer a significant improvement over the use of TMSI alone. In addition, this system is touted to be superior to other TMSI ester deprotections because the triphenylphosphine increases the selectivity of the process by reducing the reactivity of the TMSI, which preferentially reacts with the ester that leads to the most stable cationic intermediate. The TMSI-PPh3 complex is also more stable than the uncomplexed Lewis acid; solutions of TMSI-PPh3 can be stored at 0 °C for several months without loss of activity. A variety of β-lactam-containing PMB esters were deprotected with this combination of reagents.78
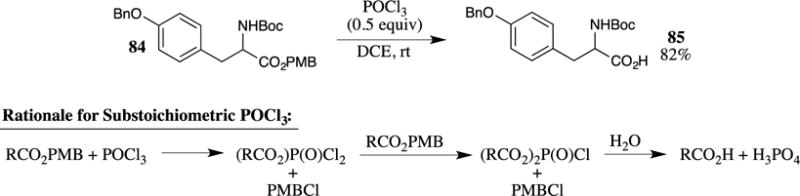
Ilangovan and his group recently demonstrated that PMB esters could be cleaved with POCl3 in dichloroethane at room temperature (Scheme 23).79 These conditions were also found to be effective in the deprotection of PMB ethers but did not affect benzyl ethers or acid-sensitive Boc-carbamates. For example, the tyrosine-based PMB ester 84 was deprotected to the carboxylic acid 85 in 82% yield under these conditions. No information was provided regarding the stability of the chiral center under these conditions, as racemic material was used in this investigation. Because the side-product of the reaction, (RCO2)P(O)Cl2, can react further with another equivalent of the PMB ester to provide (RCO2)2P(O)Cl, only 0.5 equiv. of POCl3 is required for the deprotection.
Scheme 23.
Silver hexafluoroantimonate has also been effectively used to deprotect PMB esters (Scheme 24).80 Although this Lewis acid is somewhat expensive, a high turnover (only 5 mol % of the Lewis acid is required) was observed in this system when electron-rich 1,3,5-trimethoxybenzene (TMB) (0.5 equiv.) was used as a cation scavenger. These conditions were also effective in cleaving PMB ethers but did not affect benzyl ethers, carbonates, acetate esters, triisopropylsilyl ethers (OTIPS), acetonides or Boc-carbamates. Other silver salts, such as AgOTf, AgBF4 and AgNTf2, were also shown to be efficient catalysts for these deprotection reactions, although the highest reaction rates were observed with AgSbF6.
Scheme 24.
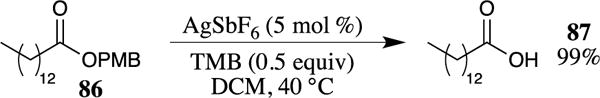
3. Deprotection Using Base-mediated Hydrolysis
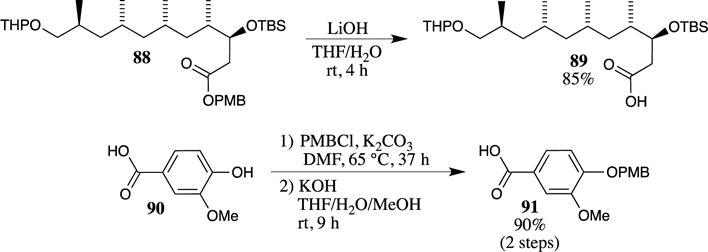
PMB esters may be cleaved using many other approaches that are not dependent on strong Brønsted or Lewis acids. Base-promoted hydrolysis has been shown to proceed uneventfully with potassium hydroxide at room temperature. For example, in a formal synthesis of (−)-borrelidin, hydrolysis using lithium hydroxide was performed on PMB ester 88 to avoid concurrent cleavage of the tetrahydropyranyl (THP) ether (Scheme 25).81 This method was also utilized by Shioiri and co-workers in their synthesis of the lignan vibsanol, which required carboxylic acid 91 as a starting material.82 Protection of vanillic acid (90) resulted in the formation of both the PMB ester and the PMB ether. The PMB ester was then selectively removed under basic conditions at room temperature to provide the desired carboxylic acid 91.
Scheme 25.
4. Deprotection by Hydrogenation
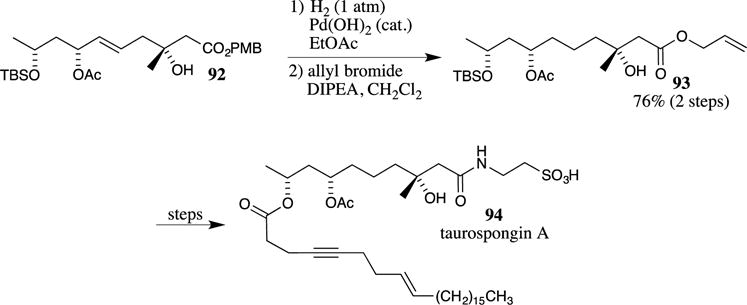
As with most benzyl esters, the PMB ester can be removed by hydrogenation utilizing a transition metal catalyst and a hydrogen source. For example, Fujino and co-workers used a PMB ester as a protecting group in their formal total synthesis of taurospongin A (94) (Scheme 26).8 Hydrogenation of PMB ester 92 with Pd(OH)2 followed by re-protection of the carboxylic acid with an allyl group afforded ester 93 in 76% yield in two steps. The reduction of alkenes is typically competitive in these transformations, as observed in the reduction of the alkene of compound 92. The removal of the tert-butyldimethylsilyl (TBS) group then completed a formal total synthesis of taurospongin A.
Scheme 26.
5. Deprotection by Oxidative Methods
Given that a common method for the deprotection of PMB ethers is oxidation with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ),83,84 one might assume that PMB esters can be cleaved in a similar manner. Indeed, this type of deprotection for PMB esters has been mentioned as an option in some studies.54,85 Attempts to cleave PMB esters with DDQ have proven fruitless, however, as even under forcing conditions (CH2Cl2/H2O, reflux, 5 h), the PMB ester remains intact (Scheme 27).86,87 The oxidation potential of the PMB ester must be too low to form the charge-transfer complex with DDQ that is essential for the oxidative deprotection to proceed; therefore, the ester is stable under these conditions. Interestingly, cleavage of the related 2,6-dimethoxybenzyl ester86 (such as 97) and the 4-methoxy-α-methylbenzyl ester87 (such as 95) have been reported to proceed in high yields with DDQ to provide the desired carboxylic acid products.
Scheme 27.

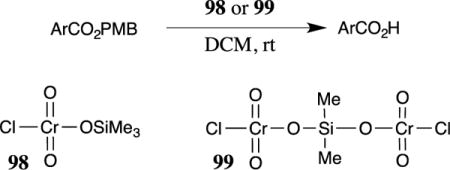
PMB esters have been cleaved with oxidizing agents that are more powerful than DDQ. The combination of chromium trioxide and a silyl chloride in dichloromethane has been reported to be effective in cleaving 4-methoxybenzyl esters.88 The active reagent for these transformations was not determined because attempts to isolate the active species led to an explosion during distillation; however, the authors suggested that silyl chromium species 98 or 99 (Table 2) may be formed from the reaction of trimethylsilyl chloride or dichlorodimethylsilane with chromium trioxide. Care was taken to remove hydrogen chloride from the active reagent by bubbling nitrogen through the solution before use; thus, the cleavage appears to proceed through an oxidative mechanism rather than an acidic cleavage. 3,4-Dimethoxybenzyl esters were also cleaved when exposed to 98 or 99 in DCM at room temperature. These reagents were also reported to oxidize alcohols to ketones; thus, functional group compatibility must be carefully matched with the oxidative cleavage conditions. Only benzoic acid esters were described as substrates for this cleavage method, and the yield appeared to vary significantly depending on the functionality present on the aromatic ring of the ester substrate (Table 2).
Table 2.
Deprotection of PMB Esters with Chromium-based Reagents
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Ar | Reagent | Time | Yield |
| 1 | Ph | 98 | 5 h | 70% |
| 2 | Ph | 99 | 5 h | 70% |
| 3 | 4-NO2-Ph | 98 | 1 h | 40% |
| 4 | 4-Cl-Ph | 98 | 5 h | 75% |
| 5 | 4-Cl-Ph | 99 | 5 h | 30% |
| 6 | 2-Me-Ph | 98 | 5 h | 30% |
| 7 | 2-Me-Ph | 99 | 5 h | 35% |
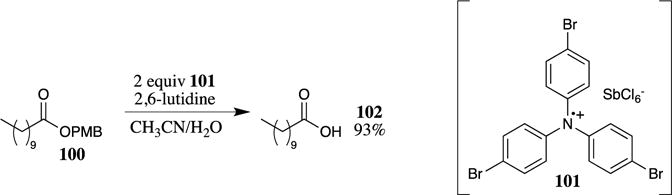
The oxidative removal of PMB esters may also be accomplished by treatment with electron-transfer reagents, specifically stable triarylamine radical cations, such as 101 (Scheme 28).89 These conditions rapidly cleave benzyl, PMB, benzhydryl and 2,4-dimethoxybenzyl esters. The selective cleavage of PMB esters in the presence of benzyl esters was achieved under these reaction conditions. This method has not found widespread application because the triarylamine radical cations are not commercially available and must be synthesized or generated in situ in a divided cell by indirect electrolysis. When produced by indirect electrolysis in situ, only 20 mol % of the triarylamine 101 is required to accomplish the cleavage. Similar conditions have also been used to cleave PMB ethers.90
Scheme 28.
6. Deprotection by Photochemical Methods
Murphy and co-workers recently showed that PMB esters can be cleaved under photochemical conditions in the presence of an organic electron-donor, such as the heterocycle 104 (Scheme 29), resulting in a metal-free reductive cleavage.91 In contrast to PMB esters, PMB ethers were found to be stable under these reaction conditions, providing a means for the deprotection of PMB esters in the presence of the related PMB ethers under near neutral reaction conditions. Although different reactivities were observed for PMB esters and PMB ethers, 2-methoxybenzyl ethers and esters were both readily cleaved in high yields using this methodology, providing a striking case of interesting and potentially useful selectivity. However, some functional groups remain incompatible this method, as aryl chlorides are also reduced to the corresponding arenes under these conditions.
Scheme 29.
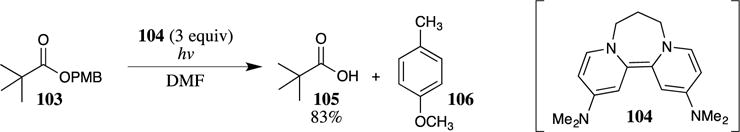
III. Examples of the Use of PMB Esters
PMB esters have found extensive use in total syntheses and have proven to be particularly valuable to synthetic organic chemists. More recently, these applications have been expanded into investigations in organic methodology and bio-organic chemistry. Because of the ability of PMB esters to be formed and cleaved under a variety of mild conditions from inexpensive starting materials, the PMB group has become one of the “workhorse” protecting groups when working with complex organic molecules that require carboxylate protection.
1. Examples of the Use of PMB Esters in Total Syntheses
PMB esters were useful in the synthesis of the diene-containing histone deacetylase inhibitor trichostatin A (111).92,93 The major carbon-carbon bond forming reaction in this synthetic sequence was the palladium-catalyzed coupling of ketone 107 with vinyl bromide 108 (Scheme 30). The removal of the PMB group from the ester followed by coupling of the newly formed carboxylic acid with a hydroxylamine equivalent provided trichostatin A (111) in excellent yield. The palladium-catalyzed coupling was equally efficient with the methyl ester, but hydrolysis of the methyl ester with hydroxide led to isomerization of the trisubstituted alkene, forming a mixture of E- and Z-alkene isomers. The removal of the PMB group with TFA provided a useful solution to this problem, with no alkene isomerization being observed under these conditions. In the case of ester 109, triethylsilane is added to the reaction as a cation scavenger to reduce the 4-methoxybenzyl cation, forming 4-methylanisole and triethylsilyl trifluoroacetate as side products while protecting the diene and the aromatic ring from side reactions with the 4-methoxybenzyl cation.
Scheme 30.
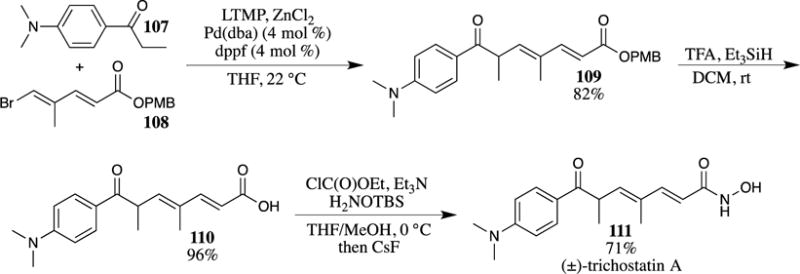
Theodorakis and his group used a PMB ester in their synthesis of (−)-borrelidin (116) (Scheme 31).94 Aldehyde 112 was utilized in an acetate aldol reaction with the silyl enol ether of 4-methoxybenzyl acetate (113) to provide β-hydroxy ester 114 in 71% yield (the other diastereomer of the alcohol was also produced in 14% yield). The stereoselectivity of this reaction is attributed to the chiral center that is present adjacent to the aldehyde in compound 112. The protection of alcohol 114 as a methoxymethyl (MOM) ether followed by deprotection of the TBS ethers and the PMB ester led to the hydroxy acid 115, which was then used in a macrolactonization. The PMB ester was stable under the HF•pyridine conditions used to remove the TBS group due to the pyridine acting as a buffer, protecting the acid-sensitive PMB ester. Notable in this example is the stability of the acid sensitive MOM and 2-methoxyethoxymethyl (MEM) ethers under the deprotection conditions of the PMB ester. After a number of subsequent steps, the natural product (−)-borrelidin (116) was accessed from hydroxy acid 115.
Scheme 31.
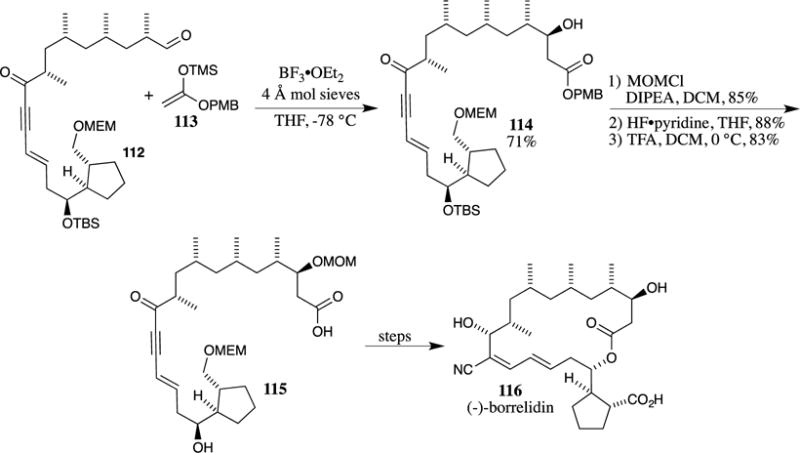
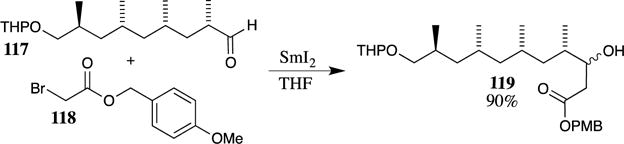
A PMB ester fragment was also used in the formal synthesis of (−)-borrelidin reported by Madduri and Minnaard (Scheme 32).81 Here, aldehyde 117 was used in a samarium iodide-promoted Reformatsky reaction with 4-methoxybenzyl 2-bromoacetate (118). A mixture of isomers resulted from this reaction, but both alcohols could be oxidized to the identical ketone and then reduced selectively utilizing a chiral ruthenium catalyst to access a single stereoisomer of the β-hydroxy ester. This synthetic sequence eventually generated an intermediate that intercepted the route used by Nagamitsu and co-workers to obtain (−)-borrelidin (116),95 thus completing a formal synthesis of this molecule.
Scheme 32.
A interesting Mannich reaction involving a PMB-protected acetate was used in a recent synthesis of the histone deacetylase inhibitor azumamide A (124) (Scheme 33).96 The reaction of the titanium enolate of PMB-protected propionate 121 with sulfinylimine 120 provided the Mannich product 122 in 46% yield with high diastereoselectivity owing to the presence of the chiral sulfinimide auxiliary. The moderate yield in this Mannich reaction is attributed to the unstable nature of the imine, which undergoes rapid hydrolysis. The deprotection of the PMB ester provided the corresponding carboxylic acid, which was not isolated; rather, the crude material was directly coupled with a dipeptide to provide 123 in 66% yield over two steps. A notable feature of this example is the ability to deprotect the PMB ester under acidic conditions with TFA without removing the acid-labile t-butylsulfinyl group (this group is cleaved later in the synthesis with 1.25 M HCl in ethyl acetate). Key intermediate 123 is then converted to azumamide A (124) after a number of subsequent steps.
Scheme 33.
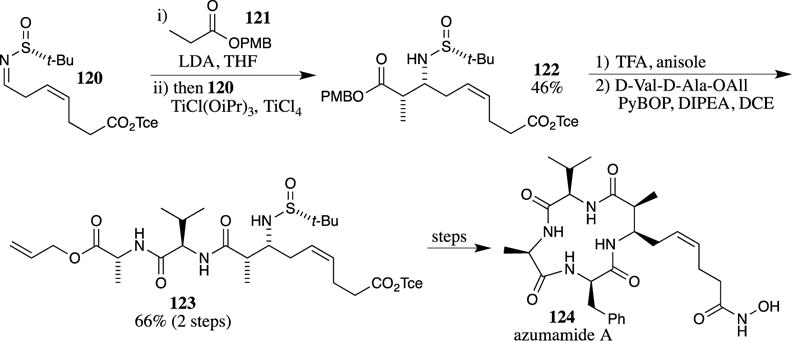
The PMB ester was utilized to excellent advantage in Hoye and Wang’s synthesis of haterumalide NA/oocydin A (128) (Scheme 34).97 The use of a PMB ester in the synthesis of this cytotoxic macrolide was significant because previous routes employing a methyl ester led to intermediates that could not be hydrolyzed under basic and/or neutral conditions without first opening the lactone ring.98–100 The sensitivity of the 14-membered macrocyclic lactone is attributed to the presence of the trans-substituted tetrahydrofuran ring, which imparts significant instability in the macrocycle, accelerating the lactone opening. In the successful synthetic route, a Nozaki-Hiyama-Kishi reaction101 was used to append the PMB ester-containing fragment 126 to the macrocyclic aldehyde 125. The PMB ester was then easily cleaved to the acid 128 with TFA. No yield was provided for the cleavage of the PMB ester, which the authors describe as uneventful. In an identical system prepared using a different synthetic sequence, Roulland reported a 71% yield for the cleavage of the PMB ester 127 with TFA and triethylsilane.102 Subsequent efforts substituted the 2,4-dimethoxybenzyl ester for the PMB ester to facilitate the deprotection in even more highly sensitive systems.103
Scheme 34.
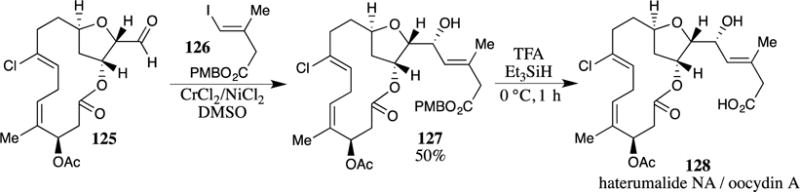
A PMB ester was critical in a synthetic study by Liu and co-workers on the C19 diterpenoid alkaloids (Scheme 35).104 Essential to this endeavor was the preparation of the lactone 131 as a key intermediate. An intramolecular Diels-Alder reaction of triene 129 afforded bicyclic system 130. At this point in the sequence, the PMB ester required cleavage without opening the lactone ring and the resulting acid needed to be reduced to the alcohol 131. The removal of the PMB ester with TFA followed by activation of the resulting carboxylic acid as the mixed carbonate and reduction with sodium borohydride provided primary alcohol 131 in 90% overall yield without any undesired opening of the lactone ring.
Scheme 35.
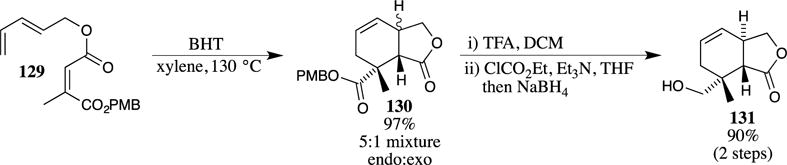
The excellent functional group compatibility of PMB esters is amply demonstrated in the synthesis of the anti-inflammatory compound EI-1941-2 (137) (Scheme 36).28,29 First, the PMB ester was installed on carboxylic acid 132 using 4-methoxybenzyl trichloroacetimidate (36). The formation of the PMB ester under basic conditions is precluded by the presence of the sensitive epoxide and the electron-poor alkene in compound 132. The use of the trichloroacetimidate avoids these difficulties, however, demonstrating the utility of these mild reaction conditions for the installation of a PMB ester in the synthesis of complex molecules. After formation of this ester, a Stille coupling reaction was employed to introduce an alkene with a Z-configuration. The PMB group was then removed using TFA in dichloromethane, providing carboxylic acid 136 in excellent yield. This sequence illustrates the utility of the TFA-promoted cleavage conditions for PMB esters in sensitive systems, as the epoxide and diene of 136 are not appreciably degraded under these deprotection conditions, and no isomerization of the newly introduced Z-alkene was observed.
Scheme 36.
The mild removal conditions associated with PMB ester cleavage were also used in the synthesis of the DEF-ring system of the antitumor agent (−)-FR182877 (138) (Scheme 37) by Nakada and his co-workers.55 The ring system of pyran 139 is highly strained due to the bridging alkyl chain that spans the unsaturated oxydecalin framework. The Nakada group used an elegant intramolecular hetero-Diels-Alder reaction with the methyl ester analog of 140 to form the core dihydropyran system of (−)-FR182877 (138). Unfortunately, the methyl ester could not be cleaved without disturbing the sensitive, strained alkene. Switching to the PMB ester in 141 allowed for the concurrent cleavage of the PMB ester and the silyl protecting group in high yield. Although OTBS groups are typically stable under deprotection conditions for PMB esters, the inclusion of water in the reaction matrix facilitates the hydrolysis of the silyl ether and may also aid in scavenging the 4-methoxybenzyl cation that is generated from cleavage of the PMB ester. Although hydroxyacid 142 could then be cyclized, the desired lactone 143 was unstable and could only be isolated as the corresponding hydrate 144.
Scheme 37.
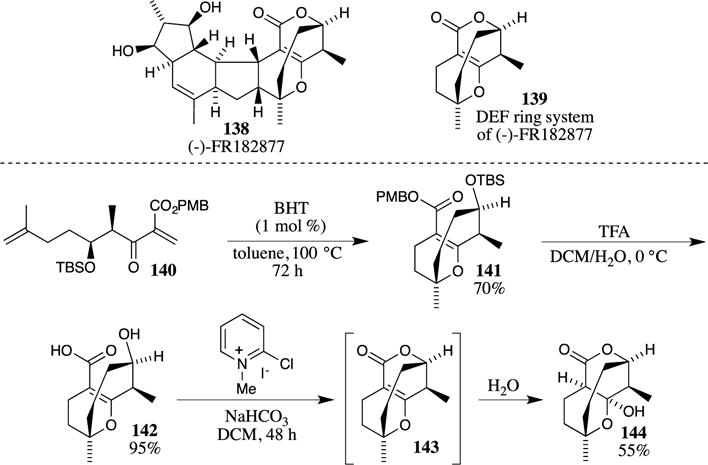
The most common acidic conditions that remove PMB esters (neat TFA or TFA in DCM) are also capable of removing Boc carbamates from amines. Joullié and co-workers exploited these simultaneous deprotections in the synthesis of ustiloxin D (147) (Scheme 38).105 After the preparation of intermediate 145, the PMB ester and the N-Boc groups were removed with TFA. Notably, the benzyl ether, benzyl ester and N-Cbz carbamate present in 145 were inert to these deprotection conditions. Intramolecular coupling of the carboxylate and the amine then provided the cyclic peptide 147 in 38% yield over the two steps. The removal of the benzyl protecting groups, along with hydrogenation of the alkyne, then provided ustiloxin D (148).
Scheme 38.
Cleavage of a PMB ester in an even more complex molecule is included in a synthetic approach to phorboxazole B (152). This example was reported in a doctoral dissertation from the Burke group at the University of Wisconsin in which the synthesis of the protected hydroxyacid 149, an advanced intermediate toward the natural product (Scheme 39), is described.85 Initial attempts to simultaneously remove the PMB ether and the PMB ester from 149 with DDQ failed, with only cleavage of the PMB ether being observed. Other methods to cleave the PMB ester were investigated; however, TFA/anisole/DCM, AlCl3/anisole and Pd(OH)2/H2/MeOH were shown to be problematic because these conditions led to extensive decomposition of the substrate. Base hydrolysis with lithium hydroxide provided the desired hydroxy acid 150, albeit in a moderate 41% yield; unfortunately, attempts to cyclize hydroxy acid 150 to the lactone were unsuccessful. The Burke group was able to complete the synthesis of the natural product through a modified route, however, which utilized a Horner-Wadsworth-Emmons reaction to close the macrocycle.106
Scheme 39.

The PMB group has also been used to protect phosphonates as phosphate esters, as shown in PMB phosphate 153 (Scheme 40). The pioneering work of Evans, Gage and Leighton on the synthesis of calyculin A (154) first demonstrated this concept.107 The PMB ester was chosen for this application because it could be removed from the phosphate under acidic conditions. In this case, the weaker hydrofluoric acid was employed to deprotect the phosphate group. This proved advantageous because HF is also effective at removing silyl protecting groups, thus providing a global deprotection strategy for the synthesis of complex molecules such as calyculin A (154).
Scheme 40.
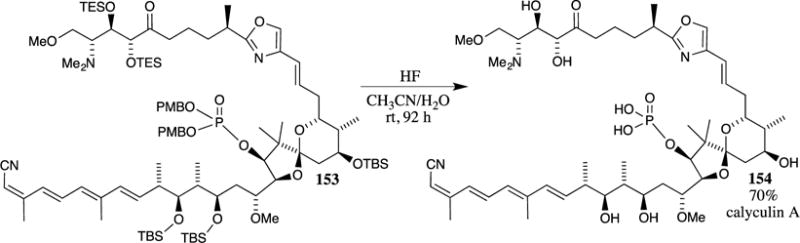
Trost and McDougall chose to use a PMB ester in the synthesis of the welwitindolinone core system 159 (Scheme 41).108 The PMB ester containing tropone 156 was treated with allylic silane 155 in a rapid Pd-catalyzed [6+3] cycloaddition. The process was rendered enantioselective utilizing the phosphoramidite L1 as a ligand, which provided the desired cyclohexanone 157 in 80% yield and a 94% ee. After isomerization of the exocyclic alkene with DMAP, the PMB ester was removed in virtually quantitative yield with TFA to provide carboxylic acid 158, a key intermediate in the synthesis of the target molecule 159.
Scheme 41.
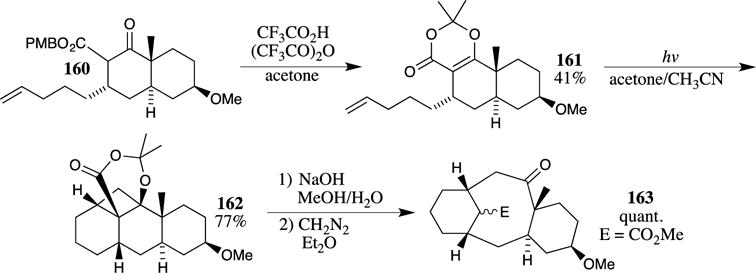
Winkler and co-workers utilized a PMB ester to advantage in their synthetic studies on taxane diterpenes (Scheme 42).109 PMB ester 160 was treated with TFA and trifluoroacetic anhydride in acetone to provide acetonide 161, which was then subjected to photocyclization. While not explicitly stated, attempts to hydrolyze the ester under basic conditions likely resulted in decarboxylation, thus leading to the employment of the PMB ester, which could be converted to the desired heterocycle under acidic conditions. After photocyclization, opening of the cyclobutane with aqueous sodium hydroxide provided the desired taxane-like ring system 163.
Scheme 42.
The Winkler group also explored the use of similar chemistry toward the ingenane diterpenes, such as ingenol (170) (Scheme 43).51 The acylation of ketone 164 with 4-methoxybenzyl cyanoformate (165) provided the PMB-protected ketoester 166. The formation of the acetonide followed by irradiation then formed the polycyclic intermediate 168. Base-mediated fragmentation then provided ketoacid 169, which possessed the requisite ring system to access the ingenane diterpenes. This route was able to provide ample material for subsequent studies on the biological activity of ingenol analogues that focused on their binding to protein kinase C;110 the route was also eventually adapted to a total synthesis of ingenol (170).111
Scheme 43.

2. Examples in Methodology Development
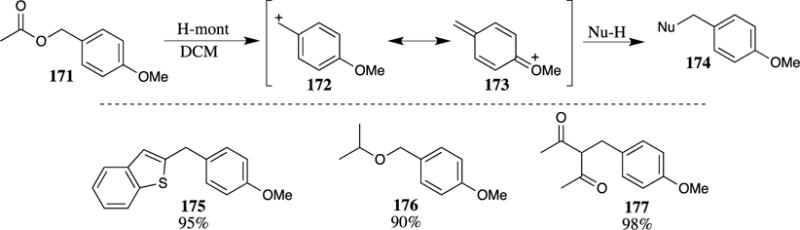
The H-Mont montmorillonite catalyst used to cleave PMB esters (see Scheme 20) has also been used to generate the corresponding 4-methoxybenzyl cation from PMB acetate (171) (Scheme 44).73 This reactive intermediate is highly stabilized, as illustrated by the O-methylated quinone methide resonance structure 173. Cation 172 could be trapped with a number of nucleophiles to provide the benzylated products. The nucleophiles include aromatic rings (such as benzothiophene), alcohols, β-diketones, β-ketoesters, allylsilanes and silyl enol ethers. These reactions provide access to many useful systems, such as 175–177.
Scheme 44.
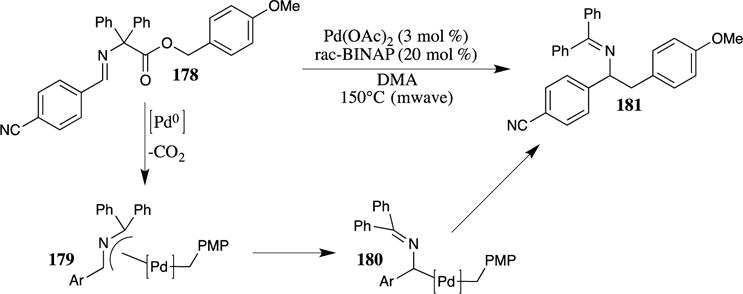
Fields and Chumra found that PMB esters were good substrates for the decarboxylative benzylation of diphenylglycinate esters (Scheme 45).112 Using a palladium catalyst and microwave heating, this reaction provides ready access to benzyl imine derivatives, such as compound 181. The mechanism is believed to proceed through the insertion of a palladium(0) catalyst (generated from Pd(OAc)2 and rac-BINAP) into the benzylic C-O bond. Rapid decarboxylation then leads to the aza-π-allyl intermediate 179, which isomerizes to the η1-complex 180. Reductive elimination then provides the observed product. The ability of the PMB ester to participate in this reaction is notable, as it expands the scope of decarboxylative C-C bond-forming reactions.
Scheme 45.
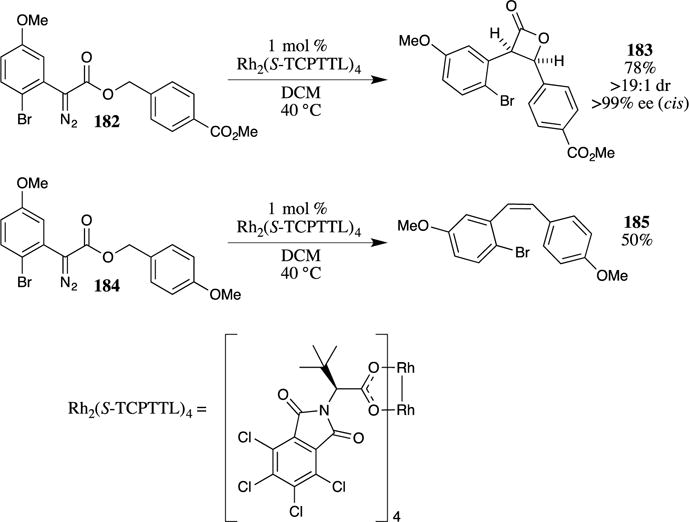
Davies and co-workers recently reported that the use of PMB esters caused an unusual divergence in the products of a rhodium-catalyzed β-lactone formation (Scheme 46).113 Utilizing a rhodium(II) catalyst derived from a modified tert-leucine (Rh2(TCPTTL)4), they observed a rapid intramolecular C-H insertion with diazoester 182 to form the β-lactone 183. When the reaction was performed on the PMB ester 184, however, the expected lactone was not formed; rather, the alkene 185 was isolated as the sole product. This result was attributed to the facile extrusion of CO2 from the 4-methoxybenzyl ester, which is consistent with previous studies.114
Scheme 46.
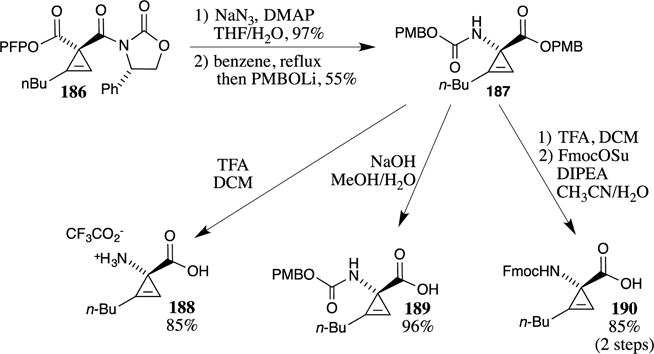
PMB esters have also been used in the synthesis of cyclopropene-containing α-amino acids. Zhang and Fox developed a useful method for the enantioselective desymmetrization of these systems by reacting symmetrical cyclopropene-1,1-diesters with an oxazolidinone chiral nucleophile.115 This reaction provided ready access to enantiomerically pure cyclopropenes, such as 186 (Scheme 47). The conversion of the pentafluorophenyl (PFP) ester to the corresponding acyl azide, subsequent rearrangement to the isocyanate, followed by the addition of the lithium salt of 4-methoxybenzyl alcohol results in the overall conversion of the PFP ester into a 4-methoxybenzyl carbamate. The PMB alkoxide also displaces the oxazolidinone, affording PMB ester 187. The PMB ester and PMB carbamate of 187 provide a flexible protecting group system to access the parent amino acid and mono-protected variants. For example, treatment with TFA removes both of the protecting groups, leading to amino acid 188. Reaction with base selectively cleaves the PMB ester, providing the free carboxylic acid 189. The free amino acid can also be treated with a protecting reagent (such as fluorenylmethyloxycarbonyl (Fmoc) succinate) to introduce other protecting groups on the amine, as shown for the synthesis of the Fmoc-protected cyclopropene 190. These efforts provide useful routes to cyclopropene containing α-amino acids that can be incorporated into novel peptide architectures.
Scheme 47.
3. Other Examples
In addition to their primary uses in peptide synthesis, medicinal chemistry, natural product synthesis and the development of new methodologies, PMB esters have also begun to find applications in bioorganic chemistry. For example, Vosmann and co-workers recently evaluated the formation of 4-methoxybenzyl esters in enzyme-catalyzed transesterification reactions with hydroxybenzoic acid derivatives.116 This methodology focused on finding an improved method for the isolation of desirable food additives (specifically 4-hydroxybenzoic acid and 4-hydroxy-3-methoxybenzoic acid) from bulk plant material. bis-PMB phosphonate esters have also been explored as pro-drugs, imparting improved oral bioavailability compared with the unprotected phosphate analogoues.117 These bis-PMB phosphonate esters were shown to have increased stability in aqueous solution over other phosphonate esters. The PMB groups are easily cleaved in vivo by cytochrome P450 enzymes, facilitating the delivery of the phosphonic acids.
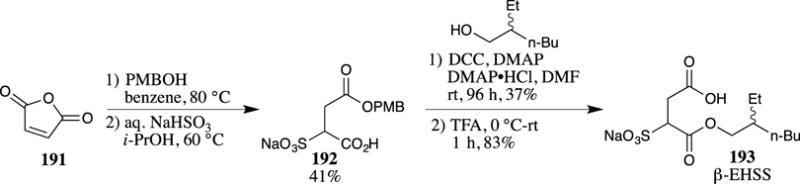
PMB esters have also found use in radiolabeling studies, for example, in the preparation of 13C-enriched ethylhexyl sulfosuccinate (EHSS, 193) (Scheme 48).118 EHSS (193) has been detected as a bio-degradation product from surfactants used to combat water contamination from oil spills in the Gulf of Mexico. An isotopically labeled standard for EHSS was required for LC-MS/MS quantification experiments; therefore, a synthesis was undertaken. PMB alcohol was added to maleic anhydride to form the corresponding PMB ester 192. The carboxylic acid was esterified to form the corresponding ethylhexyl ester 193. Cleavage of the PMB group was achieved with TFA without disturbing the ethylhexyl ester or the pendant sulfonate to provide β-EHSS (193). The use of [13C]4-maleic anhydride in this route provided access to isotopically labeled EHSS.
Scheme 48.
Summary and Conclusion
The PMB ester has become a dependable and extremely useful protecting group in organic synthesis. Although early work in peptide-type chemistry provided an indication that PMB esters could be introduced and cleaved under mild conditions without disturbing sensitive functionalities, the adoption of the PMB ester by synthetic organic chemists has demonstrated the full potential of this versatile carboxylate protecting group. The stability of the PMB ester in many common carbon-carbon bond-forming reactions (such as the aldol condensation, Diels-Alder reactions, and palladium-catalyzed cyclizations), as well as the diverse procedures for the cleavage of the ester (acid, base, photochemical, and hydrogenolysis), provides a flexible solution for carboxylate group protection that is extremely attractive when planning a complex synthesis. In addition, the starting 4-methoxybenzyl alcohol is inexpensive, with many options (through the corresponding halides, trichloroacetimidate and urea reagents, or transesterification) for the formation of the desired PMB ester. PMB esters are also only mildly invasive in NMR spectra, with simple signals due to the inherent symmetry of the 4-methoxybenzyl structure. These NMR signals are typically readily identified and separate from other peaks, thus facilitating the interpretation of the spectra. Therefore, PMB esters have found use in the synthesis of many complex systems and in numerous methodology studies. Given the increasing complexity of synthetic targets and the large number of synthetic organic groups, the use of the PMB ester will almost certainly continue to increase in the future. In addition, PMB esters are finding more applications in medicinal and biological chemistry, further increasing the importance of this group. Future studies on PMB esters will likely include the development of more environmentally friendly and inexpensive methods for their introduction and deprotection.
Acknowledgments
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for a recent New Directions award (54823-ND1) in support of our work with trichloroacetimidates. Syracuse University is thanked for a sabbatical leave for J.D.C., during which time this review was completed.
References
- 1.Wuts PGM. In: Protective Groups in Organic Synthesis. 5th. Greene TW, editor. John Wiley & Sons; Hoboken, NJ: 2014. [Google Scholar]
- 2.Kocienski PJ. Protecting Groups. 3rd. Thieme Verlag; Stuttgart: 2005. [Google Scholar]
- 3.Schelhaas M, Waldmann H. Angew Chem Int Ed Engl. 1996;35:2056. [Google Scholar]
- 4.Maclaren JA. Australian J Chem. 1971;24:1695. [Google Scholar]
- 5.Hiskey RG, Adams JB., Jr J Am Chem Soc. 1965;87:3969. doi: 10.1021/ja01095a033. [DOI] [PubMed] [Google Scholar]
- 6.Haack T, Kurtkaya S, Snyder JP, Georg GI. Org Lett. 2003;5:5019. doi: 10.1021/ol036007d. [DOI] [PubMed] [Google Scholar]
- 7.Fürstner A. Chem Rev. 1999;99:991. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]
- 8.Fujino A, Sugai T. Adv Synth Catal. 2008;250:1712. [Google Scholar]
- 9.Stelakatos GC, Argyropoulos N. J Chem Soc C. 1970:964. [Google Scholar]
- 10.Matsumoto A, Tanaka T, Oka K. Synthesis. 2005:1479. [Google Scholar]
- 11.Ruder SM, Ronald RC. Tetrahedron Lett. 1987;28:135. [Google Scholar]
- 12.Wuts PGM. p-Methoxybenzyl Chloride. In: Paquette LA, editor. e-EROS Encycl Reagents Org Synth. John Wiley & Sons, Ltd; Chichester, UK: 2001. [Google Scholar]
- 13.Patterson LD, Miller MJ. J Org Chem. 2010;75:1289. doi: 10.1021/jo902406b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siewert B, Pianowski E, Obernauer A, Csuk R. Bioorg Med Chem. 2014;22:594. doi: 10.1016/j.bmc.2013.10.047. [DOI] [PubMed] [Google Scholar]
- 15.Berry NG, Iddon L, Iqbal M, Meng X, Jayapal P, Johnson CH, Nicholson JK, Lindon JC, Harding JR, Wilson ID, Stachulski AV. Org Biomol Chem. 2009;7:2525. doi: 10.1039/b822777b. [DOI] [PubMed] [Google Scholar]
- 16.Hutchinson JH, Riendeau D, Brideau C, Chan C, Delorme D, Denis D, Falgueyret JP, Fortin R, Guay J, Hamel P, Jones TR, Macdonald D, McFarlane CS, Piechuta H, Scheigetz J, Tagari P, Thérien M, Girard Y. J Med Chem. 1993;36:2771. doi: 10.1021/jm00071a008. [DOI] [PubMed] [Google Scholar]
- 17.Walker MA. Tetrahedron. 1997;53:14591. [Google Scholar]
- 18.Barlos K, Gatos D, Kallitsis I, Papaioannou D, Sotiriou P. Liebigs Ann Chem. 1988:1079. [Google Scholar]
- 19.Iranpoor N, Firouzabadi H, Khalili D. Org Biomol Chem. 2010;8:4436. doi: 10.1039/c004357e. [DOI] [PubMed] [Google Scholar]
- 20.Burkett BA, Huleatt PB. The Mitsunobu Reaction: From bench to batch. Current Perspectives and Future Challenges. In: Luque R, editor. Green Chem. Nova Science Publishers; Hauppauge, NY: 2014. p. 149. [Google Scholar]
- 21.But TYS, Toy PH. J Am Chem Soc. 2006;128:9636. doi: 10.1021/ja063141v. [DOI] [PubMed] [Google Scholar]
- 22.Büchi H, Steen K, Eschenmoser A. Angew Chem. 1963;75:1176. [Google Scholar]
- 23.Brechbühler H, Büchi H, Hatz E, Schreiber J, Eschenmoser A. Helv Chim Acta. 1965;48:1746. [Google Scholar]
- 24.Waddell ST, Santorelli GM. Tetrahedron Lett. 1996;37:1971. [Google Scholar]
- 25.Wang MF, Golding BT, Potter GA. Synth Commun. 2000;30:4197. [Google Scholar]
- 26.Schmidt E, Däbritz E, Thulke K. Justus Liebigs Ann Chem. 1965;685:161. [Google Scholar]
- 27.Finke PE, Ashe BM, Knight WB, Maycock AL, Navia MA, Shah SK, Thompson KR, Underwood DJ, Weston H, Zimmerman M, Doherty JB. J Med Chem. 1990;33:2522. doi: 10.1021/jm00171a029. [DOI] [PubMed] [Google Scholar]
- 28.Jones JH, Young GT. Chem Ind. 1966:1722. [Google Scholar]
- 29.Shoji M, Uno T, Hayashi Y. Org Lett. 2004;6:4535. doi: 10.1021/ol0481479. [DOI] [PubMed] [Google Scholar]
- 30.Shoji M, Uno T, Kakeya H, Onose R, Shiina I, Osada H, Hayashi Y. J Org Chem. 2005;70:9905. doi: 10.1021/jo0516436. [DOI] [PubMed] [Google Scholar]
- 31.Shah JP, Russo CM, Howard KT, Chisholm JD. Tetrahedron Lett. 2014;55:1740. [Google Scholar]
- 32.Weygand F, Hunger K. Chem Ber. 1962;95:1. [Google Scholar]
- 33.Weygand F, Hunger K. Chem Ber. 1962;95:7. [Google Scholar]
- 34.Sheikh MC, Takagi S, Yoshimura T, Morita H. Tetrahedron. 2010;66:7272. [Google Scholar]
- 35.Feldman KS, Eastman KJ, Lessene G. Org Lett. 2002;4:3525. doi: 10.1021/ol026694t. [DOI] [PubMed] [Google Scholar]
- 36.Moore JD, Byrne RJ, Vedantham P, Flynn DL, Hanson PR. Org Lett. 2003;5:4241. doi: 10.1021/ol0352759. [DOI] [PubMed] [Google Scholar]
- 37.Karade NN, Shirodkar SG, Potrekar RA, Karade HN. Synth Commun. 2004;34:391. [Google Scholar]
- 38.Roof AAM, van Woerden HF, Cerfontain H. Tetrahedron. 1976;32:2967. [Google Scholar]
- 39.Yang Z, Zhou J. J Am Chem Soc. 2012;134:11833. doi: 10.1021/ja304099j. [DOI] [PubMed] [Google Scholar]
- 40.Biel M, Kretsovali A, Karatzali E, Papamatheakis J, Giannis A. Angew Chem Int Ed. 2004;43:3974. doi: 10.1002/anie.200453879. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto H, Matsuda T, Nakata S, Mitoguchi T, Kimura T, Hayashi Y, Kiso Y. Bioorg Med Chem. 2001;9:417. doi: 10.1016/s0968-0896(00)00261-3. [DOI] [PubMed] [Google Scholar]
- 42.Williams JB, Chapman TM, Hercules DM. Macromolecules. 2003;36:3898. [Google Scholar]
- 43.Nefkens GHL, Thuring JWJF, Zwanenburg B. Synthesis. 1997:290. [Google Scholar]
- 44.Bolm C, Schiffers I, Atodiresei I, Hackenberger CPR. Tetrahedron: Asymmetry. 2003;14:3455. [Google Scholar]
- 45.Stewart FHC. Australian J Chem. 1968;21:2543. [Google Scholar]
- 46.Stewart FHC. Australian J Chem. 1965;18:887. [Google Scholar]
- 47.Goodman M, Stueben KC. J Am Chem Soc. 1959;81:3980. [Google Scholar]
- 48.Heller ST, Fu T, Sarpong R. Org Lett. 2012;14:1970. doi: 10.1021/ol300339q. [DOI] [PubMed] [Google Scholar]
- 49.Heller ST, Sarpong R. Org Lett. 2010;12:4572. doi: 10.1021/ol1018882. [DOI] [PubMed] [Google Scholar]
- 50.Heller ST, Sarpong R. Tetrahedron. 2011;67:8851. [Google Scholar]
- 51.Winkler JD, Henegar KE, Hong BC, Williard PG. J Am Chem Soc. 1994;116:4183. [Google Scholar]
- 52.Dharma Rao GB, Acharya BN, Kaushik MP. Tetrahedron Lett. 2013;54:6644. [Google Scholar]
- 53.Orita A, Sakamoto K, Hamada Y, Mitsutome A, Otera J. Tetrahedron. 1999;55:2899. [Google Scholar]
- 54.Otera J, Dan-oh N, Nozaki H. J Org Chem. 1991;56:5307. [Google Scholar]
- 55.Kobayakawa Y, Mori Y, Okajima H, Terada Y, Nakada M. Org Lett. 2012;14:2086. doi: 10.1021/ol300615w. [DOI] [PubMed] [Google Scholar]
- 56.Jägel J, Schmauder A, Binanzer M, Maier ME. Tetrahedron. 2007;63:13006. [Google Scholar]
- 57.Webber MJ, Warren SA, Grainger DM, Weston M, Clark S, Woodhead SJ, Powell L, Stokes S, Alanine A, Stonehouse JP, Frampton CS, White AJP, Spivey AC. Org Biomol Chem. 2013;11:2514. doi: 10.1039/c3ob27187k. [DOI] [PubMed] [Google Scholar]
- 58.Grant SW, Zhu K, Zhang Y, Castle SL. Org Lett. 2006;8:1867. doi: 10.1021/ol0604264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Street LJ, Baker R, Castro JL, Chambers MS, Guiblin AR, Hobbs SC, Matassa VG, Reeve AJ, Beer MS, Middlemiss DN, Noble AJ, Stanton JA, Scholey K, Hargreaves RJ. J Med Chem. 1993;36:1529. doi: 10.1021/jm00063a003. [DOI] [PubMed] [Google Scholar]
- 60.Schultz AG, Dai M, Khim SK, Pettus L, Thakkar K. Tetrahedron Lett. 1998;39:4203. [Google Scholar]
- 61.Khim SK, Dai M, Zhang X, Chen L, Pettus L, Thakkar K, Schultz AG. J Org Chem. 2004;69:7728. doi: 10.1021/jo0490853. [DOI] [PubMed] [Google Scholar]
- 62.Molina MT, del Valle C, Escribano AM, Ezquerra J, Pedregal C. Tetrahedron. 1993;49:3801. [Google Scholar]
- 63.Schoenfelder A, Mann A. Synth Commun. 1990;20:2585. [Google Scholar]
- 64.Gagosz F, Zard SZ. Org Lett. 2002;4:4345. doi: 10.1021/ol0270024. [DOI] [PubMed] [Google Scholar]
- 65.Sibi MP, Kawashima K, Stanley LM. Org Lett. 2009;11:3894. doi: 10.1021/ol901504p. [DOI] [PubMed] [Google Scholar]
- 66.Groselj U, Golobic A, Svete J, Stanovnik B. Chirality. 2013;25:541. doi: 10.1002/chir.22166. [DOI] [PubMed] [Google Scholar]
- 67.Ma G, Deng J, Sibi MP. Angew Chem Int Ed. 2014;53:11818. doi: 10.1002/anie.201406684. [DOI] [PubMed] [Google Scholar]
- 68.Ma G, Sibi MP. Org Chem Front. 2014;1:1152. [Google Scholar]
- 69.Li Y, Du W, Deng WP. Tetrahedron. 2012;68:3611. [Google Scholar]
- 70.Torii S, Tanaka H, Taniguchi M, Kameyama Y, Sasaoka M, Shiroi T, Kikuchi R, Kawahara I, Shimabayashi A, Nagao S. J Org Chem. 1991;56:3633. [Google Scholar]
- 71.Sutton AE, Clardy J. Org Lett. 2000;2:319. doi: 10.1021/ol991264x. [DOI] [PubMed] [Google Scholar]
- 72.Goodacre J, Ponsford RJ, Stirling I. Tetrahedron Lett. 1975;42:3609. [Google Scholar]
- 73.Chen D, Xu C, Deng J, Jiang C, Wen X, Kong L, Zhang J, Sun H. Tetrahedron. 2014;70:1975. [Google Scholar]
- 74.Young SD, Tamburini PP. J Am Chem Soc. 1989;111:1933. [Google Scholar]
- 75.Ohtani M, Watanabe F, Narisada M. J Org Chem. 1984;49:5271. [Google Scholar]
- 76.Tsuji T, Kataoka T, Yoshioka M, Sendo Y, Nishitani Y, Hirai S, Maeda T, Nagata W. Tetrahedron Lett. 1979;20:2793. [Google Scholar]
- 77.Fisher JW, Trinkle KL. Tetrahedron Lett. 1994;35:2505. [Google Scholar]
- 78.Cha KH, Kang TW, Cho DO, Lee HW, Shin J, Jin KY, Kim KW, Kim JW, Hong CI. Synth Commun. 1999;29:3533. [Google Scholar]
- 79.Ilangovan A, Saravanakumar S, Malayappasamy S, Manickam G. RSC Adv. 2013;3:14814. [Google Scholar]
- 80.Kern N, Dombray T, Blanc A, Weibel JM, Pale P. J Org Chem. 2012;77:9227. doi: 10.1021/jo301787v. [DOI] [PubMed] [Google Scholar]
- 81.Madduri AVR, Minnaard AJ. Chem Eur J. 2010;16:11726. doi: 10.1002/chem.201001284. [DOI] [PubMed] [Google Scholar]
- 82.Sakai A, Aoyama T, Shioiri T. Heterocycles. 2000;52:643. [Google Scholar]
- 83.Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021. [Google Scholar]
- 84.Nakajima N, Abe R, Yonemitsu O. Chem Pharm Bull. 1988;36:4244. [Google Scholar]
- 85.Wysocki LM. Ph D Thesis. Univ of Wisconsin; Madison: 2008. The Total Synthesis of Trilobacin and Investigation of the Synthesis of Phorboxazole B. Dissertation Abstracts, 3349109. [Google Scholar]
- 86.Kim CU, Misco PF. Tetrahedron Lett. 1985;26:2027. [Google Scholar]
- 87.Yoo SE, Kim HR, Yi KY. Tetrahedron Lett. 1990;31:5913. [Google Scholar]
- 88.Aizpurua JM, Juaristi M, Lecea B, Palomo C. Tetrahedron. 1985;41:2903. [Google Scholar]
- 89.Dapperheld S, Steckhan E. Angew Chem, Int Ed. 1982;21:780. [Google Scholar]
- 90.Schmidt W, Steckhan E. Angew Chem, Int Ed. 1978;17:673. [Google Scholar]
- 91.Doni E, O’Sullivan S, Murphy JA. Angew Chem Int Ed. 2013;52:2239. doi: 10.1002/anie.201208066. [DOI] [PubMed] [Google Scholar]
- 92.Cosner CC, Bhaskara Reddy Iska V, Chatterjee A, Markiewicz JT, Corden SJ, Löfstedt J, Ankner T, Richer J, Hulett T, Schauer DJ, Wiest O, Helquist P. Eur J Org Chem. 2013;2013:162. [Google Scholar]
- 93.Cosner CC, Helquist P. Org Lett. 2011;13:3564. doi: 10.1021/ol200964m. [DOI] [PubMed] [Google Scholar]
- 94.Vong BG, Kim SH, Abraham S, Theodorakis EA. Angew Chem Int Ed. 2004;43:3947. doi: 10.1002/anie.200460203. [DOI] [PubMed] [Google Scholar]
- 95.Nagamitsu T, Takano D, Marumoto K, Fukuda T, Furuya K, Otoguro K, Takeda K, Kuwajima I, Harigaya Y, Omura S. J Org Chem. 2007;72:2744. doi: 10.1021/jo062089i. [DOI] [PubMed] [Google Scholar]
- 96.Wen S, Carey KL, Nakao Y, Fusetani N, Packham G, Ganesan A. Org Lett. 2007;9:1105. doi: 10.1021/ol070046y. [DOI] [PubMed] [Google Scholar]
- 97.Hoye TR, Wang J. J Am Chem Soc. 2005;127:6950. doi: 10.1021/ja051749i. [DOI] [PubMed] [Google Scholar]
- 98.Kigoshi H, Kita M, Ogawa S, Itoh M, Uemura D. Org Lett. 2003;5:957. doi: 10.1021/ol0341804. [DOI] [PubMed] [Google Scholar]
- 99.Gu Y, Snider BB. Org Lett. 2003;5:4385. doi: 10.1021/ol0356789. [DOI] [PubMed] [Google Scholar]
- 100.Yu G. Ph D Thesis. Brandeis University; 2004. Total Synthesis of (−)- and (+)-Dysibetaine Synthesis of (−)- and of (+)-Haterumalide NA Methyl Ester. Dissertation Abstracts, 3127118. [Google Scholar]
- 101.Takai K. Org React. 2004;64:1. [Google Scholar]
- 102.Roulland E. Angew Chem Int Ed. 2008;47:3762. doi: 10.1002/anie.200800585. [DOI] [PubMed] [Google Scholar]
- 103.Ueda M, Yamaura M, Ikeda Y, Suzuki Y, Yoshizato K, Hayakawa I, Kigoshi H. J Org Chem. 2009;74:3370. doi: 10.1021/jo802806z. [DOI] [PubMed] [Google Scholar]
- 104.Liu ZG, Xu L, Chen QH, Wang FP. Tetrahedron. 2012;68:159. [Google Scholar]
- 105.Li P, Evans CD, Wu Y, Cao B, Hamel E, Joullié MM. J Am Chem Soc. 2008;130:2351. doi: 10.1021/ja710363p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lucas BS, Gopalsamuthiram V, Burke SD. Angew Chem Int Ed. 2007;46:769. doi: 10.1002/anie.200603656. [DOI] [PubMed] [Google Scholar]
- 107.Evans DA, Gage JR, Leighton JL. J Am Chem Soc. 1992;114:9434. [Google Scholar]
- 108.Trost BM, McDougall PJ. Org Lett. 2009;11:3782. doi: 10.1021/ol901499b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Winkler JD, Lee CS, Rubo L, Muller CL, Squattrito PJ. J Org Chem. 1989;54:4491. [Google Scholar]
- 110.Winkler JD, Kim S, Harrison S, Lewin NE, Blumberg PM. J Am Chem Soc. 1999;121:296. [Google Scholar]
- 111.Winkler JD, Rouse MB, Greaney MF, Harrison SJ, Jeon YT. J Am Chem Soc. 2002;124:9726. doi: 10.1021/ja026600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fields WH, Chruma JJ. Org Lett. 2010;12:316. doi: 10.1021/ol902651j. [DOI] [PubMed] [Google Scholar]
- 113.Fu L, Wang H, Davies HML. Org Lett. 2014;16:3036. doi: 10.1021/ol5011505. [DOI] [PubMed] [Google Scholar]
- 114.Guptill DM, Cohen CM, Davies HML. Org Lett. 2013;15:6120. doi: 10.1021/ol4028978. [DOI] [PubMed] [Google Scholar]
- 115.Zhang F, Fox JM. Org Lett. 2006;8:2965. doi: 10.1021/ol060847l. [DOI] [PubMed] [Google Scholar]
- 116.Vosmann K, Wiege B, Weitkamp P, Weber N. Appl Microbiol Biotechnol. 2008;80:29. doi: 10.1007/s00253-008-1534-y. [DOI] [PubMed] [Google Scholar]
- 117.Dang Q, Liu Y, Rydzewski RM, Brown BS, Robinson E, van Poelje PD, Colby TJ, Erion MD. Bioorg Med Chem Lett. 2007;17:3412. doi: 10.1016/j.bmcl.2007.03.089. [DOI] [PubMed] [Google Scholar]
- 118.Barsamian AL, Perkins MJ, Field JA, Blakemore PR. J Labelled Compd Radiopharm. 2014;57:397. doi: 10.1002/jlcr.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]