

Abstract
Background:
Mitochondrial diseases are a group of energy metabolic disorders with multisystem involvements. Variable clinical features present a major challenge in pediatric diagnoses. We summarized the clinical spectrum of m.3243A>G mutation in Chinese pediatric patients, to define the common clinical manifestations and study the correlation between heteroplasmic degree of the mutation and clinical severity of the disease.
Methods:
Clinical data of one-hundred pediatric patients with symptomatic mitochondrial disease harboring m.3243A>G mutation from 2007 to 2013 were retrospectively reviewed. Detection of m.3243A>G mutation ratio was performed by polymerase chain reaction (PCR)-restriction fragment length polymorphism. Correlation between m.3243A>G mutation ratio and age was evaluated. The differences in clinical symptom frequency of patients with low, middle, and high levels of mutation ratio were analyzed by Chi-square test.
Results:
Sixty-six patients (66%) had suffered a delayed diagnosis for an average of 2 years. The most frequent symptoms were seizures (76%), short stature (73%), elevated plasma lactate (70%), abnormal magnetic resonance imaging/computed tomography (MRI/CT) changes (68%), vomiting (55%), decreased vision (52%), headache (50%), and muscle weakness (48%). The mutation ratio was correlated negatively with onset age (r = −0.470, P < 0.001). Myopathy was more frequent in patients with a high level of mutation ratio. However, patients with a low or middle level of m.3243A>G mutation ratio were more likely to suffer hearing loss, decreased vision, and gastrointestinal disturbance than patients with a high level of mutation ratio.
Conclusions:
Our study showed that half of Chinese pediatric patients with m.3243A>G mutation presented seizures, short stature, abnormal MRI/CT changes, elevated plasma lactate, vomiting, and headache. Pediatric patients with these recurrent symptoms should be considered for screening m.3243A>G mutation. Clinical manifestations and laboratory abnormalities should be carefully monitored in patients with this point mutation.
Keywords: Clinical Symptom, Heteroplasmy, Mitochondrial A3243G Mutation, Mitochondrial Disease
INTRODUCTION
Mitochondrial diseases are a group of energy metabolism disorders with multisystem involvement. They can be caused by mutations in nuclear genes either relating to mitochondrial components or mitochondrial DNA (mtDNA). The prevalence of mitochondrial diseases due to mutations in mtDNA and related nuclear genes is estimated to be 1 in 5000.[1] Mutations in mtDNA are much higher than those in the related nuclear genes, probably because that mitochondrial genome has been exposed in reactive oxygen species(ROS) and lacked protection structure.
In 1990, the adenine to guanine transition at the mtDNA position of 3243 encoding tRNALeu (UUR) was found to be the molecular basis for mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS).[2,3] Epidemiological studies showed that m.3243A>G mutation is the most frequent pathogenic mutation in mtDNA. The prevalence of m.3243A>G mutation in mtDNA is 3.5/100,000 adults in the North East of England[4] and approximately 1/6000 in an adult population in Finland.[5] The clinical features of m.3243A>G mutation are highly variable.[6,7,8] Seizures, encephalopathy, and stroke-like episodes were found in about 80% patients with MELAS, and short stature, deafness, cognitive impairment, exercise intolerance, migraines, depression, cardiomyopathy, cardiac conduction defects, and diabetes mellitus were also found in some MELAS cases.
Here, we summarized the clinical spectrum of m.3243A>G mutation in Chinese pediatric patients, to define the common clinical manifestations and study the correlation between heteroplasmic degree of the mutation and clinical severity of the disease.
METHODS
Subjects
Clinical data of 100 Chinese pediatric patients who were first diagnosed as mitochondrial diseases through gene test for m.3243A>G mutation in Peking University First Hospital from 2007 to 2013 were retrospectively reviewed. Informed consent was obtained from patient or patient's guardian. Patients without complete clinical information and laboratory data were excluded. This study was approved by the Medical Ethics Committee of Peking University First Hospital (No. 2009 (23)).
A total of ten clinical characteristics including vision impairment, hearing loss, encephalopathy, myopathy, and gastrointestinal disturbances were collected from these patients. Short stature was defined as the body height less than two standard deviations below the mean height of normal children. Myopathy was manifested as exercise intolerance, weakness, gait instability, etc. Exercise intolerance was defined as poor endurance in sports.
Mitochondrial DNA analysis
The peripheral blood samples were collected from these patients when they were first screened for the gene mutation. Total DNA was extracted from blood sample using a routine method.[9] Detection of m.3243A>G mutation ratio was performed by polymerase chain reaction (PCR)-restriction fragment length polymorphism method. The PCR product was digested with restriction enzyme Apa I, and then separated in 2% agarose gel. Finally, the density of electrophoretic band was measured and calculated for mutation ratio.[10] The patients were divided into three groups based on mutation ratio (low level: 0–30%; middle level: 31–60%; and high level: 61–100%).
Statistic analysis
SPSS version 16.0 (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. The normality of quantitative data was tested by Kolmogorov-Smirnov test. Nonparametric test was used if the data did not conform to Gaussian distribution. Age and mutation ratio inconsistent with Gaussian distribution were presented as median (Q1, Q3). Correlation between m.3243A>G mutation ratio and age was evaluated with Spearman's rank correlation method. The differences in clinical symptom frequency of patients with low, middle, and high levels of mutation ratio were analyzed by Chi-square test. A P < 0.05 was considered statistically significant.
RESULTS
The age of the diagnosis of mitochondrial disease ranged from 2 months to 18 years, with the median age of 9 years (5.8 years, 12.0 years). The median onset age was 7 years (4.0 years, 9.0 years). A delayed diagnosis for an average of 2 years was found in 66% patients. Forty patients were males and sixty were females (ratio = 1.0:1.5). The m.3243A>G mutation ratio varied from 5% to 93%, with a median level of 44% (36%, 54%). There was no significant difference in m.3243A>G mutation ratio between males and females (t = 0.691, P = 0.491).
Clinical features of patients
Patients at the onset had one or more symptoms, including seizures (54%), muscle weakness (29%), headache complicated with vomiting (25%), decreased vision (18%), hearing loss (10%), impaired verbal communication (6%), and heart preexcitation syndrome (5%). The most prevalent symptom of these patients was seizures (76%), followed by short stature (73%), elevated plasma lactate (70%), abnormal magnetic resonance imaging/computed tomography (MRI/CT) changes (68%), vomiting (55%), decreased vision (52%), headache (50%), and muscle weakness (48%). Most of the patients were multisymptomatic, only two patients had one symptom, and five patients manifested two symptoms.
Seizures were present in 76 patients (76%), of which 13 patients had stroke during or shortly after seizures. Recurrent headaches were found in 50 patients (50%), of which most were complicated with vomiting and/or vision loss. Sixty-eight patients (68%) were found to have MRI/CT abnormalities, including abnormal asymmetric signals in occipital area (51/68, 75%), temporal area (21/68, 31%), parietal area (20/68, 29%), bilateral basal ganglia calcification (16/68, 24%), cerebral atrophy (10/68, 15%), and thalamus and brainstem lesions (5/68, 7%). Twenty-one patients (21%) had verbal communication difficulties. Slurred speech was present in 16 patients, of whom 3 had progressively worsening speech, and 2 had delayed speech development (at 1.5 and 3.0 years of age).
Growth impairment was found on height or/and weight in 77 patients. Seventy-three patients (73%) had a short stature and 69 patients (69%) had a weight loss. Gastrointestinal disturbance was also a common symptom for m.3243A>G patients. Fifty-five patients (55%) had experienced recurrent vomiting and 38 patients (38%) had diarrhea or/and constipation.
Plasma lactate ranged from 1.4 to 19.0 mmol/L (normal range: 0.7–2.0 mmol/L). Elevated plasma lactate was detected in 70 patients (70%), of which 90% had a plasma lactate >5 mmol/L. Hyperglycemia (>6.1 mmol/L) was found in ten patients (10%).
Vision impairment was found in 52 patients (52%), of which 33 experienced blurred vision and 16 had decreased visual acuity. Failure to track object or light with their eyes was detected in 3 infants.
Reduced muscle strength was reported in 48 patients (48%), of which 31 had minimal muscle weakness in upper limbs and shoulders and 17 manifested muscle weakness in lower limbs, and 2 had walk difficulties. Thirty-seven patients (37%) complained of difficulties in maintaining stability, of them 18 experienced frequent tumbling, and 38 patients (38%) had compromised exercise tolerance, which appeared during running or going upstairs in most cases and during walk on flat places in only two cases. Eighteen patients (18%) had bilateral ptosis.
Heart diseases were detected in 35 patients (35%), of which 17 were found to have abnormal electrocardiogram such as ST-T changes and arrhythmias. Left ventricular hypertrophy was found in eight patients, right ventricular hypertrophy in five patients, and preexcitation syndrome in five patients.
Twenty-one patients (21%) had hearing impairment, presenting tinnitus or hearing loss (mild deafness in 14 patients, moderate deafness in 5 patients, and severe deafness in 2 patients).
Correlation between m.3243A>G mutation ratio and onset age
The m.3243A>G mutation ratio in peripheral leukocytes was determined in all the patients, and 32% of them had a mutation ratio above 50%. The relationship between m.3243A>G mutation ratio and onset age was analyzed by Spearman's rank correlation method, which showed that m.3243A>G mutation ratio was correlated negatively with onset age [r = −0.470, P < 0.001; Figure 1].
Figure 1.
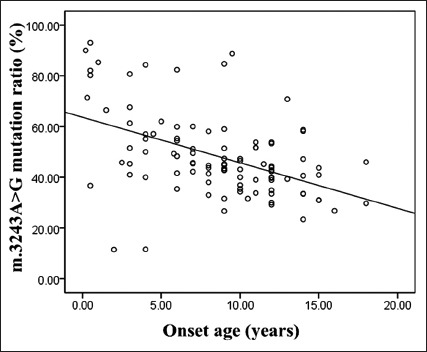
Correlation between m.3243A>G mutation ratio in peripheral leukocytes and onset age (r=−0.470, n=100).
Clinical symptom frequency of patients with different mutation ratio
The differences in clinical symptom frequency among patients with low, middle, and high levels of mutation ratio were analyzed by Chi-square test [Table 1]. There were significant differences in the frequencies of hearing loss, decreased vision, myopathy, and gastrointestinal disturbance among three groups. Myopathy was more frequent in patients with a high level of mutation ratio. However, patients with a low or middle level of m.3243A>G mutation ratio were more likely to suffer from hearing loss, decreased vision, and gastrointestinal disturbance than high level group.
Table 1.
Analysis of clinical symptom frequency in different distributions of 3243A>G mutation ratio, n
| Clinical symptom frequencies | Low level of mutation ratio (n = 8) | Middle level of mutation ratio (n = 75) | High level of mutation ratio (n = 17) | χ2 | P |
|---|---|---|---|---|---|
| Decreased vision | 3 | 45 | 4 | 6.423 | 0.040 |
| Hearing loss | 3 | 18 | 0 | 6.239 | 0.044 |
| Encephalopathy | 5 | 63 | 13 | 2.444 | 0.295 |
| Gastrointestinal disturbance | 6 | 56 | 5 | 13.089 | 0.001 |
| Diabetes | 1 | 8 | 1 | 0.413 | 0.813 |
| Growth impairment | 6 | 59 | 12 | 0.530 | 0.767 |
| Myopathy | 4 | 56 | 16 | 6.097 | 0.047 |
| Heart disease | 4 | 26 | 5 | 1.028 | 0.598 |
| MRI/CT changes | 5 | 52 | 11 | 0.257 | 0.879 |
| Elevated lactate | 5 | 55 | 10 | 1.622 | 0.444 |
Low level: A mutation ratio range 0–30%; Middle level: A mutation ratio range 31–60%; High level: A mutation ratio range 61–100%; MRI: Magnetic resonance imaging; CT: Computed tomography.
DISCUSSION
The m.3243A>G mutation has been shown to lead to reduced levels of the tRNALeu (UUR), decrease in aminoacylation, and absence of the normal modification with 5-taurinomethyl group at the wobble base.[11,12] Mitochondrial disease caused by this mutation may result from the reduction of mtDNA-encoded proteins and oxidative phosphorylation activity in mitochondrial translation. The m.3243A>G mutation is usually present as heteroplasmic state.[13] Its phenotype is highly variable, ranging from asymptomatic to mild or severe phenotype.[14] Several clinical syndromes including MELAS, myoclonic epilepsy with ragged-red fibers, chronic progressive external ophthalmoplegia, and Leigh's syndrome may associate with m.3243A>G mutation.[6,15,16,17,18] Patients with m.3243A>G mutation but without clinical symptoms are not uncommon.[19] To define the spectrum of clinical manifestations due to m.3243A>G mutation, we retrospectively reviewed clinical data of 100 Chinese pediatric patients with m.3243A>G mutation regardless of their clinical presentations.
Central nervous system (CNS) is frequently involved in mitochondrial diseases because of its higher energy demand. Seizures may result from neuronal energy depletion, oxidative stress, impaired calcium signaling, immune disturbances, and deficiencies of vitamins, cofactors, and amino acids.[20] In this study, the CNS symptoms of seizures and headache were found in more than 50% patients. The prevalence of seizures was as high as 76%, and was nearly 100% in our previous study,[21] similar to the prevalence of 70.5% in a report of pediatric mitochondrial disease from Taiwan, China.[22] Brain consumes more energy in children than adults. CNS symptoms are, therefore, more common and severe in children than adults. The prevalence of seizures ranged from 9% to 20% in adults.[23,24] In MELAS patients, recurrent stroke-like episodes were more frequent in those with m.3243A>G mutation than those without the mutation.[25,26] We found stroke-like episodes in 22% patients, higher than the prevalence from patients with mitochondrial mutations other than m.3243A>G mutation.[22]
Most of our patients showed abnormal brain image findings were probably not due to vascular injuries. Bilateral basal ganglia were the most vulnerable site in this disease, followed by temporal, parietal, and occipital area, similar to the previous report.[27] Chae et al.[25] also observed the tendency that basal ganglia were frequently involved in patients without m.3243A>G mutation.
Gastrointestinal symptoms were common (67%) in this series of patients. Vomiting was more frequent than constipation or/and diarrhea, and 68% vomiting cases were complicated with headaches. A higher prevalence (42%) of these symptoms was also reported in a recent study.[24] Therefore, mitochondrial diseases should be considered clinically in children with unexplained vomiting and headaches.
The prevalence of cardiac involvement was highly variable in patients with mtDNA mutations, ranging from 17% to 40%.[28,29] In this study, 35% patients exhibited cardiac involvement with the main manifestations of abnormal electrocardiogram (17%) and left ventricular hypertrophy (8%). The prevalence of left ventricular hypertrophy was less in our patients than the patients with m.3243A>G mutation (56%) reported by Majamaa-Voltti et al.[30] Pediatric patients may be the reason of that lower prevalence of cardiac involvement was present in this study. For patients suspected of mitochondrial disease, routine electrocardiogram, and ultrasonocardiography should be performed.
The m.3243A>G mutation ratio in peripheral leukocytes was negatively correlated with patients’ onset age. Previous studies have also described a decrease of mutant mtDNA with increasing age.[21,31] Prospective studies are needed to confirm this phenomenon. Among these symptoms we described, myopathy was frequently seen in patients with high level of m.3243A>G mutation ratio. However, patients with a low or middle level of m.3243A>G mutation ratio were more likely to suffer from hearing loss, decreased vision, and gastrointestinal disturbance.
In conclusion, this study summarized the common symptoms and onset symptoms in a large cohort of Chinese pediatric patients with m.3243A>G mutation. In view of the fact that the diagnosis of 66% of patients was delayed an average of 2 years, we suggested that examination of m.3243A>G mutation in mtDNA should be considered in children with one or more of the symptoms including seizures, short stature, muscle weakness, headache complicated with vomiting, decreased vision, and hearing loss. Although the mutation ratio in blood sample is an available test for diagnosis of mitochondrial disease, the m.3243A>G mutation ratio is usually higher in muscle and urine sample. Therefore, the mutation ratio in other tissues, such as muscle and urine cell, should be included in the future study.
Financial support and sponsorship
This study was supported by grants from National Natural Science Foundation of China (No. 81271256 and No. 81471153) and the Capital Characteristic Clinical Application Research Projects (No. Z131107002213062).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Xin Chen
REFERENCES
- 1.Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: A metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu) (UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–3. doi: 10.1038/348651a0. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi Y, Momoi MY, Tominaga K, Momoi T, Nihei K, Yanagisawa M, et al. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes) Biochem Biophys Res Commun. 1990;173:816–22. doi: 10.1016/s0006-291x(05)80860-5. doi: 10.1016/S0006-291X(05)80860-5. [DOI] [PubMed] [Google Scholar]
- 4.Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–9. doi: 10.1002/ana.24362. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Kärppä M, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: Prevalence of the mutation in an adult population. Am J Hum Genet. 1998;63:447–54. doi: 10.1086/301959. doi: 10.1086/301959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moraes CT, Ciacci F, Silvestri G, Shanske S, Sciacco M, Hirano M, et al. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul Disord. 1993;3:43–50. doi: 10.1016/0960-8966(93)90040-q. doi: 10.1016/0960-8966(93)90040-Q. [DOI] [PubMed] [Google Scholar]
- 7.Uusimaa J, Moilanen JS, Vainionpää L, Tapanainen P, Lindholm P, Nuutinen M, et al. Prevalence, segregation, and phenotype of the mitochondrial DNA 3243A>G mutation in children. Ann Neurol. 2007;62:278–87. doi: 10.1002/ana.21196. doi: 10.1002/ana.21196. [DOI] [PubMed] [Google Scholar]
- 8.Majamaa K, Turkka J, Kärppä M, Winqvist S, Hassinen IE. The common MELAS mutation A3243G in mitochondrial DNA among young patients with an occipital brain infarct. Neurology. 1997;49:1331–4. doi: 10.1212/wnl.49.5.1331. doi: 10.1212/WNL.49.5.1331. [DOI] [PubMed] [Google Scholar]
- 9.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Fang F, Yang Y, Zou L, Zhang Y, Wang S, et al. The study of mitochondrial A3243G mutation in different samples. Mitochondrion. 2009;9:139–43. doi: 10.1016/j.mito.2009.01.004. doi: 10.1016/j.mito.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Park H, Davidson E, King MP. The pathogenic A3243G mutation in human mitochondrial tRNALeu(UUR) decreases the efficiency of aminoacylation. Biochemistry. 2003;42:958–64. doi: 10.1021/bi026882r. doi: 10.1021/bi026882r. [DOI] [PubMed] [Google Scholar]
- 12.Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K, et al. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc Natl Acad Sci U S A. 2004;101:15070–5. doi: 10.1073/pnas.0405173101. doi: 10.1073/pnas.0405173101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deschauer M, Chinnery PF, Schaefer AM, Turnbull DM, Taylor RW, Zierz S, et al. No association of the mitochondrial DNA A12308G polymorphism with increased risk of stroke in patients with the A3243G mutation. J Neurol Neurosurg Psychiatry. 2004;75:1204–5. doi: 10.1136/jnnp.2003.026278. doi: 10.1136/jnnp.2003.026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torroni A, Campos Y, Rengo C, Sellitto D, Achilli A, Magri C, et al. Mitochondrial DNA haplogroups do not play a role in the variable phenotypic presentation of the A3243G mutation. Am J Hum Genet. 2003;72:1005–12. doi: 10.1086/373936. doi: 10.1086/373936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368–71. doi: 10.1038/ng0892-368. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- 16.Bosbach S, Kornblum C, Schröder R, Wagner M. Executive and visuospatial deficits in patients with chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome. Brain. 2003;126(Pt 5):1231–40. doi: 10.1093/brain/awg101. doi: 10.1093/brain/awg101. [DOI] [PubMed] [Google Scholar]
- 17.Koga Y, Akita Y, Takane N, Sato Y, Kato H. Heterogeneous presentation in A3243G mutation in the mitochondrial tRNA(Leu(UUR)) gene. Arch Dis Child. 2000;82:407–11. doi: 10.1136/adc.82.5.407. doi: 10.1136/adc.82.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabrizi GM, Cardaioli E, Grieco GS, Cavallaro T, Malandrini A, Manneschi L, et al. The A to G transition at nt 3243 of the mitochondrial tRNALeu(UUR) may cause an MERRF syndrome. J Neurol Neurosurg Psychiatry. 1996;61:47–51. doi: 10.1136/jnnp.61.1.47. doi: 10.1136/jnnp.61.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finsterer J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol Scand. 2007;116:1–14. doi: 10.1111/j.1600-0404.2007.00836.x. doi: 10.1111/j.1600-0404.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- 20.Rahman S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015;49:71–5. doi: 10.1016/j.yebeh.2015.05.003. doi: 10.1016/j.yebeh.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Ma Y, Fang F, Cao Y, Yang Y, Zou L, Zhang Y, et al. Clinical features of mitochondrial DNA m.3243A>G mutation in 47 Chinese families. J Neurol Sci. 2010;291:17–21. doi: 10.1016/j.jns.2010.01.012. doi: 10.1016/j.jns.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Chi CS, Lee HF, Tsai CR, Lee HJ, Chen LH. Clinical manifestations in children with mitochondrial diseases. Pediatr Neurol. 2010;43:183–9. doi: 10.1016/j.pediatrneurol.2010.04.015. doi: 10.1055/s-2006-943661. [DOI] [PubMed] [Google Scholar]
- 23.Chin J, Marotta R, Chiotis M, Allan EH, Collins SJ. Detection rates and phenotypic spectrum of m.3243A>G in the MT-TL1 gene: A molecular diagnostic laboratory perspective. Mitochondrion. 2014;17:34–41. doi: 10.1016/j.mito.2014.05.005. doi: 10.1016/j.mito.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 24.de Laat P, Koene S, van den Heuvel LP, Rodenburg RJ, Janssen MC, Smeitink JA. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A>G mutation. J Inherit Metab Dis. 2012;35:1059–69. doi: 10.1007/s10545-012-9465-2. doi: 10.1007/s10545-012-9510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chae JH, Hwang H, Lim BC, Cheong HI, Hwang YS, Kim KJ. Clinical features of A3243G mitochondrial tRNA mutation. Brain Dev. 2004;26:459–62. doi: 10.1016/j.braindev.2004.01.002. doi: 10.1016/j.braindev.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Wang YX, Le WD. Progress in Diagnosing Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episodes. Chin Med J. 2015;128:1820–5. doi: 10.4103/0366-6999.159360. doi: 10.4103/0366-6999.159360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sue CM, Crimmins DS, Soo YS, Pamphlett R, Presgrave CM, Kotsimbos N, et al. Neuroradiological features of six kindreds with MELAS tRNA(Leu) A2343G point mutation: Implications for pathogenesis. J Neurol Neurosurg Psychiatry. 1998;65:233–40. doi: 10.1136/jnnp.65.2.233. doi: 10.1136/jnnp.65.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmgren D, Wåhlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M. Cardiomyopathy in children with mitochondrial disease;clinical course and cardiological findings. Eur Heart J. 2003;24:280–8. doi: 10.1016/s0195-668x(02)00387-1. doi: 10.1016/s0195-668x(02)00387-1. [DOI] [PubMed] [Google Scholar]
- 29.Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–31. doi: 10.1542/peds.2004-0718. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- 30.Majamaa-Voltti K, Peuhkurinen K, Kortelainen ML, Hassinen IE, Majamaa K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc Disord. 2002;2:12. doi: 10.1186/1471-2261-2-12. doi: 10.1093/brain/awh515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frederiksen AL, Andersen PH, Kyvik KO, Jeppesen TD, Vissing J, Schwartz M. Tissue specific distribution of the 3243A->G mtDNA mutation. J Med Genet. 2006;43:671–7. doi: 10.1136/jmg.2005.039339. doi: 10.1136/jmg.2005.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]