

Abstract
BACKGROUND
Prenatal exposure to inorganic arsenic (iAs) is associated with dysregulated gene and protein expression in the fetus, both evident at birth. Potential epigenetic mechanisms that underlie these changes include but are not limited to the methylation of cytosines (CpG).
OBJECTIVE
The aim of the present study was to compile datasets from studies on prenatal arsenic exposure to identify whether key genes, proteins, or both and their associated biological pathways are perturbed.
METHODS
We compiled datasets from 12 studies that analyzed the relationship between prenatal iAs exposure and fetal changes to the epigenome (5-methyl cytosine), transcriptome (mRNA expression), and/or proteome (protein expression changes).
FINDINGS
Across the 12 studies, a set of 845 unique genes was identified and found to enrich for their role in biological pathways, including those signaled by peroxisome proliferator-activated receptor, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, and the glucocorticoid receptor. Tumor necrosis factor was identified as a putative cellular regulator underlying most (n = 277) of the identified iAs-associated genes or proteins.
CONCLUSIONS
Given their common identification across numerous human cohorts and their known toxicologic role in disease, the identified genes and pathways may underlie altered disease susceptibility associated with prenatal exposure to iAs.
Keywords: CpG methylation, gene expression, inorganic arsenic, microRNA expression, protein expression
INTRODUCTION
Inorganic arsenic (iAs) is ranked as the highest priority toxic agent by the Agency for Toxic Substances and Disease Registry.1 Exposure to iAs primarily occurs via contaminated drinking water. In addition to the detrimental health consequences associated with chronic exposure in adults, prenatal exposure to iAs is associated with adverse birth outcomes and susceptibility to diseases later in life.2 Specifically, prenatal iAs exposure has been associated with lower birth weight (BW), preterm birth, reduced height and head circumference, increased susceptibility to infection, including inflammation and infectious disease, and later life cancers.3
A single molecular mechanism is unlikely to underlie these diverse health outcomes, and many changes at the cellular level in circulating cord leukocytes in infants have been observed. For example, prenatal iAs exposure has been associated with changes to the fetal cord leukocyte epigenome, including changes to CpG methylation, altered transcription profiles, and changes in protein signaling.4-15 Thus, the pathophysiological consequences of prenatal iAs exposure are likely linked to complex dysregulation at the level of the epigenome, transcriptome, and proteome.
A growing body of literature supports the relationship between prenatal iAs exposure and changes to the fetal epigenome. With cord leukocytes as the primary target site of study, alterations in CpG methylation patterns in relation to prenatal iAs exposure have been observed in human population-based studies. Specifically, prenatal iAs exposure has been associated with global CpG methylation changes and 2 specific tumor suppressors in peripheral blood leukocytes in a cohort in Bangladesh.10 Gene-specific CpG methylation has been associated with iAs in Bangladesh and Thailand6,9 and genome-wide gene-specific changes observed in a pregnancy cohort in Mexico.14 Some alterations in CpG methylation profiles have been shown to be sex-specific: one study identified 3 CpG sites that altered significantly associated with iAs exposure in boys, but none in girls.6
Studies focusing on changes at the level of the fetal cord leukocyte transcriptome analyzing altered gene expression have identified many target genes that are altered in relationship to prenatal iAs exposure. Altered patterns of gene expression related to inflammatory signaling pathways were observed in cord blood leukocyte samples collected from newborns exposed prenatally to varying levels of iAs in Thailand8 and Mexico.13,14 Similarly, genes have been shown to be altered in cord blood and placental tissues from a pregnancy cohort in New Hampshire.7,11,12,15
In terms of protein expression studies, 2 studies have identified changes in protein abundance in relationship to prenatal iAs exposure. In a pregnancy cohort in Bangladesh, 18 proteins were analyzed in cord blood for their associations with maternal urinary iAs and 3 differentially expressed proteins were identified.4 Using the largest proteome assessment to date related to prenatal iAs exposure, a panel of 507 proteins was assessed in newborns exposed to iAs prenatally in Gómez Palacio, Mexico. A total of 111 proteins were found to be associated with prenatal iAs exposure, which are known to be regulated by tumor necrosis factor (TNF) and are enriched in functionality related to immune/inflammatory response and cellular development/proliferation.5
Most of these aforementioned studies have examined the identified genes or proteins for biological pathways that are altered when exposed to prenatal iAs. However, the datasets have not been systematically evaluated to identify whether there are common genes or pathways that are dysregulated across multiple birth cohorts. Additionally, the genes altered at the level of the epigenome, transcriptome, and proteome have yet to be integrated across studies. Given that these genes and proteins are likely important contributors to environmental exposure-induced disease from prenatal iAs exposure, the identification of potential common biological pathways will aid in determining biomarkers of exposure or disease associated with iAs. We performed an analysis of datasets that assessed the molecular endpoints of epigenetic alterations (CpG methylation), gene expression, and protein expression in relationship to prenatal iAs exposure in human pregnancy cohort studies to identify key biological pathways that are perturbed as a result of prenatal exposure to iAs.
METHODS
Identifying Genes and Proteins Associated with iAs Exposure
To assess the relationship between prenatal iAs exposure from previous human cohort studies that evaluated CpG methylation, gene expression or protein expression changes, a literature review was performed in PubMed searching the terms, prenatal arsenic exposure plus one of the following terms, gene expression, epigenetics, DNA methylation, CpG methylation, and protein expression. From this search, 12 studies and their corresponding datasets were identified. Genes were represented if they were found to be significantly associated with prenatal iAs exposure as identified within the respective studies. Common gene identifiers were converted to an Entrez Gene number as an identifier for each gene and 845 unique genes and proteins were identified across the 12 datasets.
Pathway Analyses
Genes or proteins were analyzed at a biological pathway level using 2 methods, namely Ingenuity Pathway Analysis (IPA; Ingenuity Systems®, Redwood City, CA, USA) and the Database for Annotation, Visualization, and Integrated Discovery (DAVID; david.ncifcrf.gov). Analyses were carried out for all genes (N = 845), as well as separately for genes that were identified as significantly associated with prenatal iAs levels from all gene expression studies (n = 731), those that were identified from studies examining CpG methylation (n = 61), and those that encode proteins from studies that assessed protein expression (n = 114). To perform these analyses, genes identified from the individual studies were analyzed using their Entrez gene number. Significant enrichment of canonical pathways was assessed using IPA and the Kyoto Encyclopedia of Genes and Genomes pathways were identified using DAVID. Benjamini-Hochberg-corrected P values were reported for all canonical pathways in IPA. The P value determined by DAVID is an absolute enrichment P value determined by a modified Fischer’s Exact Test.16 Pathways were prioritized based on the overlap between IPA and DAVID and top hits between them. The enrichment of cellular regulators for all proteins and genes (N = 845) was carried out using IPA.
RESULTS
Generation of a Cross-study Database of Gene/Proteins Modified in Relation to Prenatal iAs Exposure
Twelve studies that assessed epigenetic, genomic, and proteomic alterations in cord leukocytes or placental tissue of newborns exposed prenatally to iAs exposure were identified. Of these studies, 5 investigated CpG methylation, 6 focused on gene expression, and 2 investigated protein expression (Table 1). One of the studies examined the relationship between changes to CpG methylation with concomitant changes in gene expression.14
Table 1.
Summary of Studies Included for Analysis (N = 12)
| Study (PMID) | Participants (n) | Geographic Location | Molecular Marker | Tissue |
|---|---|---|---|---|
| Ahmed et al., 2011 (20940111) | 130 | Matlab, Bangladesh | Protein expression | Placenta/cord blood |
| Bailey et al., 2014 (24675094) | 50 | Gómez Palacio, Mexico | Protein expression | Cord blood |
| Broberg et al., 2014 (24965135) | 127 | Matlab, Bangladesh | CpG methylation | Cord blood |
| Fei et al., 2013 (23866971) | 133 | New Hampshire, United States | Gene expression | Placenta |
| Fry et al., 2007 (18039032) | 32 | Thailand | Gene expression | Cord blood |
| Intarasunanont et al., 2012 (22551203) | 71 | Thailand | CpG methylation | Cord blood |
| Kile et al., 2012 (22466225) | 113 | Sirajdikhan Upazila, Bangladesh | CpG methylation | Cord blood |
| Koestler et al., 2013 (23757598) | 134 | New Hampshire, United States | CpG methylation | Cord blood |
| Nadeau et al., 2014 (25229165) | 70 | New Hampshire, United States | Gene expression | Placenta |
| Rager et al., 2014 (24327377) | 40 | Gómez Palacio, Mexico | Gene expression | Cord blood |
| Rojas et al., 2014 (25304211) | 38 | Gómez Palacio, Mexico | CpG methylation/Gene expression | Cord blood |
| Winterbottom et al., 2015 (26288817) | 133 | New Hampshire, United States | Gene expression | Placenta |
The following studies investigated changes to CpG methylation in response to prenatal iAs exposure: Broberg et al. carried out a genome-wide methylation analysis of cord blood leukocytes from 127 infants from a pregnancy cohort in Matlab, Bangladesh.6 Intarasunanont et al. conducted a global CpG methylation and TP53-specific analysis of cord blood leukocytes from 71 newborns with prenatal iAs exposure from southern Thailand.9 Kile et al. analyzed global CpG methylation and 2 gene-specific tumor suppressors of cord blood leukocytes in response to prenatal iAs in 113 mothere–-baby pairs from Sirajdikhan Upazila, Bangladesh.10 Koestler et al. performed a genome-wide gene-specific CpG methylation study of cord blood leukocytes 134 infants nested in the NHBCS (New Hampshire Birth Cohort Study).11 Rojas et al. performed a genome-wide gene-specific study of cord leukocytes in a cohort of 40 mother–baby pairs from the BEAR (Biomarkers of Exposure to ARsenic) pregnancy cohort in Gómez Palacio, Mexico. Unlike previous studies, genes identified in the BEAR cohort (N = 54) were altered both at the gene expression level and in CpG methylation status.14
The following studies were identified that investigated gene expression profiles in response to prenatal iAs exposure: Fei et al. investigated the expression of 9 predicted iAs-associated genes in placental tissue from 133 pregnant women from New Hampshire7; Fry et al. identified 447 differentially expressed genes from a genome-wide analysis in fetal cord blood leukocytes of 32 infants in Thailand8; Nadeau et al. analyzed immune-specific gene expression in placental tissues from 70 women who were exposed to iAs in New Hampshire.12 Additionally, Winterbottom et al. assessed placenta gene-specific expression in 133 placental tissues for associations with iAs exposure and fetal growth.15 Rager et al. identified 334 differentially expressed mRNAs from a genome-wide analysis in cord blood leukocytes in the BEAR pregnancy cohort of 40 mother–baby pairs.13
Two of the 12 studies analyzed protein expression associated with prenatal iAs exposure. Ahmed et al. performed a targeted analysis of 18 inflammation-associated cytokines in cord blood leukocytes in a cohort of 130 pregnant women from Bangladesh.4 Bailey et al. investigated iAs-associated changes to the prenatal proteome with prenatal iAs exposure in the cord blood serum of 50 newborns from the BEAR cohort in Gómez Palacio, Mexico.5
Assessing Gene/Protein Expression Trends Across Datasets
Across the 12 studies that assessed in the present study, 845 genes were identified as being associated with prenatal iAs exposure (Supplementary Table 1). Of these genes, 731 were from studies that assessed gene expression, 61 were identified from studies of CpG methylation, and 114 from studies that assessed protein expression. Additionally, 61 genes were identified to be altered for 2 molecular endpoints where 54 genes had altered levels of gene expression as well as altered levels of CpG methylation in relation to prenatal iAs exposure, and 7 genes had altered gene expression levels as well as protein expression levels (Fig. 1; Supplementary Table 2). Additionally, when examining the overlap of genes and proteins across studies independent of the molecular endpoint assessed, 67 genes were identified in at least 2 studies (Supplementary Table 2). Of these, 17 were common to studies from different pregnancy cohorts (Table 1). Specifically these genes were interleukin (IL)-1 beta (IL1β), IL-8 (IL8), chemokine (C-C motif) receptor 2 (CCR2), chemokine (C-C motif) receptor 3 (CCR3), chemokine (C-X-C motif) ligand 1 (melanoma growth-stimulating activity alpha) (CXCL), chemokine (C-X-C motif) receptor 4 (CXCR4), epiregulin (EREG), chemokine (C-X-C motif) ligand 16 (CXCL16), ferritin, heavy polypeptide 1; ferritin, heavy polypeptide-like 16; similar to ferritin, heavy polypeptide 1; ferritin, heavy polypeptide-like 3 pseudogene (FTH1), muscleblind-like 2 (MBNL2), nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (NFKBIA), triggering receptor expressed on myeloid cells 1 (TREM1), casein kinase 1, delta (CSNK1D), HLA complex group 27 (HCG27), tribbles homolog 1 (TRIB1), GLI family zinc finger 2 (GLI2), and porcupine homolog (PORCN) (Table 2).
Figure 1.
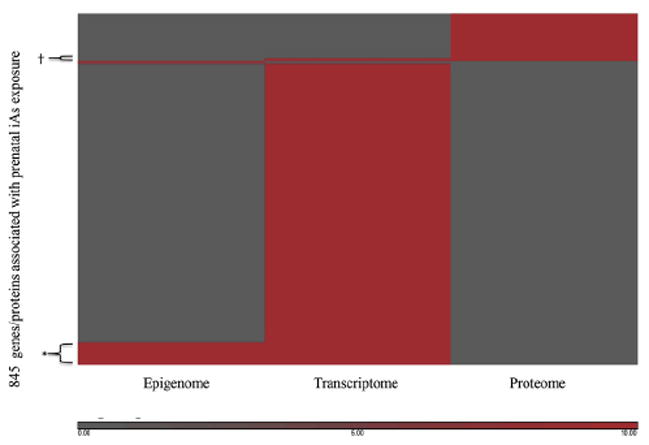
The heatmap depicts genes (N = 845) identified as changed in relationship to prenatal iAs in studies focusing on the epigenome (CpG methylation), the transcriptome (gene expression), and the proteome (protein expression) representing 12 studies. Red shading indicates that the gene was significantly associated with prenatal iAs exposure. Gray shading indicates that the gene was not significantly associated with prenatal iAs exposure. *The 54 genes that had altered levels of gene expression as well as altered levels of CpG methylation in relation to prenatal iAs exposure. †The 7 genes that had altered gene expression levels as well as protein expression levels.
Table 2.
Genes/Proteins (n = 17) Associated with Prenatal iAs Exposure Common to Different Study Cohorts
| Gene | Official Gene Name | Source |
|---|---|---|
| IL1B | Interleukin 1, beta | (Fry 2007)*, (Nadeau 2014)* |
| IL8 | Interleukin 8 | (Fry 2007)*, (Ahmed 2011)† |
| CCR2 | Chemokine (C-C motif) receptor 2 | (Fry 2007)*, (Bailey 2014)† |
| CCR3 | Chemokine (C-C motif) receptor 3 | (Fry 2007)*, (Bailey 2014)† |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) | (Fry 2007)*, (Bailey 2014)† |
| CXCR4 | Chemokine (C-X-C motif) receptor 4 | (Fry 2007)*, (Bailey 2014)† |
| EREG | Epiregulin | (Fry 2007)*, (Bailey 2014)† |
| CXCL16 | Chemokine (C-X-C motif) ligand 16 | (Fry 2007)*, (Rager 2014)*, (Bailey 2014)† |
| FTH1 | Ferritin, heavy polypeptide 1; ferritin, heavy polypeptide-like 16; similar to ferritin, heavy polypeptide 1; ferritin, heavy polypeptide-like 3 pseudogene | (Fry 2007)*, (Rager 2014)* |
| MBNL2 | Muscleblind-like 2 (Drosophila) | (Fry 2007)*, (Rager 2014)* |
| NFKBIA | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha | (Fry 2007)*, (Rager 2014)* |
| TREM1 | Triggering receptor expressed on myeloid cells 1 | (Fry 2007)*, (Rager 2014)* |
| CSNK1D | Casein kinase 1, delta | (Fry 2007)*, (Rager 2014)*, (Rojas 2014)*,‡ |
| HCG27 | HLA complex group 27 | (Fry 2007)*, (Rager 2014)*, (Rojas 2014)*, ‡ |
| TRIB1 | Tribbles homolog 1 (Drosophila) | (Fry 2007)*, (Rager 2014)*, (Rojas 2014)*, ‡ |
| GLI2 | GLI family zinc finger 2 | (Rager 2014)*, (Rojas 2014)*,‡, (Winterbottom 2015)* |
| PORCN | Porcupine homolog (Drosophila) | (Rager 2014)*, (Winterbottom 2015)* |
Gene expression.
Protein expression.
CpG methylation.
Identified Enriched Biological Pathways
Pathway analysis was performed for all genes within the database (N = 845), and separately for genes derived from studies that focused on a specific molecular endpoint (ie, CpG methylation; n = 61 genes), gene expression (n = 731 genes), or protein expression (n = 113 genes/proteins). Among the set of 845 genes identified to be associated with prenatal iAs exposure, there was an enrichment for pathways associated with known transcription factors including peroxisome proliferator-activated receptor (PPAR), NF-κB, transforming growth factor (TGF)-β, glucocorticoid receptor, among others (Supplementary Table 3).
The genes identified with differential CpG methylation (n = 61), were enriched for pathways known to play a role in cell cycle and apoptosis including Myc-mediated apoptosis, G1/S checkpoint regulation, tumor protein p53 (Supplementary Table 3). The genes with altered expression in relationship to prenatal iAs (n = 731) were enriched for the following immune and inflammatory response pathways: toll-like receptor (TLR), glucocorticoid receptor, inducible nitric oxide synthase (iNOS), and IL-6 (Supplementary Table 3). Of the 113 genes with altered protein expression, there was an enrichment for the growth factors that play a role in inflammation including TGF-β, bone morphogenetic protein (BMP) signaling pathway, and fibroblast growth factors (FGF; Supplementary Table 3).
In Silico Identification of Regulatory Molecules Reveals the TNF Pathway as a Master Regulator
Analysis was performed on the 845 genes and proteins to identify key cellular regulators predicted to control gene and protein expression. These include signaling molecules, transcription factors, or both. This analysis identified TNF as a key cellular regulator of the genes/proteins across all studies (Fig. 2; Supplementary Table 4). These proteins include those belonging to the following pathways: ILB, interferon (IFN)-γ, jun proto-oncogene (JUN), NF-κB complex, NF-κBIA, NF-κB1, prostaglandin E2, v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA), SMAD family member 4 (SMAD4), signal transducers and activators of transcription-1 (STAT1), signal transducers and activators of transcription-3 (STAT3), and TGFB1. Interestingly, the 17 genes that were common across cohorts were also enriched for TNF as a common regulator (P = 1.06 × 10−9).
Figure 2.
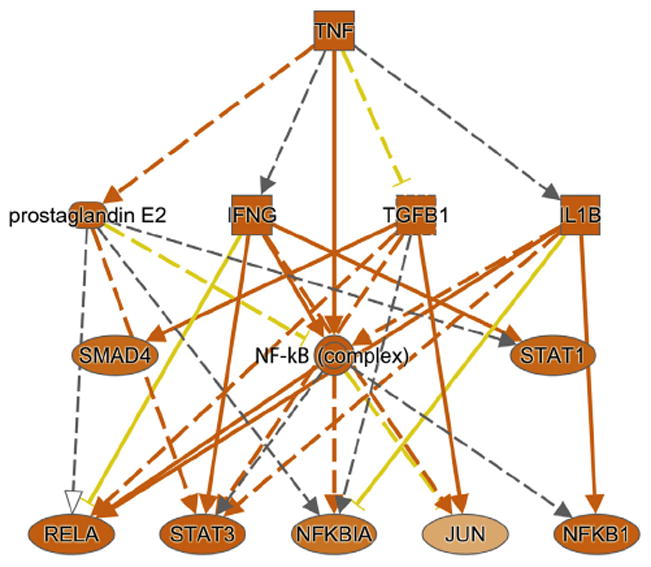
Top upstream regulator, tumor necrosis factor (TNF), identified as enriched among the iAs-associated genes (N = 845).
DISCUSSION
Prenatal exposure to iAs is associated with detrimental health outcomes at birth as well as later life disease. In the hunt for the molecular basis for these outcomes, prior research has focused on examining gene/pathway disruption in fetal cord leukocytes or in placental tissues associated with prenatal iAs exposure. Previous studies have demonstrated that prenatal exposure to iAs is associated with alterations to the fetal epigenome including altered levels of CpG methylation, as well as changes in gene expression, and altered protein expression in cord leukocytes or placental tissue. However, it is currently not known whether there are common pathways targeted by iAs exposure across these studies. In the present study, we used a systems toxicologic approach to assess the relationship between prenatal iAs exposure and several molecular endpoints. We generated a database from 12 studies examining prenatal iAs-associated changes to the epigenome (CpG methylation), the transcriptome (mRNA expression), or proteome (protein expression changes). A set of 845 genes that were enriched for pathways regulated by TNF were identified as key regulators of prenatal iAs exposure and disease. Interestingly, given the vastly different geographic locations of the pregnancy cohorts from which these data are derived, 17 genes were common across cohorts independent of the molecular endpoint under study and known to be regulated by TNF. Given the known relationship between prenatal exposure to iAs and several adverse birth outcomes, early-life health outcomes, and disease later in life, the identification of common pathways could provide insight into disease etiology.
Across all 845 genes identified here, transcription factor regulator-associated pathways and immune/inflammation-response pathways were identified. These included PPAR, NF-κB, gluco-corticoid receptor, and TLRs. Many of these pathways have been previously associated with exposure to iAs, iAs-associated diseases, or both. For example, it has been suggested that decreased expression of PPAR-γ and altered glucocorticoid receptor-mediated transcription are pathologic events associated with iAs-induced diabetes.17 The finding in the present study of NF-κB as a dysregulated pathway associated with prenatal iAs exposure was previously reported.8 This is relevant because many cellular effects of iAs have been shown to be mediated through arsenic’s ability to modulate NF-κB activity.18-21 Toll-like receptors, identified here, are known to play an important role in innate immune signaling. Supporting our findings, these receptors have been shown to have altered expression in animal studies where a subset of genes involved in innate immune response, including IL-1 and TLR, were identified to have altered expression within lungs of mice exposed to iAs.22 Taken together, these results highlight the importance of PPAR, NF-κB, TLRs, that likely play an important role in determining how prenatal iAs exposure influences cellular signaling at the pathway level.
Interestingly, the top cellular regulator for all identified genes and proteins and for the set of 17 genes common to multiple pregnancy cohorts was TNF, which plays a critical role in inflammation and cellular growth/development-related signaling.23 Our results are consistent with previous findings that proteomic shifts from prenatal iAs exposure is associated with TNF-receptor superfamily members.5,10 Additionally, an animal study of iAs exposed mice suggest that TNF is involved in inflammatory reactions in the liver and subsequent tissue injury and fibrosis.24 Furthermore, TNF has been implicated to be important in iAs-associated disease, where previous studies of iAs-exposed individuals have identified polymorphisms within the TNF promoter are associated with increased levels of TNF and a higher incidence of skin lesions, respiratory diseases, and conjunctivitis.25 These results, coupled with evidence from prior studies, demonstrate that TNF may be an important signaling molecule mediating the relationship between prenatal iAs exposure and iAs-associated disease.
The search for genes that are altered in different ways in the cell, either through CpG methylation or gene expression or protein expression modification, resulted in the identification of 61 common genes and proteins. This finding highlights the diverse ways that iAs can affect gene regulation with targeted CpG methylation influencing both the expression of genes and proteins. Interestingly, we identified 17 genes that were altered in response to prenatal iAs exposure and were commonly derived from multiple studies of different cohorts. Many of these genes play a role in inflammation, including interleukins, chemokines, FTH1, NFKBIA, TREM1, and TRIB1. As these cohorts are geographically diverse based in Bangladesh, Thailand, Mexico, and the United States, the fact that there are commonalities at the gene and pathway level highlight the conservation in the response of the fetus to prenatal exposure to iAs.
The findings here are significant because they provide a hypothesis for specific biological mechanisms by which prenatal exposure to iAs can disrupt cell signaling observed across numerous prenatal cohort studies. However, this study is not without limitations. The methodologies of the studies used to compile the database differed, including statistical methodologies for gene set identification that could affect the genes included in the current analysis. Nevertheless, to our knowledge, this is the first study to perform an integrated assessment of changes to fetal cell signaling and integration of epigenetic, transcriptomic, and proteomic datasets.
CONCLUSION
The present study used a systems toxicologic approach to identify a set of genes from multiple studies that examined molecular markers of prenatal iAs exposure. Interestingly, many genes were altered across studies, even those focused on different molecular endpoints with the largest overlap between studies of gene expression and CpG methylation. However, there was some additional overlap between gene and protein expression studies. Additionally, a unique set of genes that is common across multiple pregnancy cohorts across different geographic locations suggested a conserved biological response to prenatal iAs exposure. TNF was identified as a potentially important regulator underlying most of the identified genes and proteins. The results from this study may inform future research on the molecular mechanisms of the relationship between prenatal iAs exposure and disease.
Supplementary Material
Acknowledgments
The authors acknowledge Elizabeth Martin for her comments on the manuscript.
Support for this research was made possible with support from the National Institute of Environmental Health Sciences (P42-ES005948, R01-ES019315).
Footnotes
SUPPLEMENTARY DATA
Supplementary tables accompanying this article can be found in the online version at http://dx.doi.org/10.1016/j.aogh.2016.01.015.
The authors declare no conflict of interest and all authors had access to the data and a role in writing the manuscript.
Contributor Information
Jessica E. Laine, Department of Epidemiology, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, NC.
Rebecca C. Fry, Department of Environmental Sciences and Engineering, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, NC.
References
- 1.ATSDR [Google Scholar]
- 2.Vahter M. Effects of arsenic on maternal and fetal health. Annu Rev Nutr. 2009;29:381–99. doi: 10.1146/annurev-nutr-080508-141102. [DOI] [PubMed] [Google Scholar]
- 3.Naujokas MF, Anderson B, Ahsan H, et al. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect. 2013;121:295–302. doi: 10.1289/ehp.1205875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed S, Mahabbate-Khoda S, Rekha RS, et al. Arsenic-associated oxidative stress, inflammation, and immune disruption in human placenta and cord blood. Environ Health Perspect. 2011;119:258–64. doi: 10.1289/ehp.1002086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey KA, Laine J, Rager JE, et al. Prenatal arsenic exposure and shifts in the newborn proteome: interindividual differences in tumor necrosis factor (TNF)-responsive signaling. Toxicol Sci. 2014;139:328–37. doi: 10.1093/toxsci/kfu053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broberg K, Ahmed S, Engström K, et al. Arsenic exposure in early pregnancy alters genome-wide DNA methylation in cord blood, particularly in boys. J Dev Orig Health Dis. 2014;5:288–98. doi: 10.1017/S2040174414000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fei DL, Koestler DC, Li Z, et al. Association between In Utero arsenic exposure, placental gene expression, and infant birth weight: a US birth cohort study. Environ Health. 2013;12:58. doi: 10.1186/1476-069X-12-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fry RC, Navasumrit P, Valiathan C, et al. Activation of inflammation/NF-kappaB signaling in infants born to arsenic-exposed mothers. PLoS Genet. 2007;3:e207. doi: 10.1371/journal.pgen.0030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Intarasunanont P, Navasumrit P, Waraprasit S, et al. Effects of arsenic exposure on DNA methylation in cord blood samples from newborn babies and in a human lymphoblast cell line. Environ Health. 2012;11:31. doi: 10.1186/1476-069X-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kile ML, Baccarelli A, Hoffman E, et al. Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord blood leukocytes. Environ Health Perspect. 2012;120:1061–6. doi: 10.1289/ehp.1104173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koestler DC, Avissar-Whiting M, Houseman EA, Karagas MR, Marsit CJ. Differential DNA methylation in umbilical cord blood of infants exposed to low levels of arsenic in utero. Environ Health Perspect. 2013;121:971–7. doi: 10.1289/ehp.1205925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nadeau KC, Li Z, Farzan S, et al. In utero aresenic exposure and fetal immune repertoire in a US pregnancy cohort. Clin Immunol. 2014;155:188–97. doi: 10.1016/j.clim.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rager JE, Bailey KA, Smeester L, et al. Prenatal arsenic exposure and the epigenome: altered microRNAs associated with innate and adaptive immune signaling in newborn cord blood. Environ Mol Mutagen. 2014;55:196–208. doi: 10.1002/em.21842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rojas D, Rager JE, Smeester L, et al. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methyl-cytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci. 2015;143:97–106. doi: 10.1093/toxsci/kfu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winterbottom EF, Fei DL, Koestler DC, et al. GLI3 links environmental arsenic exposure and human fetal growth. EBioMedicine. 2015;2:536–43. doi: 10.1016/j.ebiom.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 17.Singh AP, Goel RK, Kaur T. Mechanisms pertaining to arsenic toxicity. Toxicol Int. 2011;18:87–93. doi: 10.4103/0971-6580.84258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Felix K, Manna SK, Wise K, Barr J, Ramesh GT. Low levels of arsenite activates nuclear factor-kappaB and activator protein-1 in immortalized mesencephalic cells. J Biochem Mol Toxicol. 2005;19:67–77. doi: 10.1002/jbt.20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh P, Banerjee M, Giri AK, Ray K. Toxicogenomics of arsennic: classical ideas and recent advances. Mutat Res. 2008;659:293–301. doi: 10.1016/j.mrrev.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Kaul D, Sharma S, Sharma M, Arora M. Arsenic programmes cellular genomic-immunity through miR-2909 RNomics. Gene. 2014;536:326–31. doi: 10.1016/j.gene.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Sharma M, Sharma S, Arora M, Kaul D. Regulation of cellular cyclin D1 gene by arsenic is mediated through miR-2909. Gene. 2013;522:60–4. doi: 10.1016/j.gene.2013.03.058. [DOI] [PubMed] [Google Scholar]
- 22.Kozul CD, Hampton TH, Davey JC, et al. Chronic exposure to arsenic in the drinking water alters the expression of immune response genes in mouse lung. Environ Health Perspect. 2009;117:1108–15. doi: 10.1289/ehp.0800199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtmann MH, Neurath MF. Differential TNF-signaling in chronic inflammatory disorders. Curr Mol Med. 2004;4:439–44. doi: 10.2174/1566524043360636. [DOI] [PubMed] [Google Scholar]
- 24.Das S, Santra A, Lahiri S, Guha Mazumder DN. Immplications of oxidative stress and hepatic cytokine (TNF-alpha and IL-6) response in the pathogenesis of hepatic collagene-sis in chronic arsenic toxicity. Toxicol Appl Pharmacol. 2005;204:18–26. doi: 10.1016/j.taap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee N, Nandy S, Kearns JK, et al. Polymorphisms in the TNF-α and IL10 gene promoters and risk of arsenic-induced skin lesions ans other nondermatological health effects. Toxicol Sci. 2011;121:132–9. doi: 10.1093/toxsci/kfr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.