

Abstract
In this study, we evaluated photosensitized chemistry at the air–sea interface as a source of secondary organic aerosols (SOA). Our results show that, in addition to biogenic emissions, abiotic processes could also be important in the marine boundary layer. Photosensitized production of marine secondary organic aerosol was studied in a custom-built multiphase atmospheric simulation chamber. The experimental chamber contained water, humic acid (1–10 mg L–1) as a proxy for dissolved organic matter, and nonanoic acid (0.1–10 mM), a fatty acid proxy which formed an organic film at the air–water interface. Dark secondary reaction with ozone after illumination resulted in SOA particle concentrations in excess of 1000 cm–3, illustrating the production of unsaturated compounds by chemical reactions at the air–water interface. SOA numbers via photosensitization alone and in the absence of ozone did not exceed background levels. From these results, we derived a dependence of SOA numbers on nonanoic acid surface coverage and dissolved organic matter concentration. We present a discussion on the potential role of the air–sea interface in the production of atmospheric organic aerosol from photosensitized origins.
Introduction
Although the dominant mass fraction of sea-spray aerosol is inorganic sea salt, organic matter can also contribute to the overall mass of aerosols in the marine boundary layer (MBL).1,2 Recent field measurements clearly documented the presence of organic matter in oceanic particles.3,4 Cavalli et al.5 and O’Dowd et al.6 found a significant and dominating fraction of organic matter in the submicrometer size range, while the supermicrometer size range predominately consisted of inorganic sea salt. During high biological activity, the organic fraction ranges from 40 to 60% of the submicrometer aerosol mass, while during low biological activity periods, this fraction decreases to 10–15%. Concentrations of organic aerosol mass in air advected over regions of high biological activity were up to 4 μg m–3 and comparable to polluted air masses.7 The concentration of organic aerosol formed by secondary processes has also been correlated with biological activity.8 Volatile sulfur species greatly impact the formation of secondary marine aerosols9−13 and are included in general circulation models predicting climate evolution.13−15 Together, these findings potentially link ocean biota with marine derived organic aerosols.16 As a result, the organic fraction of the marine aerosols as well as the trace gas composition over the ocean are controlled by the chemical and physical properties of the sea-surface microlayer (SML).17−19 Indeed, recent studies reported the use of natural seawater to generate sea spray aerosol (SSA) in order to evaluate how SML composition drives the composition and associated properties of freshly emitted SSA.20,21
Organic material present at the sea surface includes amphiphiles derived from oceanic biota (fatty acids, fatty alcohols, sterols, amines, etc.) and more complex colloids and aggregates exuded by phytoplankton, which mainly consist of lipopolysaccharides.22−31 All of these compounds can be highly enriched in this microlayer.32,33 The presence of complex and potentially photoactive compounds, such as a fatty acid film at the air–sea interface and therefore in the primary marine aerosol, was reported on the surface of continental and marine aerosols.34−36 This could give rise to the assumption that new processes affect the chemistry in the MBL. Indeed, Reeser et al.37,38 showed that photoexcited chlorophyll can oxidize halide anions at the salt water surface, producing atomic halogens. A similar chemistry is expected for nitrate and nitrite anions, suggesting a rich new source of oxidants in the MBL. These studies stress the need for a better understanding of the chemistry and potential photochemistry of the surface microlayer. Indeed, the photochemistry at the air–sea interface has not been adequately considered over the past years.39 Previous works from our group have shown that such photochemical processing of a surfactant in the presence of a photosensitizer led to the formation of unsaturated and highly functionalized volatile organic compounds (VOCs).40,41 The use of humic acid as a photosensitizer initiates chemical transformation of surfactants, such as nonanoic acid40 and octanol,41 through multiple pathways. The initial step is H-abstraction on the alkyl chain, possibly by OH reaction, leading to the formation of α-carboxyalkyl radicals (R-CHCOOH) which further react with of O2, forming an α-peroxyl carboxylic acid (R–CH(O2)COOH). The unimolecular rearrangement of α-peroxyl radical revealed the production of α-oxygenated VOCs (i.e., saturated carbonyls, saturated acids). In addition, due to the high concentration of surfactants at the interface, disproportionation or dimerization reactions of the α-carboxyalkyl radical has been suggested, yielding functionalized and unsaturated products (i.e., alkenes, dienes, C2–C9 unsaturated aldehydes, unsaturated acids).40,41 These observations of a double-bond formation (i.e., water elimination) highlight the fact that the mechanism occurred preferentially in an organic-enriched environment rather than in a bulk aqueous media. Furthermore, the reaction between excited triplet ketones and hydroxyl acid forming an unsaturated diol, followed by decarboxylation and hydroxyalkyl-hydroxy elimination, may explain the formation of observed isoprene, which represents a new abiotic source in marine environments.42 The formation of these compounds from photosensitized interfacial chemistry likely contributes to SOA formation. In order to address this question, we propose that photosensitized reactions, initiated by a substance capable of absorbing light and transferring the energy to the desired reactants at the air–sea interface, are a key process leading to the formation of marine SOA in the marine boundary layer.
Here, we present an experimental study using a multiphase atmospheric simulation chamber which aimed at investigating aerosol formation from photoinduced chemical processes at the air–water interface. Photosensitized reactions were based on the photodegradation of nonanoic acid (NA) as a surfactant in the presence of humic acid (HA) used as a proxy for dissolved organic matter. Particle concentrations were measured and the gas phase composition was monitored over the experimental time scale. The relationship between the observed particle and chemical processes that occurred at the air–water interface is presented and the atmospheric implications of these photosensitized reactions as a source of SOA loading in the marine boundary layer are discussed.
Experimental section
Multiphase Simulation Chamber
Air–sea interface experiments were performed in a multiphase atmospheric simulation chamber (Figure 1). The 2 m3 rectangular chamber (length (1 m) × width (1 m) × height (2 m)) was made of FEP film (fluorinated ethylene propylene) and equipped with a glass water container with a capacity of 89 L and an exposed surface of 0.64 m2. The chamber was continuously filled with purified air. Photochemical reactions were initiated through UV irradiation (centered at 365 nm) of an aqueous solution of HA used as a photosensitizer and NA as a surfactant with water. We positioned 12 UV lamps (OSRAM lamps, Eversun L80W/79-R) in two banks as the light source, with six by six lamps mounted on opposite sides. The spectrum of the UV lamps is shown in Supporting Information (Figure S1).
Figure 1.

Scheme of the multiphase atmospheric simulation chamber used for the investigation of chemical processes at the air–sea interface.
Particle number and volume size distribution was measured by a scanning mobility particle sizer (SMPS model 3936, TSI) consisting of a long differential mobility analyzer (DMA 3081, TSI) and a condensation particle counter (CPC 3772, TSI, d50 > 10 nm). Sheath and sample flow rates were 6 and 0.6 L min–1, respectively, scanning a particle size range of 9.8–414 nm. The density of measured particles was assumed to be 1 g cm–3. Ultrafine particle formation was monitored using an ultrafine condensation particle counter (UCPC 3776, TSI, d50 > 2.5 nm). The UCPC sampling inlet was positioned at the bottom of the chamber, 30 cm above the liquid surface. The SMPS inlet was placed an additional 150 cm higher in order to observe particle growth. The gas phase concentration of volatile organic compounds (VOCs) in the chamber was monitored using a high-resolution proton transfer reaction mass spectrometer (PTR-TOF-MS 8000, Ionicon Analytik) operated with a 2.2 mbar drift tube pressure and 600 V drift tube voltage at 60 °C. Hydronium ions (H3O+) were used as ionization source to protonate VOCs. The atmospheric simulation chamber was also equipped with trace gas analyzers continuously measuring NOX (Thermo Environmental Instruments Model 42C and Eco Physics CLD88p coupled to a Photolytic NO2 Converter PLC860) and ozone (Thermo Environmental Instruments Model 49i). In order to compensate for leaks and sampling flow rates from the connected analytical instruments, a dilution flow rate of 3 L min–1 was continuously added, maintaining a slight overpressure in the chamber. Temperature and relative humidity were measured using a combined sensor (Vaisala HUMICAP Humidity and Temperature Probe HMP110), and the differential pressure was monitored using a sensor (Vaisala Differential Pressure Transmitter PDT101). Prior to any experiment, NA was purified by bubbling ozone (generated via O2 photolysis using a Hg lamp) in the liquid phase for few hours before use. For the purification of NA, ozone was generated via O2 photolysis using a Hg lamp. Under these experimental conditions, the use of commercialized NA (unpurified) produces particles when ozone is added in contrast to the use of purified NA, where no particle formation was observed.
A known volume of NA was injected using a syringe through a septum installed on a Teflon line, in which ultrapure water (18.2 MΩ cm resistivity at 25 °C) circulated. HA solutions were prepared in an amber glass bottle mounted on a magnetic stirrer where an aliquot was dissolved in ultrapure water. The volume of ultrapure water introduced in the glass container was time-controlled using a peristaltic pump running at a known liquid flow rate. The final volume in the glass container was 30 L. Surfactant concentration in water ranged from 0.1 mM to 10 mM and the concentration of HA from 1 to 10 mg L–1. Based on Henry’s law, the estimated concentrations of NA in the gas phase ranged from ∼100 ppb to ∼2.0 ppm (using Henry’s law constant for NA, KH = 0.101 Pa m3 mol–1).43,44 Water solubility of NA is 210 mg L–1 (1.8 mM) at 25 °C.45 Bulk acidity, in agreement with the acid strength of NA in water, was measured at pH ∼ 4.9 and did not change over the experimental time scale. Although the pH at the air–water interface is still under discussion,46 previous work based on theoretical calculations and experimental observations has shown accumulation of hydronium ions at the surface at high concentrations, leading to increased acidity, whereas in the bulk, higher pH values were observed.47,48 This observation is consistent with the acidic properties of the experimental conditions used in this study. As the bulk acidity of the ocean has an average pH value of about 8, a lower pH at the interface might be observed due to the migration properties of hydronium ions to the interface, which is also consistent with the interface properties of this study.
After introducing about 15 L of ultrapure water (during 20 min) in the glass container, NA was injected. With the introduction of NA, the surface exhibited some small “organic islands” of NA that were minimized, but not avoided, by our experimental procedure. Over the experimental time scale, these islands tended to agglomerate, leading to a larger size, but decreasing in terms of number. At the lowest concentration of NA used (0.1 mM), these islands disappeared rapidly after the end of the introduction of NA and were almost invisible to the naked eye. HA was usually injected around 25 min after NA. This time interval corresponded to the equilibrium time of NA between the gas and liquid phases. Lamps were switched on 90 min after the introduction of the liquid components for a homogeneous distribution of NA and HA over the surface exposure. The experiments were conducted as follows: solutions were introduced into the chamber; lighting was the switched on and off prior to ozone injection (avoiding OH production in the chamber from ozone photolysis). At each step, the gas and particle phases were monitored.
Exposure of the liquid mixture to UV light lasted for at least 14 h. During the irradiation period, temperature and relative humidity were stable at about 300 K and 84%, respectively. After the irradiation period, ozone was generated using a commercial generator (Biozone corporation, USA) based on electric discharge from oxygen flow and introduced in the chamber in the dark (avoiding OH production in the chamber from ozone photolysis). Care was taken that no seed particles were introduced simultaneously. This allowed a fast introduction of ozone in the chamber, reaching the desired concentration within a few minutes. After the experiments, the chamber was flushed with purified air for at least 24 h under maximum irradiation in the presence of ozone at several ppm levels. The glass container was evacuated and filled with ultrapure water several times, in which sodium hydroxide ([NaOH] ∼ 10 mM) was also added in order to promote the dissolution of organic materials.
Chemicals
Chemicals were purchased with the following purities from: nonanoic acid (Alfa Aesar, 97%) and humic acid (Fluka, Technical grade), NaOH (Chimie-Plus laboratoire, Solution at 30%). Oxygen used for ozone generation was purchased from Linde (O2, 99.9991%).
Results
Particle Formation
Figure 2 illustrates a typical experiment where photosensitized production of aerosol was observed. Table 1 shows a complete list of experiments and corresponding initial conditions. Particles in the chamber were subject to dilution, wall loss, and coagulation processes, which represented sink processes in the multiphase simulation chamber. Reported particle concentrations were not corrected for these losses. Background concentrations of particles before the irradiation period were found in the range of 150–500 cm–3. It should be noted that the presence of particles as background level did not affect our observations on the aerosol concentration number. As shown in Figure 2, a significant production of SOA was observed about 30 min after the ozone injection, while the initial composition of the gas phase did not carry any chemical functionality that was expected to react with ozone that is, the photosensitized chemistry produced some SOA precursors. These observations differed strongly from our blank experiments, which were performed by injecting either HA or NA solutions in separate experiments while monitoring particle concentrations under irradiation and after ozone addition in the gas phase.
Figure 2.
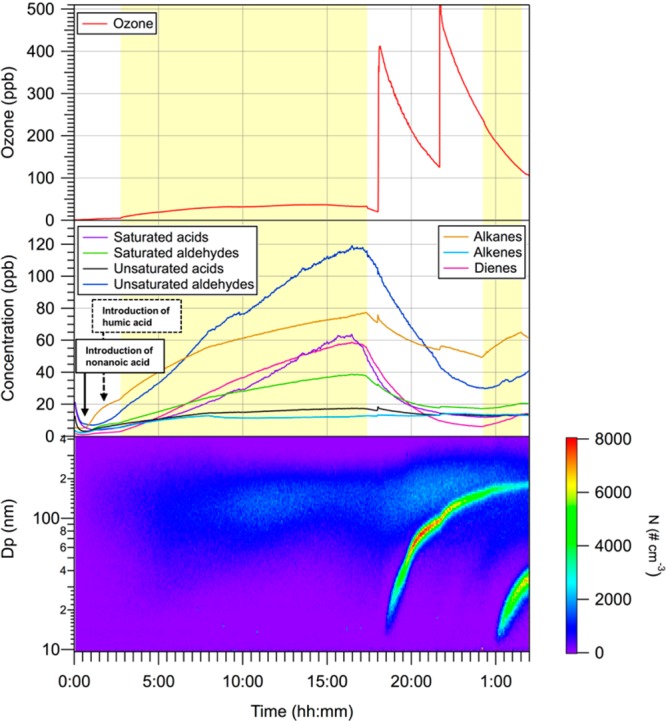
Multiphase simulation chamber experiments conducted in the presence of ([HA] = 10 mg L–1 and NA = 1 mM). Top graph: time–concentration profile of ozone; Middle graph: time concentration profiles of VOC identified and measured by the PTR-ToF-MS; Bottom graph: Corresponding time series of particle number size distributions. The yellow part represents the period when the lights were on (Exp. 9 in Table 1).
Table 1. Initial Experimental Conditions: Dark Ozone Reaction after UV Light Processing of the Liquid Mixture of Nonanoic Acid (NA) and Humic Acid (HA).
| exp. | [NA] | [HA] | [O3] | particle numbera |
|---|---|---|---|---|
| mM | mg L–1 | ppb | cm–3 | |
| 1b | 20 | 829 | 112 | |
| 2 | 0.1 | 10 | 250 | 285 |
| 3b | 0.1 | 526 | 64 | |
| 4 | 0.1 | 10 | 500 | 396 |
| 5 | 0.1 | 1 | 301 | 68 |
| 6b | 2 | 534 | 84 | |
| 7 | 2 | 10 | 461 | 3057 |
| 8 | 0.5 | 10 | 476 | 568 |
| 9 | 1 | 10 | 391 | 887 |
Particle number concentrations were subtracted from the particle background.
Control experiments.
With [HA] = 20 mg L–1 and in the absence of any surfactant, particles did not exceed background concentrations. It is important to note that the HA concentration used in the blank experiment was higher than that for mixed HA and NA experiments. When introducing NA only, in the absence of any photosensitizer, no significant particle formation was observed during irradiation and from the gas phase reaction with ozone in the dark (as compared to the results shown below) during the course of these experiments (Supporting Information Figure S2). This highlights the involvement of HA in the photochemical transformation of NA and thus demonstrates that particle formation originated from the photosensitized reaction.
The temporal profiles of NA and gas phase volatile organic compounds were continuously monitored using PTR-ToF-MS. Numerous VOCs were identified from the light-induced reaction of NA in the presence of HA: saturated aldehydes (C7–C9), unsaturated aldehydes (C6–C9), alkanes (C7–C9), alkenes (C5–C9), and dienes (C6–C9). The concentration of VOCs was estimated using the reaction rate coefficient between H3O+ and VOCs, using the simple kinetic calculation reported in Lindinger et al.49 For the compounds with rate coefficient data available, an estimated value of 2 × 10–9 cm3 s–1 was used. Uncertainties in our data may arise from systematic errors associated with the reaction rate constant, accurate within ±30%. Typical detection limits, precisions, and accuracies for the measured VOCs are 5 ppt, 3%, and 6%, respectively. Measured concentrations of the VOC identified for 1 mM of NA and 1 mg L–1 of HA during irradiation before the introduction of ozone were: ∼74 ppb for unsaturated aldehydes, ∼27 ppb for saturated aldehydes, ∼32 ppb for saturated acids, ∼14 ppb for unsaturated acids, ∼57 ppb for alkanes, ∼11 ppb for alkenes, ∼34 ppb for dienes. Typical time–concentration profiles of VOC from the simulation chamber experiments conducted in the presence of humid acid and nonanoic acid ([HA] = 10 mg L–1 and NA = 1 mM) are presented in Supporting Information Figure S3. A list of the identified VOCs during the photochemistry of NA in the presence of HA can be found in the Supporting Information (Table S1). As the observed products are arising from an interfacial process, degassing and solubilization are in competition. Therefore, the solubility of products may affect their gas phase yield. In order to bring more comprehension into the distribution of the identified VOCs, ranges of Henry’s law constants in water at 298.15 K (in unit of mol m–3 Pa–1) taken from Sander et al.43 are given: saturated aldehydes, (1.0–7.5) × 10–2; unsaturated aldehydes, (5.8–20) × 10–2; saturated acids (1.2–2.3) × 101; alkanes, (1.8–12) × 10–6; alkenes, (1.0–4.4) × 10–5; dienes, (4.6–7.3) × 10–5; isoprene, (2.3–3.4) × 10–4. According to these values, saturated acids exhibit the highest solubility in water among the detected products, followed by unsaturated and saturated aldehydes. Henry’s law solubilities of nonoxygenated compounds are several orders of magnitude lower than those of oxygenated compounds. Alkanes, alkenes, and dienes were mostly partitioned in the gas phase.
The mechanistic pathways leading to the observed gas phase products from an NA-HA system are presented in Figure 3 and have been reported in detail in previous work.40,41 A detailed description of the mechanism can be found in the Supporting Information.
Figure 3.

Proposed mechanism for the photochemical degradation of nonanoic acid at the interface in the presence of humic acid as a photosensitizer.
Light processing, without the subsequent ozonolysis of the NA–HA system, did not lead to a significant fraction of SOA under our experimental conditions. Particle concentrations during the irradiation period were 150–700 cm–3 and similar to the background concentrations of particles before irradiation. These results demonstrate that the photochemical processes did not represent a direct source of SOA under our experimental conditions.
After irradiation, ozone was added in the dark 30 min after the lights were turned off. For all the experiments conducted with NA in the presence of HA, particle formation was observed after ozone addition, supporting the presence of SOA precursors among the VOCs formed during irradiation. Maximum observed particle number concentrations (subtracted from the background concentration) ranged from 70 to 3060 cm–3. As shown in Figure 2, particle formation was observed when ozone was added in the presence of NA and HA, whereas the addition of ozone in the presence of NA only did not lead to any increase of particle concentration. Although performed at [NA] = 0.1 mM, which is below the limit of solubility, the observation of SOA production demonstrated the presence of a surface film. New particles were produced after an induction time ranging from 1.4 and 11.5 min, corresponding to the time when the increase of particle levels was quantitatively significant. In addition, about 2 h after the last introduction of ozone, UV lamps were turned on. As shown in Figure 2, a new particle formation event was observed when photochemistry occurred for the second time in the presence of ozone. This increase of particle number concentration was caused by a reactivation of the photosensitized reaction. Indeed, under these experimental conditions, the concentrations of OH radicals may be very limited since the photolysis of ozone in the presence of water is inefficient. In addition, based on simple kinetic calculation, more than 55% of OH radicals formed through irradiation would be scavenged by NA, which represents a lower limit, since numerous saturated VOCs present in the gas phase would also deplete OH radicals. We can therefore consider that OH chemistry is a minor process in SOA formation, where ozonolysis of additional formed SOA precursors in the gas phase is the main pathway of particle formation. Although temperature increased by about 4 °C and reached a stable value of 26 °C after 1.5 h, the formation of new particles was not believed to originate from the increase of temperature. The observed delay between the beginning of irradiation and the observable increase of particle concentration resulted from a combination of the transport and kinetic processes in the gas chamber. A bar graph in Figure 4 displayed the particle formation rate obtained during the blanks and the experiments. Values of formation rate originated from the average of individual blanks and experiments.
Figure 4.
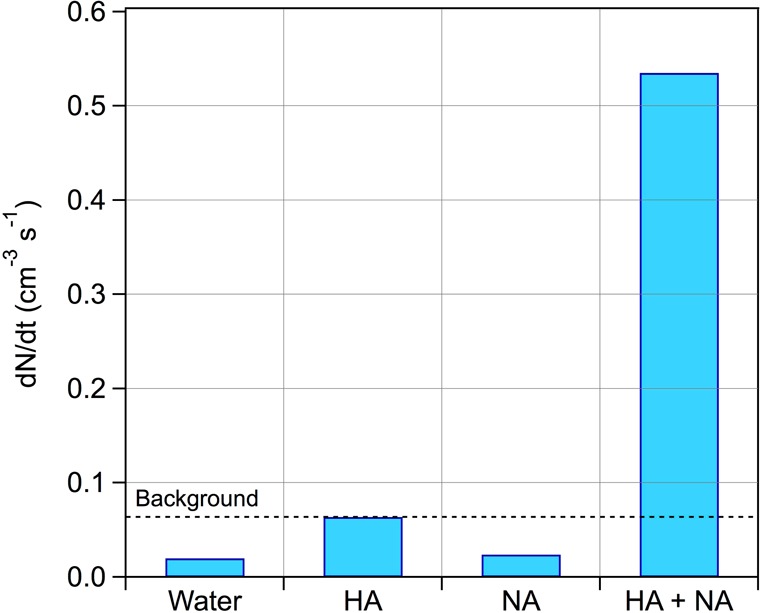
Bar graph representing particle formation rate (dN/dt) under different experimental conditions followed by ozone addition.
Representative time series of the number size distributions over the experimental time scale are presented in Figure 2. Monomodal distributions were measured. Particle number concentrations measured by the scanning mobility particle sizer (SMPS) were systematically lower than those measured with the ultrafine condensation particle counter (UCPC) due to different cutoff diameters and the position of the SMPS sampling inlet, positioned one meter above the CPC. As ozone concentration ranged from 250 to 500 ppb, we observed no dependence on ozone concentration.
Minimum concentration of particles formed (68 cm–3) was obtained with the lowest concentrations of NA (0.1 mM) and HA (1 mg L–1), and maximum particle concentration (3057 cm–3) was obtained with the highest concentrations of NA (2 mM) and HA (10 mg L–1). This shows that the concentration of the surface film as well as the concentration of dissolved organic matter in the bulk impacts SOA formation. In addition, the total particle mass concentration (ΔM0) formed under these experimental conditions did not exceed 1 μg m–3 during the dark ozonolysis reaction. These observations suggest that the dark ozonolysis reaction of VOCs formed through the light induced reaction of NA in the presence of HA led to the formation of condensable organic vapors,50−52 hereby promoting the formation of new particles as seen by the increase of particle numbers, also followed by some condensation processes (promoting an increase of the volume/mass of particles). This observation was experimentally supported by adding ozone in the gas phase after irradiation, leading to a low mass concentration of particles (Supporting Information Figure S4). It is well established that aqueous phase OH radicals are generated through light absorption of humic substances,53−55 but are not expected to degas into the gas phase. In addition, under these experimental conditions, the high concentration of NA in gas and liquid phases combined with the high reactivity of OH radicals (kOH+NA = 9.76 × 10–12 cm3 molecule–1 s–1, EPI suite)45 would make NA an efficient OH scavenger, acting as an inhibitor of any interfering OH reaction. This assumption has been verified, since no particle formation event was observed during the photosensitization period, underlying the absence of any contribution of OH reaction in particle formation. SOA production from the dark addition of ozone sustained the presence of unsaturated VOCs, bearing one or several unsaturations, which are potential VOC candidates in SOA formation.
Discussion
SOA Formation Potential from Photosensitized Reactions Followed by Ozone Addition
In this section, the relationship between the observed particle formation and the light processing of NA in the presence of HA at the air–water interface followed by ozone addition will be tentatively established. The modification of the surface tension at the interface induced by addition of surface-enriched organic compounds is described by the Gibbs adsorption isotherm, where the surface excess concentration of NA is as follows:56
| 1 |
with ΓNA, the surface excess concentration of NA (in molecule cm–2), which represents the amount of NA at the air–sea interface, CNA, the concentration of NA (in mol cm–3), γ, the surface tension (in N m–1), R, the gas constant (8.314 J K–1 mol–1), and T, the temperature in Kelvin. The corresponding surface tensions were measured previously using a Krüss K6 (Krüss GmbH) tensiometer by varying the concentration of NA.40 In this work, the estimated surface excess concentration ranged from 1.66 × 1014 to 3.99 × 1014 molecule cm–2.
As HAs are fully soluble under slightly acidic conditions used in this study,57 we can then easily assume that the humic acids were homogeneously distributed in the water tank in the chamber. In addition, as the surface concentration of the surfactant drives the chemical transformation rate, a correlation between the measured number of particles and the production rate of gaseous organic material is expected,58 as shown in Figure 5. In fact, assuming that both NA and HA were present in excess, that is, neglecting the influence of mass transport or dilution in the chamber, can be simplified as
| 2 |
where k can be regarded as a global rate coefficient for the photosensitized reaction of NA in the presence of HA (including reactions kinetics, product yield and phase transfer kinetics, etc.) (in cm3 molecule–1 s–1), A is the surface area of the liquid (in m2), and V is the internal volume of the chamber (in m3). The amount of condensable vapor is then related to the ozonolysis of unsaturated products (functionalized alkenes). Assuming that the ozonolysis reaction occurred under pseudo-first-order conditions, the amount of condensable products (in molecule cm–3) can be expressed as
| 3 |
where kO3 is the bimolecular ozone reaction rate coefficient with the alkenes (in cm3 molecule–1 s–1), tirr is the irradiation time (in s), and tind is the induction time (in s) corresponding to the time interval between the introduction of ozone and the formation of particles. However, the concentration of condensable vapors was not determined accurately, since many factors remained unknown. Indeed, the SOA precursor concentration in the gas phase could not be measured in this work. Modeling such condensable vapor concentrations would be possible, but would be implicitly associated with numerous assumptions, increasing the level of their uncertainties. Jokinen et al.51 reported the formation yields of ELVOCs from the ozonolysis of terpenes, but with very high uncertainties (−50/+100%). Furthermore, as they are extremely low-volatility organic compounds, loss on the surface of the chamber would be highly expected. All combined assumptions would lead to a very uncertain estimate of ELVOC or condensable vapor concentrations in these experiments.
Figure 5.
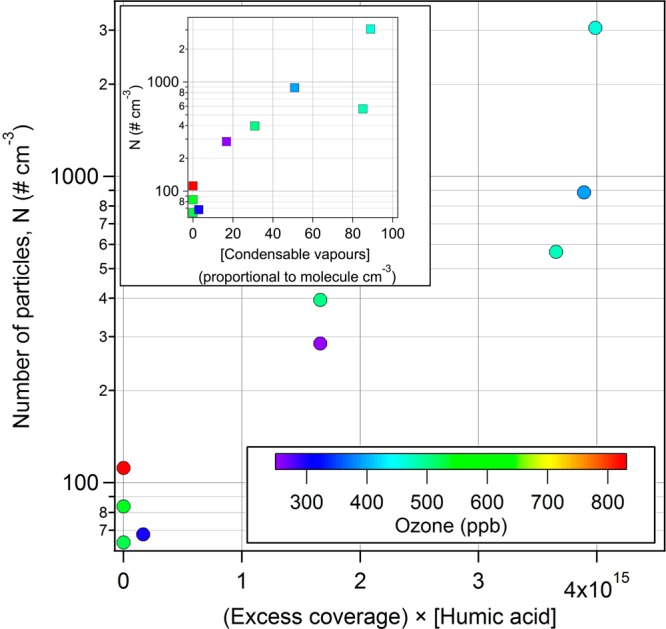
Correlation between the light-induced reaction of surface excess nonanoic acid (NA) in the presence of humic acid (HA) and the number of particles (N) formed through the addition of ozone. In addition, correlation between the number of particles (N) and estimated levels of condensable vapors (proportional to molecules cm–3) is presented.
Thereby, we assumed that the ratio of the SOA precursor concentration to the total amount of products is similar whatever the initial liquid phase concentrations are. As displayed in Figure 5, correlations were indeed observed, similar to the one reported by Boulon et al.58 between the particle production rate and the surface excess concentration and also the concentration of condensable vapors, highlighting the direct role of the air–water interface in the production of SOA particles. It is also underlined that, under our experimental conditions, the formation of particles was dictated by the surface process and not by ozone concentration, which was in large excess and simply titrated away all unsaturations.
Atmospheric Implications
Secondary organic aerosol production from a photosensitized reaction was evaluated in a new multiphase atmospheric simulation chamber. Obviously, the experiments described were not necessarily conducted strictly under atmospheric conditions, but they clearly highlight the fact that the light-induced reaction of a simple fatty acid (i.e., NA) in the presence of dissolved organic matter (HA) led to a significant production of aerosol precursors, which upon ozonolysis was the main pathway leading to SOA production. We found evidence that unsaturated compounds were the aerosol precursors among the other VOCs formed during the photochemical process. We are aware that the concentration of ozone necessary in this work is equivalent to several hundreds of ppb (250–830 ppb), which is an order of magnitude higher than the concentration in the marine boundary layer (i.e., ∼ 0–50 ppb). High ozone concentrations compared to atmospheric conditions were used to compensate: (1) the low light intensity of the UV lamps compared to solar radiation (about seven times lower at 365 nm) and (2) the possible loss of reactive VOC through heterogeneous loss on the chamber wall, reducing the amount of condensable vapors in the gas phase, therefore lowering SOA particle production.59 Overall, the high ozone concentration compensated for these effects and led to rapid SOA formation. Furthermore, the low particle mass yield observed in those experiments confirmed the formation of extremely low-volatility oxidation products, preferentially leading to an increase in particle number in higher proportion than the concentration of semivolatile oxidation products. Numerous other VOCs characterized by a low vapor pressure originating from light processing likely participated in aerosol growth. Furthermore, the approach used in this study focused on an unexplored photochemical process and its contribution to the production of marine SOA. Considering the studies reported by Prather and co-workers,20,21,60 who used a quasi-realistic approach to describe the chemical complexity of SSA, an evaluation of the contribution of the photochemistry in the formation of VOCs and SOA would be of interest, especially at low wind speed where mechanical SSA production is reduced while the SML are still covering the air–sea interface. This work highlights the need to conduct future experiments more closely mimicking marine conditions.
While our study was not directly performed under realistic conditions, the results presented here may still be related to real environmental issues. In fact, Wurl et al.,61 by studying the organic enrichment in the sea surface microlayer (SML) in different regions of the ocean (subtropical, temperate, polar), observed a much larger coverage than previously assumed and showed that part of the oceans can be assumed as fully covered by an organic layer, mostly during fall, with wind speeds of up to 10 m s–1. These conditions are similar to those reproduced in our chamber by simulating an almost molecular coverage of the air–sea interface with a fatty acid (i.e., nonanoic acid). In the case of such a C9 carboxylic acid, and assuming a homogeneous distribution throughout a 100 μm thick water layer at the interface (matching operationally defined SML thickness), the enrichment factors simulated in the chamber were between 10 and 100 and therefore higher than typical marine values. While DOM is present at low concentrations in the open oceans (0.5–1.0 mg L–1),19 with concentrations possibly peaking near estuaries, the high range of reported values is overlapping the conditions used in this study. Under such conditions, the lowest values of (Excess coverage × [Humic acid]), as reported in Figure 5, are nevertheless approaching real marine conditions in which photosensitized SOA production could lead to a particle increase of ca. 100–500 # cm–3. For instance, Covert et al.62 reported new particle formation (assigned at that time to SO2 processing) in the marine boundary layer along the coast of Washington State, with an increase of a few hundreds of particles per cm3 within a few hours. While their measurements could not distinguish between changes in number concentration caused by particle nucleation versus advection or vertical mixing, they underlined a continuous particle production on a mesoscale in the air mass. Such changes are in the same range as those estimated above; therefore, one could hypothesize that SML photoprocessing could contribute significantly to such events, especially in view of the recent advances in the description of nucleation in which not only sulfuric acid is the key, but also its interaction with complex organic molecules (even if present at very low mixing ratios).63
This study clearly points toward a possible impact of photochemistry of the organic films at the air/water interface on the production of organic aerosols (and their gaseous precursors). As such chemistry involves only fatty acids as surfactants and dissolved organic matter as photosensitizers, both being ubiquitous in the marine environment, its quantitative importance will have to be assessed during further studies.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement 290852 - AIRSEA.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.6b03520.
Table S1 lists the volatile organic compounds identified during air sea interface experiments. Figure S1 shows the spectrum of the lamps used. Figure S2 illustrates particle formation during the blank experiment (Humic acid only). Figure S3 displays typical concentration–time profiles of ozone, VOC, and particles. In addition, an example of particle mass distribution obtained during the experiments is shown in Figure S4 (PDF)
Author Present Address
§ Earth System Research Laboratory, Chemical Sciences Division, National Oceanic and Atmospheric Administration, Boulder, Colorado, USA and Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, Colorado, USA.
Author Present Address
⊥ UMR ECOSYS, INRA, AgroParisTech, Université Paris-Saclay, 78850, Thiverval-Grignon, France.
The authors declare no competing financial interest.
Supplementary Material
References
- Blanchard D. C. Sea-to-air transport of surface active material. Science 1964, 146 (3642), 396–397. 10.1126/science.146.3642.396. [DOI] [PubMed] [Google Scholar]
- Hoffman E. J.; Duce R. A. Factors influencing the organic carbon content of marine aerosols: A laboratory study. J. Geophys. Res. 1976, 81 (21), 3667–3670. 10.1029/JC081i021p03667. [DOI] [Google Scholar]
- Middlebrook A. M.; Murphy D. M.; Thomson D. S. Observations of organic material in individual marine particles at Cape Grim during the First Aerosol Characterization Experiment (ACE 1). J. Geophys. Res. 1998, 103 (D13), 16475–16483. 10.1029/97JD03719. [DOI] [Google Scholar]
- Putaud J. P.; Van Dingenen R.; Mangoni M.; Virkkula A.; Raes F.; Maring H.; Prospero J. M.; Swietlicki E.; Berg O. H.; Hillamo R.; Mäkelä T. Chemical mass closure and assessment of the origin of the submicron aerosol in the marine boundary layer and the free troposphere at Tenerife during ACE-2. Tellus, Ser. B 2000, 52 (2), 141–168. 10.1034/j.1600-0889.2000.00056.x. [DOI] [Google Scholar]
- Cavalli F.; Facchini M. C.; Decesari S.; Mircea M.; Emblico L.; Fuzzi S.; Ceburnis D.; Yoon Y. J.; O’Dowd C. D.; Putaud J. P.; Dell’Acqua A. Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic. J. Geophys. Res. 2004, 109 (D24), D24215. 10.1029/2004JD005137. [DOI] [Google Scholar]
- O’Dowd C. D.; Facchini M. C.; Cavalli F.; Ceburnis D.; Mircea M.; Decesari S.; Fuzzi S.; Yoon Y. J.; Putaud J.-P. Biogenically driven organic contribution to marine aerosol. Nature 2004, 431 (7009), 676–680. 10.1038/nature02959. [DOI] [PubMed] [Google Scholar]
- Ovadnevaite J.; O’Dowd C.; Dall’Osto M.; Ceburnis D.; Worsnop D. R.; Berresheim H. Detecting high contributions of primary organic matter to marine aerosol: A case study. Geophys. Res. Lett. 2011, 38 (2), L02807. 10.1029/2010GL046083. [DOI] [Google Scholar]
- Ovadnevaite J.; Manders A.; de Leeuw G.; Ceburnis D.; Monahan C.; Partanen A. I.; Korhonen H.; O’Dowd C. D. A sea spray aerosol flux parameterization encapsulating wave state. Atmos. Chem. Phys. 2014, 14 (4), 1837–1852. 10.5194/acp-14-1837-2014. [DOI] [Google Scholar]
- Shaw G. Bio-controlled thermostasis involving the sulfur cycle. Clim. Change 1983, 5 (3), 297–303. 10.1007/BF02423524. [DOI] [Google Scholar]
- Charlson R. J.; Lovelock J. E.; Andreae M. O.; Warren S. G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 1987, 326 (6114), 655–661. 10.1038/326655a0. [DOI] [Google Scholar]
- Clarke A. D.; Davis D.; Kapustin V. N.; Eisele F.; Chen G.; Paluch I.; Lenschow D.; Bandy A. R.; Thornton D.; Moore K.; Mauldin L.; Tanner D.; Litchy M.; Carroll M. A.; Collins J.; Albercook G. Particle nucleation in the tropical boundary layer and its coupling to marine sulfur sources. Science 1998, 282 (5386), 89–92. 10.1126/science.282.5386.89. [DOI] [PubMed] [Google Scholar]
- Vallina S. M.; Simó R.; Gassó S. What controls CCN seasonality in the Southern Ocean? A statistical analysis based on satellite-derived chlorophyll and CCN and model-estimated OH radical and rainfall. Global Biogeochem. Cycles. 2006, 20 (1), GB1014. 10.1029/2005GB002597. [DOI] [Google Scholar]
- Myriokefalitakis S.; Vignati E.; Tsigaridis K.; Papadimas C.; Sciare J.; Mihalopoulos N.; Facchini M. C.; Rinaldi M.; Dentener F. J.; Ceburnis D.; Hatzianastasiou N.; O’Dowd C. D.; van Weele M.; Kanakidou M. Global modeling of the oceanic source of organic aerosols. Adv. in Met. 2010, 2010, 1–16. 10.1155/2010/939171. [DOI] [Google Scholar]
- Gunson J. R.; Spall S. A.; Anderson T. R.; Jones A.; Totterdell I. J.; Woodage M. J.. Climate sensitivity to ocean dimethylsulphide emissions. Geophys. Res. Lett. 2006, 33 ( (7), ), n/a-n/a. 10.1029/2005GL024982 [DOI] [Google Scholar]
- Kloster S.; Feichter J.; Maier-Reimer E.; Six K. D.; Stier P.; Wetzel P. DMS cycle in the marine ocean-atmosphere system? a global model study. Biogeosci. Disc. 2005, 3 (1), 1067–1126. 10.5194/bgd-2-1067-2005. [DOI] [Google Scholar]
- Lewis E. R.; Schwartz S. E.. Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models. Geophys. Monogr. Ser, AGU, 2004; 413 pp. [Google Scholar]
- Cunliffe M.; Engel A.; Frka S.; Gašparović B.; Guitart C.; Murrell J. C.; Salter M.; Stolle C.; Upstill-Goddard R.; Wurl O. Sea surface microlayers: A unified physicochemical and biological perspective of the air–ocean interface. Prog. Oceanogr. 2013, 109, 104–116. 10.1016/j.pocean.2012.08.004. [DOI] [Google Scholar]
- Law C. S.; Brévière E.; de Leeuw G.; Garçon V.; Guieu C.; Kieber D. J.; Kontradowitz S.; Paulmier A.; Quinn P. K.; Saltzman E. S.; Stefels J.; von Glasow R. Evolving research directions in Surface Ocean–Lower Atmosphere (SOLAS) science. Environ. Chem. 2013, 10 (1), 1–16. 10.1071/EN12159. [DOI] [Google Scholar]
- Carpenter L. J.; Nightingale P. D. Chemistry and release of gases from the surface ocean. Chem. Rev. 2015, 115 (10), 4015–4034. 10.1021/cr5007123. [DOI] [PubMed] [Google Scholar]
- Quinn P. K.; Collins D. B.; Grassian V. H.; Prather K. A.; Bates T. S. Chemistry and related properties of freshly emitted sea spray aerosol. Chem. Rev. 2015, 115 (10), 4383–4399. 10.1021/cr500713g. [DOI] [PubMed] [Google Scholar]
- Prather K. A.; Bertram T. H.; Grassian V. H.; Deane G. B.; Stokes M. D.; DeMott P. J.; Aluwihare L. I.; Palenik B. P.; Azam F.; Seinfeld J. H.; Moffet R. C.; Molina M. J.; Cappa C. D.; Geiger F. M.; Roberts G. C.; Russell L. M.; Ault A. P.; Baltrusaitis J.; Collins D. B.; Corrigan C. E.; Cuadra-Rodriguez L. A.; Ebben C. J.; Forestieri S. D.; Guasco T. L.; Hersey S. P.; Kim M. J.; Lambert W. F.; Modini R. L.; Mui W.; Pedler B. E.; Ruppel M. J.; Ryder O. S.; Schoepp N. G.; Sullivan R. C.; Zhao D. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (19), 7550–7555. 10.1073/pnas.1300262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss P. S.; Duce R. A.. The Sea Surface and Global Change; Cambridge University Press: Cambridge, U. K., 1997. [Google Scholar]
- Hunter K. A.; Liss P. S. The input of organic material to the oceans: air-sea interactions and the organic chemical composition of the sea surface. Mar. Chem. 1977, 5 (4–6), 361–379. 10.1016/0304-4203(77)90029-9. [DOI] [Google Scholar]
- Bayliss N.; Bucat R. The photolysis of aqueous nitrate solutions. Aust. J. Chem. 1975, 28 (9), 1865–1878. 10.1071/CH9751865. [DOI] [Google Scholar]
- Liss P. S., The chemistry of near-surface seawater. In Dynamic processes in the chemistry of the upper ocean, Burton J. D.; Brewer P. G.; Chesselet R., Eds.; Springer, 1986; Vol. 17, pp 41–51. [Google Scholar]
- Hardy J. T. The sea surface microlayer: biology, chemistry and anthropogenic enrichment. Prog. Oceanogr. 1982, 11 (4), 307–328. 10.1016/0079-6611(82)90001-5. [DOI] [Google Scholar]
- Garabetian F.; Romano J.-C.; Paul R.; Sigoillot J.-C. Organic matter composition and pollutant enrichment of sea surface microlayer inside and outside slicks. Mar. Environ. Res. 1993, 35 (4), 323–339. 10.1016/0141-1136(93)90100-E. [DOI] [Google Scholar]
- Williams P. M.; Carlucci A. F.; Henrichs S. M.; Van Vleet E. S.; Horrigan S. G.; Reid F. M. H.; Robertson K. J. Chemical and microbiological studies of sea-surface films in the Southern Gulf of California and off the West Coast of Baja California. Mar. Chem. 1986, 19 (1), 17–98. 10.1016/0304-4203(86)90033-2. [DOI] [Google Scholar]
- Schneider J. K.; Gagosian R. B. Particle size distribution of lipids in aerosols off the coast of Peru. J. Geophys. Res. 1985, 90 (D5), 7889–7898. 10.1029/JD090iD05p07889. [DOI] [Google Scholar]
- Gershey R. M. Characterization of seawater organic matter carried by bubble-generated aerosols1. Limnol. Oceanogr. 1983, 28 (2), 309–319. 10.4319/lo.1983.28.2.0309. [DOI] [Google Scholar]
- Guitart C.; García-Flor N.; Dachs J.; Bayona J. M.; Albaigés J. Evaluation of sampling devices for the determination of polycyclic aromatic hydrocarbons in surface microlayer coastal waters. Mar. Pollut. Bull. 2004, 48 (9–10), 961–968. 10.1016/j.marpolbul.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Facchini M. C.; Rinaldi M.; Decesari S.; Carbone C.; Finessi E.; Mircea M.; Fuzzi S.; Ceburnis D.; Flanagan R.; Nilsson E. D.; de Leeuw G.; Martino M.; Woeltjen J.; O’Dowd C. D.. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys. Res. Lett. 2008, 35 ( (17), ), n/a-n/a. 10.1029/2008GL034210 [DOI] [Google Scholar]
- Kovac N.; Bajt O.; Faganeli J.; Sket B.; Orel B. Study of macroaggregate composition using FT-IR and 1H-NMR spectroscopy. Mar. Chem. 2002, 78 (4), 205–215. 10.1016/S0304-4203(02)00033-6. [DOI] [Google Scholar]
- Tervahattu H.; Juhanoja J.; Vaida V.; Tuck A. F.; Niemi J. V.; Kupiainen K.; Kulmala M.; Vehkamäki H.. Fatty acids on continental sulfate aerosol particles. J. Geophys. Res. 2005, 110 ( (D6), ), 10.1029/2004JD005400 [DOI] [Google Scholar]
- Tervahattu H.; Juhanoja J.; Kupiainen K. Identification of an organic coating on marine aerosol particles by TOF-SIMS. J. Geophys. Res. 2002, 107 (D16), ACH 18–1–ACH 18–7. 10.1029/2001JD001403. [DOI] [Google Scholar]
- Tervahattu H.; Hartonen K.; Kerminen V.-M.; Kupiainen K.; Aarnio P.; Koskentalo T.; Tuck A. F.; Vaida V. New evidence of an organic layer on marine aerosols. J. Geophys. Res. 2002, 107 (D7), AAC 1–1–AAC 1–8. 10.1029/2000JD000282. [DOI] [Google Scholar]
- Reeser D. I.; George C.; Donaldson D. J. Photooxidation of halides by chlorophyll at the air–salt water interface. J. Phys. Chem. A 2009, 113 (30), 8591–8595. 10.1021/jp903657j. [DOI] [PubMed] [Google Scholar]
- Reeser D. I.; Jammoul A.; Clifford D.; Brigante M.; D’Anna B.; George C.; Donaldson D. J. Photoenhanced reaction of ozone with chlorophyll at the seawater surface. J. Phys. Chem. C 2009, 113 (6), 2071–2077. 10.1021/jp805167d. [DOI] [Google Scholar]
- George C.; Ammann M.; D’Anna B.; Donaldson D. J.; Nizkorodov S. A. Heterogeneous photochemistry in the atmosphere. Chem. Rev. 2015, 115 (10), 4218–4258. 10.1021/cr500648z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuraru R.; Fine L.; van Pinxteren M.; D’Anna B.; Herrmann H.; George C. Photosensitized production of functionalized and unsaturated organic compounds at the air-sea interface. Sci. Rep. 2015, 5, 12741. 10.1038/srep12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H.; Ciuraru R.; Dupart Y.; Passananti M.; Tinel L.; Rossignol S.; Perrier S.; Donaldson D. J.; Chen J.; George C. Photosensitized production of atmospherically reactive organic compounds at the air/aqueous interface. J. Am. Chem. Soc. 2015, 137 (26), 8348–8351. 10.1021/jacs.5b04051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuraru R.; Fine L.; Pinxteren M. v.; D’Anna B.; Herrmann H.; George C. Unravelling new processes at interfaces: photochemical isoprene production at the sea surface. Environ. Sci. Technol. 2015, 49 (22), 13199–13205. 10.1021/acs.est.5b02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15 (8), 4399–4981. 10.5194/acp-15-4399-2015. [DOI] [Google Scholar]
- Hilal S. H.; Ayyampalayam S. N.; Carreira L. A. Air–liquid partition coefficient for a diverse set of organic compounds: Henry’s law constant in water and hexadecane. Environ. Sci. Technol. 2008, 42 (24), 9231–9236. 10.1021/es8005783. [DOI] [PubMed] [Google Scholar]
- US Environmental Protection Agency. Estimation Program Interface (EPI) Suite. Version EPI 4.10. http://www.epa.gov/opptintr/exposure/pubs/episuite.htm (accessed May 25, 2015).
- Vacha R.; Buch V.; Milet A.; Devlin J. P.; Jungwirth P. Autoionization at the surface of neat water: is the top layer pH neutral, basic, or acidic?. Phys. Chem. Chem. Phys. 2007, 9 (34), 4736–4747. 10.1039/b704491g. [DOI] [PubMed] [Google Scholar]
- Enami S.; Stewart L. A.; Hoffmann M. R.; Colussi A. J. Superacid Chemistry on Mildly Acidic Water. J. Phys. Chem. Lett. 2010, 1 (24), 3488–3493. 10.1021/jz101402y. [DOI] [Google Scholar]
- Mishra H.; Enami S.; Nielsen R. J.; Stewart L. A.; Hoffmann M. R.; Goddard W. A.; Colussi A. J. Brønsted basicity of the air–water interface. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (46), 18679–18683. 10.1073/pnas.1209307109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindinger W.; Hansel A.; Jordan A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Processes 1998, 173 (3), 191–241. 10.1016/S0168-1176(97)00281-4. [DOI] [Google Scholar]
- Donahue N. M.; Kroll J. H.; Pandis S. N.; Robinson A. L. A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution. Atmos. Chem. Phys. 2012, 12 (2), 615–634. 10.5194/acp-12-615-2012. [DOI] [Google Scholar]
- Jokinen T.; Berndt T.; Makkonen R.; Kerminen V.-M.; Junninen H.; Paasonen P.; Stratmann F.; Herrmann H.; Guenther A. B.; Worsnop D. R.; Kulmala M.; Ehn M.; Sipilä M. Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (23), 7123–7128. 10.1073/pnas.1423977112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehn M.; Thornton J. A.; Kleist E.; Sipila M.; Junninen H.; Pullinen I.; Springer M.; Rubach F.; Tillmann R.; Lee B.; Lopez-Hilfiker F.; Andres S.; Acir I.-H.; Rissanen M.; Jokinen T.; Schobesberger S.; Kangasluoma J.; Kontkanen J.; Nieminen T.; Kurten T.; Nielsen L. B.; Jorgensen S.; Kjaergaard H. G.; Canagaratna M.; Maso M. D.; Berndt T.; Petaja T.; Wahner A.; Kerminen V.-M.; Kulmala M.; Worsnop D. R.; Wildt J.; Mentel T. F. A large source of low-volatility secondary organic aerosol. Nature 2014, 506 (7489), 476–479. 10.1038/nature13032. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Schindelka J.; Herrmann H.; George C.; Rosell M.; Herrero-Martín S.; Klán P.; Richnow H. H. Investigation of humic substance photosensitized reactions via carbon and hydrogen isotope fractionation. Environ. Sci. Technol. 2015, 49 (1), 233–242. 10.1021/es502791f. [DOI] [PubMed] [Google Scholar]
- al Housari F.; Vione D.; Chiron S.; Barbati S. Reactive photoinduced species in estuarine waters. Characterization of hydroxyl radical, singlet oxygen and dissolved organic matter triplet state in natural oxidation processes. Photochem. Photobiol. Sci. 2010, 9 (1), 78–86. 10.1039/B9PP00030E. [DOI] [PubMed] [Google Scholar]
- Aguer J. P.; Richard C.; Andreux F. Effect of light on humic substances: production of reactive species. Analusis 1999, 27 (5), 387–389. 10.1051/analusis:1999270387. [DOI] [Google Scholar]
- Donaldson D. J.; Anderson D. Adsorption of atmospheric gases at the air–water interface. 2. C1–C4 alcohols, acids, and acetone. J. Phys. Chem. A 1999, 103 (7), 871–876. 10.1021/jp983963h. [DOI] [Google Scholar]
- Klaviņš M.; Purmalis O. Surface activity of humic acids depending on their origin and humification degree. Proc. Latv. Acad. Sci., Sect. B 2008, 67 (6), 493–499. 10.2478/prolas-2013-0083. [DOI] [Google Scholar]
- Boulon J.; Sellegri K.; Katrib Y.; Wang J.; Miet K.; Langmann B.; Laj P.; Doussin J. F. Sub-3 nm particles detection in a large photoreactor background: possible implications for new particles formation studies in a smog chamber. Aerosol Sci. Technol. 2013, 47 (2), 153–157. 10.1080/02786826.2012.733040. [DOI] [Google Scholar]
- La Y. S.; Camredon M.; Ziemann P. J.; Valorso R.; Matsunaga A.; Lannuque V.; Lee-Taylor J.; Hodzic A.; Madronich S.; Aumont B. Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: explicit modeling of SOA formation from alkane and alkene oxidation. Atmos. Chem. Phys. Discuss. 2015, 15 (17), 23893–23930. 10.5194/acpd-15-23893-2015. [DOI] [Google Scholar]
- Stokes M. D.; Deane G. B.; Prather K.; Bertram T. H.; Ruppel M. J.; Ryder O. S.; Brady J. M.; Zhao D. A Marine Aerosol Reference Tank system as a breaking wave analogue for the production of foam and sea-spray aerosols. Atmos. Meas. Tech. 2013, 6 (4), 1085–1094. 10.5194/amt-6-1085-2013. [DOI] [Google Scholar]
- Wurl O.; Wurl E.; Miller L.; Johnson K.; Vagle S. Formation and global distribution of sea-surface microlayers. Biogeosciences 2011, 8 (1), 121–135. 10.5194/bg-8-121-2011. [DOI] [Google Scholar]
- Covert D. S.; Kapustin V. N.; Quinn P. K.; Bates T. S. New particle formation in the marine boundary layer. J. Geophys. Res. 1992, 97 (D18), 20581–20589. 10.1029/92JD02074. [DOI] [Google Scholar]
- Riccobono F.; Schobesberger S.; Scott C. E.; Dommen J.; Ortega I. K.; Rondo L.; Almeida J.; Amorim A.; Bianchi F.; Breitenlechner M.; David A.; Downard A.; Dunne E. M.; Duplissy J.; Ehrhart S.; Flagan R. C.; Franchin A.; Hansel A.; Junninen H.; Kajos M.; Keskinen H.; Kupc A.; Kürten A.; Kvashin A. N.; Laaksonen A.; Lehtipalo K.; Makhmutov V.; Mathot S.; Nieminen T.; Onnela A.; Petäjä T.; Praplan A. P.; Santos F. D.; Schallhart S.; Seinfeld J. H.; Sipilä M.; Spracklen D. V.; Stozhkov Y.; Stratmann F.; Tomé A.; Tsagkogeorgas G.; Vaattovaara P.; Viisanen Y.; Vrtala A.; Wagner P. E.; Weingartner E.; Wex H.; Wimmer D.; Carslaw K. S.; Curtius J.; Donahue N. M.; Kirkby J.; Kulmala M.; Worsnop D. R.; Baltensperger U. Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles. Science 2014, 344 (6185), 717–721. 10.1126/science.1243527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.