

Abstract
Spondyloarthritides comprise a group of inflammatory conditions which have in common an association with the MHC class I molecule, HLA-B27. Given this association, these diseases are classically considered disorders of adaptive immunity. However, recent data are challenging this assumption and raising the possibility that innate immunity may play a more prominent role in pathogenesis than previously suspected. In this review, the concept of autoinflammation will be discussed and evidence will be presented from human and animal models to support a critical role for innate immunity in HLA-B27 associated disorders.
Keywords: HLA-B27, autoinflammation, innate, IL-1, spondyloarthritis
INTRODUCTION
AUTOINFLAMMATION
Autoinflammation is a term coined by Dan Kastner over 10 years ago when he described episodes of “seemingly unprovoked inflammation in the absence of auto-antibodies or auto-reactive T cells”1. Since this initial description, autoinflammatory mechanisms have been better elucidated and the impact of autoinflammation on common diseases, even those traditionally felt to be due to adaptive immunity, is increasingly appreciated2.
The immune response can be broadly characterized into innate and adaptive responses. Adaptive responses involve antigen specific B and T cell activation resulting in auto-antibody and antigen-specific T cell responses. Following activation, a highly specific memory response is generated. As these mechanisms were becoming increasingly understood, Polly Matzinger described an alternate hypothesis, the “danger theory”, which theorized that the immune system developed to respond to danger rather than antigen-specifc self vs. non-self3. While this was largely criticized at the time, it has since become accepted as the initial description of the innate immune system4.
The innate immune response is the first line of defense to an insult to an organism such as a break in skin or mucosal integrity or the introduction of a pathogen. This occurs before the adaptive immune response has time to amplify and contain the insult and is important in directing the adaptive immune response5. Innate immune cells respond to danger through recognition of specific motifs on danger signals by highly conserved pattern recognition receptors (PRRs). The molecular patterns from microorganisms are termed MAMPS (microbe-associated molecular patterns) and damage patterns are termed damage- or danger- associated molecular patterns (DAMPs)6. The expanding group of PRRs includes Toll-like receptors (TLRs), Nod-leucine-rich repeat-containing receptors (NLRs), C-type lectin receptors (CLRs), RIG-1-like receptors (RLRs), and AIM-2 like receptors. Much focus has been placed on understanding the signals which activate MAMPs and DAMPs with known MAMPs including LPS, flagellin, dsRNA, and unmethylated CpG motifs and known DAMPs including DNA, RNA, uric acid, ATP, and adenosine. The net result is the activation of innate immune cells including monocytes, macrophages, dendritic cells, natural killer cells, and neutrophils with a resultant inflammatory cascade. Figure 1.
Figure 1. Activation of Innate Immunity.
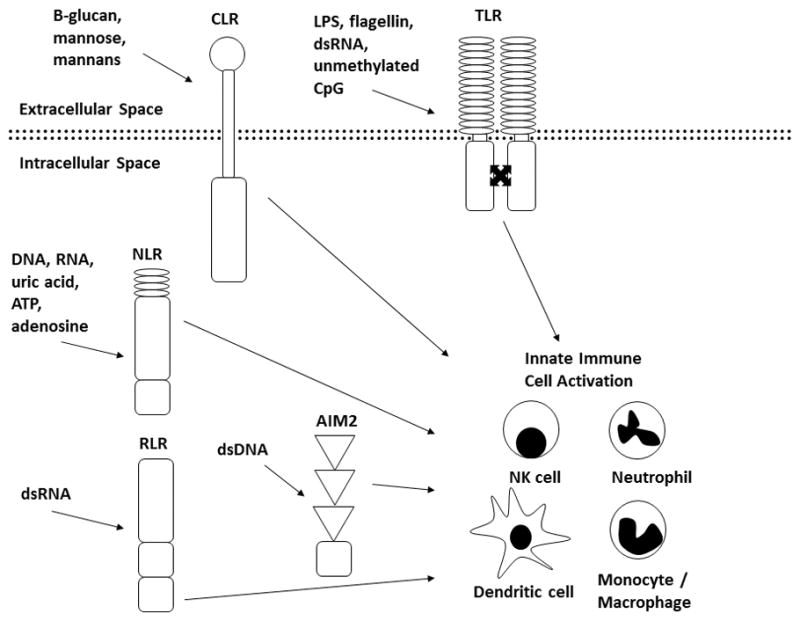
MAMPs and DAMPs activate CLRs, TLRs, NLRs, RLRs, and AIM-2 resulting in activation of innate immune cells including NK cells, neutrophils, monocytes/macrophages, and dendritic cells.
The clearest examples of autoinflammation come from the study of monogenic autoinflammatory diseases7. These are genetic disorders resulting from disruption of key mediators of the innate immune response. They are characterized by excessive inflammation, periods of relapse and remission, the lack of an antigen-specific response, and frequently neutrophilic infiltration8. The hallmark of these diseases are the cryopyrin associated periodic syndromes (CAPS) which are due to autosomal dominant mutations in NLRP3 (also known as NALP3 or cryopyrin) resulting in a constitutively active NLPR3 inflammasome causing the conversion of pro-IL1β to active IL-1β9,10. This results in excessive IL-1 signaling and a characteristic pattern of damage to affected organs11. Inflammation in CAPS and organ damage can be completely abrogated by IL-1 inhibitors12,13 and a response to IL-1 blockade can be used as a therapeutic test of the presence of an IL-1 mediated autoinflammatory disorder.
Other monogenic autoinflammatory disorders demonstrate the consequences of excessive activation of other key components of the innate immune response. This growing list includes familial Mediterranean fever (FMF due to mutations in MEFV leading to overactive pyrin)14,15, TNF-receptor associated periodic syndrome (TRAPS due to mutations in TNFRSF1A which encodes the TNF receptor and leads to its sequestration in the endoplasmic reticulum and excessive signaling through activation of MAPKs)1, PAPA (Pyogenic arthritis with pyoderma gangrenosum and acne (PAPA) syndrome due to mutations in PSTPIP1 leading to eventual activation of pyrin)16, and deletion of the IL-1 receptor antagonist (DIRA due to deletion IL1RN which encodes the IL-1 receptor antagonist leading to unopposed IL-1 signaling)17,18. Of note, the majority of these disorders respond to IL-1 blockade even in the absence of a direct role for IL-1 activation. While these disorders result in constitutive activation of inflammatory pathways, patients have periods of relapse and remission suggesting that there are important activation triggers which are largely not well understood.
HLA-B27 ASSOCIATED DISORDERS
HLA-B27 associated disorders include HLA-B27 related uveitis and members of the spondyloarthritides which include reactive arthritis, psoriatic arthritis, and ankylosing spondylitis (AS). The spondyloarthritides are characterized by axial spine, peripheral joint, and enthesial inflammation in addition to extra-articular manifestations including uveitis, psoriasis, aortic root inflammation, and gut inflammation19. The association with HLA-B27 is as high as over 90% of patients in the United Kingdom with AS20 although not all subtypes are associated with disease21. Other HLA-B27 associations include reactive arthritis (67%)22, IBD-associated spondyloarthritis (72%)23, psoriatic arthritis – both peripheral and axial (24–90%)23, juvenile enthesitis-related arthritis (76%)24, and acute anterior uveitis (52%)25.
The major histocompatibility complex (MHC) is located on chromosome 6 and encodes the human leukocyte antigens (HLAs). These complexes are responsible for antigen presentation to the immune system with class I molecules presenting antigen primarily to CD8+ T cells. MHC molecules are formed in the endoplasmic reticulum and then travel to the cellular surface after proper assembly. The HLA-B27 structure includes a groove which binds arginine at the second position of bound peptide which may be important in directing the bound antigen to immune cells26. The proportion of individuals carrying HLA-B27 varies widely but is approximately 7–8% in Caucasians and as low as 2% in African Americans27 although only a minority of carriers develop a HLA-B27 related disease.
HLA-B27 PATHOGENIC MECHANISMS : AUTOIMMUNE OR AUTOINFLAMMATORY?
Despite the clearly recognized association of HLA-B27 with spondyloarthritis, the mechanism by which disease occurs remains incompletely understood. The robust association with HLA-B27, an antigen presenting complex, is the strongest argument that adaptive immunity is important in these diseases. However, mounting evidence suggests that the role of HLA-B27 may not be so simple and that it may in fact activate innate immune mechanisms. Three main theories to explain how HLA-B27 causes disease have been proposed: the arthrogenic peptide, homodimerization, and endoplasmic reticulum (ER) misfolding theories.
HLA-B27 TRANSGENIC RAT MODEL
The HLA-B27 transgenic rat model has been critical in understanding the pathogenesis of HLA-B27 related diseases28,29. In this model, HLA-B27 is overexpressed together with varying amounts of human β2-microglobulin resulting in bowel inflammation, peripheral arthritis, and spondyloarthritis. Interesingly, additional overexpression of β2-microglobulin results in more severe arthritis and is necessary for the development of high frequency spondyoarthritis29. Through this model, the three primary theories for pathogenesis have been studied.
ARTHROGENIC PEPTIDE THEORY
One of the earliest theories on how HLA-B27 causes disease is the “arthrogenic peptide” hypothesis. Given the role of MHC I in antigen presentation, was proposed that HLA-B27 presents a specific peptide to the immune system which then results in CD8+ T cells activation30. However, the arthrogenic peptide theory has fallen out of favor when it was found that CD8+ T cells are not in fact necessary for the disease phenotype in the HLA-B27 transgenic rat model31,32. This suggests that other mechanisms may be more important in disease pathogenesis and that HLA-B27 does not likely exert its effect by antigen presentation alone.
An additional argument challenging the arthrogenic peptide theory has been that despite investigations, no specific peptide(s) targeted by autoreative T cells have been identified. Even autoantibodies have not been reproducibly found in various forms of spondyloarthritis. However, this has been challenged recently with reports of autoantibodies to class II-associated invariant chain peptide (CLIP) in 85% of patients with axial spondyloarthrtitis compared to 8% of controls 33 and higher amounts of immune complexes containing anti-noggin and antisclerostin antibodies in patients with ankylosing spondylitis when compared to controls34. The significance and reproducibility of both of these findings remains to be determined.
HOMODIMERIZATION THEORY
HLA-B27 molecules are unusual in that they can form homodimers on the surface of cells35. Based on this finding, an alternate theory was proposed that these homodimers are recognized by the immune system resulting in activation and inflammation. Innate immune receptors on both natural killer and T cells, KIRs and LILRs, respectively, recognize HLA class I monomers as well as some homodimers resulting in cytokine production36. Further supporting this theory is that KIR genetic polymorphisms have been associated with AS in some populations37,38 although notably, this has not been found consistently 39. While this theory supports a role for innate immunity in disease pathogenesis through recognition of homodimers by KIRs and/or LILRs, it is unlikely that this is the sole mechanism for pathogenesis as homodimerization is not unique to disease associated HLA-B27 variants or even to HLA-B27 itself40.
ER MISFOLDING THEORY
An alternate theory was proposed after recognition that HLA-B27 misfolds in the endoplasmic reticulum (ER) causing some heavy chains to undergo ER-associated degradation41. In HLA-B27 transgenic rats, this occurs in bone marrow macrophages following cytokine stimulation, is independent of antigen presentation, and is associated with the degree of HLA-B27 upregulation42,43. The net effect is ER stress resulting in IL-2343, IFN-β44, and IL-1α45 production. Attempts to reproduce these experiments in humans have yielded mixed results. One study found upregulation of the UPR target gene Hspa5/BiP in synovial macrophages46 and there are conflicting reports regarding the role of the UPR in peripheral blood mononuclear cells47,48.
HUMAN GENETICS
Genome-wide association studies (GWAS) allow for the examination of thousands of polymorphisms in the human genome and their association with the presence of disease. These studies have provided for powerful, cost-effective, unbiased studies of the genetic predisposition to disease shedding light on the pathogenic mechanisms.
Several GWAS have been completed in ankylosing spondylitis, psoriatic arthritis, and anterior uveitis confirming the polygenic nature of these diseases49–57. Given that these are B27 related diseases, not unexpectedly the region consistently most closely associated with disease is that of the MHC locus. However, HLA-B27 is responsible for only a minority of the genetically attributable risk in twin studies of ankylosing spondylitis indicating that other genetic factors are essential58. Perhaps more surprising is that many of these other associated variants have importance in innate as well as adaptive immunity.
In support of a role of adaptive immunity, ERAP1 is associated with AS50 and is also associated with acute anterior uveitis53. ERAP1 is important in peptide trimming prior to MHC loading which suggests a role for antigen presentation. However, it is also possible that peptide processing by ERAP1 is important for proper folding of HLA-B27 and that ERAP1 variants impact protein misfolding independent of antigen presentation. Other adaptive immune genes implicated in psoriatic arthritis include TRAF3IP2 encoding ACT154, important in the regulation of B cell signaling, and PTPN22, important in the regulation of T cell activation59. Shared to AS and psoriatic arthritis is the association with RUNX3 important in CD8+ lymphocyte activation60.
IL-23 has a role in both innate and adaptive immunity and several genes in the IL-23/IL-17 pathway have shown associations with HLA-B27 diseases including IL-23R, IL-23A, IL-12B, TYK2, and STAT350,52,61. This fits well with the known role of IL-23 in the development of these diseases (to be discussed)
However, several other variants have a more prominent role in innate immunity. These include those important in NFκB signaling in psoriatic arthritis including TNIP1, REL and TYK262 and in ankylosing spondylitis including TRADD, TKBP1, TYK2, and CARD949–52. CARD9 in ankylosing spondylitis is of particular interest given its known role in the innate immune response by mediating signals through Dectin-1, a pattern recognition protein. Similarly, in acute anterior uveitis, variants in a gene known to be important in innate immunity, IL18R1, are associated with disease53.
IMPORTANCE OF THE MICROBIOME IN HLA-B27 DISEASES
The importance of the microbiome, the collection of bacteria on and within our bodies, is becoming increasingly appreciated in many complex diseases including those of HLA-B27 related disorders63. The interplay between the innate immune system and the microbiome may be an important in both initiation and propagation of inflammation. The microbiome is critical in educating the immune system and in a pathogenic state, may be responsible for immune activation at distant sites.
One of the strongest lines of evidence that the microbiome is important in disease pathogenesis comes again from the HLA-B27 rat model. When raised in germ free conditions, bowel and joint inflammation is much reduced indicating that bacterial colonization of the gut is necessary for the disease phenotype64. Re-introduction of normal flora, specifically Bacteroides, results in the return of inflammation65.
The mechanisms by which bacteria are associated with B27-related diseases remain unclear but an active area of research. An attractive hypothesis is that HLA-B27 may shape the gut microbiome or alter the way that antigen is presented to an individual. In fact, 16S sequencing revealed differences between HLA-B27 rats when compared to control in cecal bacterial composition with an increase in Prevotella and a decrease in Rikenellaceae relative abundance in the transgenic animals compared to the wild type animals66. This is beginning to be studied in humans where differences in terminal ileum microbiota are seen in patients with ankylosing spondylitis when compared to controls67.
These findings are not surprising as it is well established that microbes may play a role in the development of certain forms of human spondyloarthritis. The clearest example of this is reactive arthritis (formerly Reiter’s syndrome), where peripheral arthritis and/or spondyloarthritis can occur following an infection, most typically Chlamydia or enteric infections68. Similarly, Yersenia and Salmonella bacterial products have been observed in reactive arthritis joints further supporting the role of bacteria in the development of disease69,70. Similar associations have been proposed in ankylosing spondylitis where colonization with Klebsiella was found more often in active AS when compared to controls71 although these results have not been reproduced.
Moreover, sub-clinical gastrointestinal inflammation has been demonstrated in the absence of symptoms in 50% of patients with HLA-B27 related arthritis72. The presence of a “leaky gut” due to mucosal barrier breakdown may then result in the interaction of gut bacteria with innate immune cells serving as an inciter of the inflammatory cascade.
IL-17/IL-23
The importance of IL-17 and IL-23 in the development of several B27-related diseases is well proven and may provide a link between innate and adaptive immunity. IL-23 is produced primarily by macrophages and dendritic cells, both innate immune cells73. These cells are active at mucosal surfaces and are produced by the gut, an area known to be inflamed in spondyloarthritis and psoriatic arthritis74. In AS, macrophages produce increased levels of IL-23 compared to controls 75 and IL-23 is found in high levels in spinal facet joints76 as well as the gut77 of spondyloarthritis patients. IL-23 is also found in higher levels in lesional compared to non-lesional skin in psoriasis78 and is associated with keratinocyte growth in mouse models of psoriasis79. IL-23 responsive cells are then activated, including innate cells such as IL-23R+ γδ T cells, which are found to be in higher levels in AS patients with active disease80. IL-23 then activates cells producing IL-17A and IL-17F which is supported by the higher proportion of circulating Th17 cells in patients with AS compared to controls81. This is also true of psoriasis and psoriatic arthritis where a higher number of Th17 cells are observed in lesional vs. non-lesional psoriatic skin82 and higher numbers of IL-17 producing CD4+ effector memory cells in psoriatic arthritis synovium compared to osteoarthritis synovium83. Further clinical data suggest that blockade of these pathways is highly effective in these diseases (to be discussed).
INNATE IMMUNE-LIKE CELLS IN ENTHESES
While the importance of IL-23 in HLA-B27 diseases is established, how this results in disease pathology is less well understood. A major advance occurred when a novel IL-23 responsive cell type residing in the entheses and aortic root was described in a mouse model of spondyloarthritis 84. A novel IL-23R+CD3+CD4−CD8− lymphocyte cell was described which was found to reside in the entheses and aortic root. These cells had innate immune characteristics in that IL-23 alone – without antigen recognition, other cytokines, or other inflammatory cells - was sufficient to induce inflammation and cytokine expression including IL-6, IL-17, IL-22, and CXCL1. Systemic expression of IL-23 resulted in an inflammatory infiltrate of macrophages and neutrophils focused in the entheses, periosteum, and aortic root with resultant new enthesial bone formation. This was not reduced by Th17 or CD4 depletion suggesting that adaptive immunity is not essential for the phenotype. Moreover, the clinical phenotype could be abrogated by IL-23 blockade. Based on these observations, it has been proposed that IL-23 may be produced at distant sites, such as the gut, and that this then results in enthesial and aortic root inflammation by activation of resident cells with innate features.
Recently, IL-23 responsive innate lymphocyte cells producing IL-17 and IL-22 were described in the gut, peripheral blood, synovial fluid, and bone marrow from patients with ankylosing spondylitis lending support to the idea that a similar phenomenon may be relevant in humans85. These cells have a different phenotype than those described in the mouse model however result in similar inflammatory cytokine production.
CLINICAL TRIALS
While animal and pre-clinical data suggest a role for inflammatory pathways in the development of disease, the ultimate proof of this principal lies in the response to therapeutic blockade. The role of adaptive immunity has been tested through studies of both rituximab (pre-B cell blockade)86,87 and abatacept (co-stimulation blockade)88. Both of these pathways are important in adaptive immunity, yet blockade in spondyloarthritis yielded disappointing results.
Mounting evidence supports that IL-23 and IL-17 blockade is effective in many of the B27 related diseases supporting the critical role of this pathway in disease pathogenesis. The first studies of IL-17 and IL-23 blockade were in psoriasis where overwhelmingly positive results were achieved89,90. These drugs were then studied in psoriatic arthritis where similarly positive results were found although the results were less robust91–93. They have been studied more recently in ankylosing spondylitis where positive results are also emerging94,95. Of great interest will be whether they are capable of blocking new bone formation to a greater extent than the most proven therapies, TNF inhibitors. Interestingly, similar results were not found with IL-17 blockade in noninfectious uveitis where three randomized studies failed to show benefit in reducing uveitis recurrence96.
A good response to IL-1 blockade is frequently used as clinical evidence for a prominent role of autoinflammation in disease pathogenesis97. A single study was performed with anakinra, the recombinant IL-1 receptor antagonist, in ankylosing spondylitis. In this open label study of a relatively small number of patients, only a minority of patients reached the primary endpoint of an ASAS20 response which is disappointing in comparison to the results of TNF inhibitors98. No changes were observed in objective measures of inflammation including acute phase reactants or MRI scores however the study was not sufficiently long to study the effects of IL-1 blockade on structural damage. The dose of anakinra was not escalated so it is not clear if patients would have responded to a higher dose which is often necessary in autoinflammatory disorders.
Less is known about the role of IL-1 blockade in uveitis. Animal studies show that mice lacking the IL-1 receptor antagonist develop a particularly severe uveitis in an intraocular LPS-induced mouse model 99. A large multi-center study is underway to test if IL-1 blockade with a long acting IL-1β blocking monoclonal antibody, gevokizumab, is effective in non-infectious uveitis100. While patients are not strictly HLA-B27+, this may lend insight into the role of IL-1 within the eye as this may differ from the effects of other organs in HLA-B27 related diseases.
CONCLUSIONS
HLA-B27 related diseases share many features with classic autoinflammatory diseases and mounting data suggests that autoinflammation is a key component of disease pathogenesis. However, ample data also exist to support the role of the adaptive immune system. The division between the innate and adaptive immune system is artificial as it is neither works in isolation and cross-talk is well described. The relative contribution of innate and adaptive immune mechanisms is under active investigation and further understanding of the role of autoinflammation in these disorders may lead to novel therapeutic directions.
Table 1.
Shared Features of Autoinflammatory, HLA-B27 Associated, and Autoimmune Diseases
| Feature | Autoinflammatory Diseases | HLA-B27 Associated Diseases | Autoimmune Diseases |
|---|---|---|---|
| Age of onset | Very young | Young | Varies |
| Sex | F = M | M > F | F > M |
| Fevers | Prominent feature | Rare | Rare |
| Disease course | Flares and remits | Flares and remits | Continuous and progressive |
| Ocular findings | Uveitis, conjunctivitis | Uveitis, conjunctivitis | Varied, scleritis, episcleritis, uveitis less common |
| Sacroiliitis | Present but not common | Prominent feature | Rare |
| Acute phase reactants | Greatly elevated | Typically normal | Elevated |
| Presence of autoantibodies | No | No | Yes |
| Histology | Neutrophils | Neutrophils and mixed inflammatory cells | Mixed inflammatory cells |
| Genetics | Typically monogenic mutations in innate pathways | Polygenic: MHC-I, ERAP1, IL-23/IL-17 | Polygenic: MHC-II, PTPN22, CTLA4, TNFAIP3 |
| Effective Treatments | Cytokine blockade, not B-cell blockade, not corticosteroids | Cytokine blockade, not B-cell blockade, not corticosteroids | Cytokine blockade, B-cell blockade, corticosteroids |
Acknowledgments
The author would like to thank Dr. Robert Colbert for his suggestions and critical review of this manuscript.
References
- 1.McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999 Apr 2;97(1):133–144. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 2.Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011 May;41(5):1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 3.Matzinger P. Tolerance, danger, and the extended family. Annual review of immunology. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 4.Pradeu T, Cooper EL. The danger theory: 20 years later. Frontiers in immunology. 2012;3:287. doi: 10.3389/fimmu.2012.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (New York, NY) 2010 Jan 15;327(5963):291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: Signal 0s that Spur Autophagy and Immunity. Immunological Reviews. 2012 Sep;249(1):158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annual review of immunology. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS medicine. 2006 Aug;3(8):e297. doi: 10.1371/journal.pmed.0030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001 Nov;29(3):301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002 Dec;46(12):3340–3348. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson C, Goldbach-Mansky R. Monogenic autoinflammatory diseases: new insights into clinical aspects and pathogenesis. Curr Opin Rheumatol. 2010 Sep;22(5):567–578. doi: 10.1097/BOR.0b013e32833ceff4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibley CH, Plass N, Snow J, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease (NOMID) treated with anakinra. Arthritis Rheum. 2012 Jan 31; doi: 10.1002/art.34409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neven B, Marvillet I, Terrada C, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010 Jan;62(1):258–267. doi: 10.1002/art.25057. [DOI] [PubMed] [Google Scholar]
- 14.Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell. 1997 Aug 22;90(4):797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 15.A candidate gene for familial Mediterranean fever. Nat Genet. 1997 Sep;17(1):25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 16.Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT. A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clinic Proceedings. 1997 Jul;72(7):611–615. doi: 10.1016/S0025-6196(11)63565-9. [DOI] [PubMed] [Google Scholar]
- 17.Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009 Jun 4;360(23):2426–2437. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009 Jun 4;360(23):2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stolwijk C, Essers I, van Tubergen A, et al. The epidemiology of extra-articular manifestations in ankylosing spondylitis: a population-based matched cohort study. Annals of the rheumatic diseases. 2015 Jul;74(7):1373–1378. doi: 10.1136/annrheumdis-2014-205253. [DOI] [PubMed] [Google Scholar]
- 20.Brown MA, Pile KD, Kennedy LG, et al. HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Annals of the rheumatic diseases. 1996 Apr;55(4):268–270. doi: 10.1136/ard.55.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan MA. Polymorphism of HLA-B27: 105 subtypes currently known. Current rheumatology reports. 2013 Oct;15(10):362. doi: 10.1007/s11926-013-0362-y. [DOI] [PubMed] [Google Scholar]
- 22.Brewerton DA, Caffrey M, Nicholls A, Walters D, Oates JK, James DC. Reiter’s disease and HL-A 27. Lancet. 1973 Nov 3;302(7836):996–998. doi: 10.1016/s0140-6736(73)91091-x. [DOI] [PubMed] [Google Scholar]
- 23.Brewerton DA, Caffrey M, Nicholls A, Walters D, James DC. HL-A 27 and arthropathies associated with ulcerative colitis and psoriasis. Lancet. 1974 May 18;1(7864):956–958. doi: 10.1016/s0140-6736(74)91262-8. [DOI] [PubMed] [Google Scholar]
- 24.Thomson W, Barrett JH, Donn R, et al. Juvenile idiopathic arthritis classified by the ILAR criteria: HLA associations in UK patients. Rheumatology (Oxford) 2002 Oct;41(10):1183–1189. doi: 10.1093/rheumatology/41.10.1183. [DOI] [PubMed] [Google Scholar]
- 25.Brewerton DA, Caffrey M, Nicholls A, Walters D, James DC. Acute anterior uveitis and HL-A 27. Lancet. 1973 Nov 3;302(7836):994–996. doi: 10.1016/s0140-6736(73)91090-8. [DOI] [PubMed] [Google Scholar]
- 26.Madden DR, Gorga JC, Strominger JL, Wiley DC. The three-dimensional structure of HLA-B27 at 2.1 A resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 1992 Sep 18;70(6):1035–1048. doi: 10.1016/0092-8674(92)90252-8. [DOI] [PubMed] [Google Scholar]
- 27.Piazza A, Menozzi P, Cavalli-Sforza LL. The HLA-A,B gene frequencies in the world: migration or selection? Human immunology. 1980 Dec;1(4):297–304. doi: 10.1016/0198-8859(80)90105-6. [DOI] [PubMed] [Google Scholar]
- 28.Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 1990 Nov 30;63(5):1099–1112. doi: 10.1016/0092-8674(90)90512-d. [DOI] [PubMed] [Google Scholar]
- 29.Tran TM, Dorris ML, Satumtira N, et al. Additional human beta2-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum. 2006 Apr;54(4):1317–1327. doi: 10.1002/art.21740. [DOI] [PubMed] [Google Scholar]
- 30.Hermann E, Yu DT, Meyer zum Buschenfelde KH, Fleischer B. HLA-B27-restricted CD8 T cells derived from synovial fluids of patients with reactive arthritis and ankylosing spondylitis. Lancet. 1993 Sep 11;342(8872):646–650. doi: 10.1016/0140-6736(93)91760-j. [DOI] [PubMed] [Google Scholar]
- 31.May E, Dorris ML, Satumtira N, et al. CD8 alpha beta T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. Journal of immunology (Baltimore, Md : 1950) 2003 Jan 15;170(2):1099–1105. doi: 10.4049/jimmunol.170.2.1099. [DOI] [PubMed] [Google Scholar]
- 32.Taurog JD, Dorris ML, Satumtira N, et al. Spondylarthritis in HLA-B27/human beta2-microglobulin-transgenic rats is not prevented by lack of CD8. Arthritis Rheum. 2009 Jul;60(7):1977–1984. doi: 10.1002/art.24599. [DOI] [PubMed] [Google Scholar]
- 33.Baraliakos X, Baerlecken N, Witte T, Heldmann F, Braun J. High prevalence of anti-CD74 antibodies specific for the HLA class II-associated invariant chain peptide (CLIP) in patients with axial spondyloarthritis. Annals of the rheumatic diseases. 2014 Jun;73(6):1079–1082. doi: 10.1136/annrheumdis-2012-202177. [DOI] [PubMed] [Google Scholar]
- 34.Tsui FW, Tsui HW, Las Heras F, Pritzker KP, Inman RD. Serum levels of novel noggin and sclerostin-immune complexes are elevated in ankylosing spondylitis. Annals of the rheumatic diseases. 2014 Oct;73(10):1873–1879. doi: 10.1136/annrheumdis-2013-203630. [DOI] [PubMed] [Google Scholar]
- 35.Allen RL, O’Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. Journal of immunology (Baltimore, Md : 1950) 1999 May 1;162(9):5045–5048. [PubMed] [Google Scholar]
- 36.Kollnberger S, Bowness P. The role of B27 heavy chain dimer immune receptor interactions in spondyloarthritis. Advances in experimental medicine and biology. 2009;649:277–285. doi: 10.1007/978-1-4419-0298-6_21. [DOI] [PubMed] [Google Scholar]
- 37.Jiao YL, Zhang BC, You L, et al. Polymorphisms of KIR gene and HLA-C alleles: possible association with susceptibility to HLA-B27-positive patients with ankylosing spondylitis. Journal of clinical immunology. 2010 Nov;30(6):840–844. doi: 10.1007/s10875-010-9444-z. [DOI] [PubMed] [Google Scholar]
- 38.Diaz-Pena R, Vidal-Castineira JR, Alonso-Arias R, et al. Association of the KIR3DS1*013 and KIR3DL1*004 alleles with susceptibility to ankylosing spondylitis. Arthritis Rheum. 2010 Apr;62(4):1000–1006. doi: 10.1002/art.27332. [DOI] [PubMed] [Google Scholar]
- 39.Fan D, Liu S, Yang T, et al. Association between KIR polymorphisms and ankylosing spondylitis in populations: a meta-analysis. Modern rheumatology/the Japan Rheumatism Association. 2014 Nov;24(6):985–991. doi: 10.3109/14397595.2014.894489. [DOI] [PubMed] [Google Scholar]
- 40.Ambarus C, Yeremenko N, Tak PP, Baeten D. Pathogenesis of spondyloarthritis: autoimmune or autoinflammatory? Curr Opin Rheumatol. 2012 Jul;24(4):351–358. doi: 10.1097/BOR.0b013e3283534df4. [DOI] [PubMed] [Google Scholar]
- 41.Mear JP, Schreiber KL, Munz C, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. Journal of immunology (Baltimore, Md : 1950) 1999 Dec 15;163(12):6665–6670. [PubMed] [Google Scholar]
- 42.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: Implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007 Jan;56(1):215–223. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 43.DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009 Sep;60(9):2633–2643. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008 May;38(5):1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Layh-Schmitt G, Yang EY, Kwon G, Colbert RA. HLA-B27 alters the response to tumor necrosis factor alpha and promotes osteoclastogenesis in bone marrow monocytes from HLA-B27-transgenic rats. Arthritis Rheum. 2013 Aug;65(8):2123–2131. doi: 10.1002/art.38001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu J, Rihl M, Marker-Hermann E, et al. Clues to pathogenesis of spondyloarthropathy derived from synovial fluid mononuclear cell gene expression profiles. J Rheumatol. 2002 Oct;29(10):2159–2164. [PubMed] [Google Scholar]
- 47.Feng Y, Ding J, Fan CM, Zhu P. Interferon-gamma contributes to HLA-B27-associated unfolded protein response in spondyloarthropathies. J Rheumatol. 2012 Mar;39(3):574–582. doi: 10.3899/jrheum.101257. [DOI] [PubMed] [Google Scholar]
- 48.Neerinckx B, Carter S, Lories RJ. No evidence for a critical role of the unfolded protein response in synovium and blood of patients with ankylosing spondylitis. Annals of the rheumatic diseases. 2014 Mar;73(3):629–630. doi: 10.1136/annrheumdis-2013-204170. [DOI] [PubMed] [Google Scholar]
- 49.Reveille JD, Sims AM, Danoy P, et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. 2010 Feb;42(2):123–127. doi: 10.1038/ng.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans DM, Spencer CCA, Pointon JJ, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 43(8):761–767. doi: 10.1038/ng.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cortes A, Hadler J, Pointon JP, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013 Jul;45(7):730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Association scan of 14,500 nsSNPs in four common diseases identifies variants involved in autoimmunity. Nat Genet. 2007 Nov;39(11):1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson PC, Claushuis TA, Cortes A, et al. Genetic dissection of acute anterior uveitis reveals similarities and differences in associations observed with ankylosing spondylitis. Arthritis & rheumatology (Hoboken, NJ) 2015 Jan;67(1):140–151. doi: 10.1002/art.38873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hüffmeier U, Uebe S, Ekici AB, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet. 2010 Nov;42(11):996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuart PE, Nair RP, Ellinghaus E, et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat Genet. 2010 Nov;42(11):1000–1004. doi: 10.1038/ng.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Helms C, Liao W, et al. A Genome-Wide Association Study of Psoriasis and Psoriatic Arthritis Identifies New Disease Loci. PLoS Genetics. 2008 Apr;4(4) doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowes J, Budu-Aggrey A, Huffmeier U, et al. Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nature Communications. 2015;6:6046. doi: 10.1038/ncomms7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown MA, Kennedy LG, MacGregor AJ, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum. 1997 Oct;40(10):1823–1828. doi: 10.1002/art.1780401015. [DOI] [PubMed] [Google Scholar]
- 59.Bowes J, Loehr S, Budu-Aggrey A, et al. PTPN22 is associated with susceptibility to psoriatic arthritis but not psoriasis: evidence for a further PsA-specific risk locus. Annals of the rheumatic diseases. 2015 Oct;74(10):1882–1885. doi: 10.1136/annrheumdis-2014-207187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Apel M, Uebe S, Bowes J, et al. Variants in RUNX3 contribute to susceptibility to psoriatic arthritis, exhibiting further common ground with ankylosing spondylitis. Arthritis Rheum. 2013 May;65(5):1224–1231. doi: 10.1002/art.37885. [DOI] [PubMed] [Google Scholar]
- 61.Smith JA. Update on ankylosing spondylitis: current concepts in pathogenesis. Current allergy and asthma reports. 2015 Jan;15(1):489. doi: 10.1007/s11882-014-0489-6. [DOI] [PubMed] [Google Scholar]
- 62.Bowes J, Budu-Aggrey A, Huffmeier U, et al. Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nature Communications. :6. doi: 10.1038/ncomms7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gill T, Asquith M, Rosenbaum JT, Colbert RA. The intestinal microbiome in spondyloarthritis. Curr Opin Rheumatol. 2015 Jul;27(4):319–325. doi: 10.1097/BOR.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taurog JD, Richardson JA, Croft JT, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. The Journal of experimental medicine. 1994 Dec 1;180(6):2359–2364. doi: 10.1084/jem.180.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rath HC, Herfarth HH, Ikeda JS, et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. The Journal of clinical investigation. 1996 Aug 15;98(4):945–953. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin P, Bach M, Asquith M, et al. HLA-B27 and human beta2-microglobulin affect the gut microbiota of transgenic rats. PloS one. 2014;9(8):e105684. doi: 10.1371/journal.pone.0105684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Costello ME, Ciccia F, Willner D, et al. Intestinal dysbiosis in ankylosing spondylitis. Arthritis & rheumatology (Hoboken, NJ) 2014 Nov 21; doi: 10.1002/art.38967. [DOI] [PubMed] [Google Scholar]
- 68.Kvien TK, Glennas A, Melby K, et al. Reactive arthritis: incidence, triggering agents and clinical presentation. J Rheumatol. 1994 Jan;21(1):115–122. [PubMed] [Google Scholar]
- 69.Merilahti-Palo R, Soderstrom KO, Lahesmaa-Rantala R, Granfors K, Toivanen A. Bacterial antigens in synovial biopsy specimens in yersinia triggered reactive arthritis. Annals of the rheumatic diseases. 1991 Feb;50(2):87–90. doi: 10.1136/ard.50.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Granfors K, Jalkanen S, Lindberg AA, et al. Salmonella lipopolysaccharide in synovial cells from patients with reactive arthritis. Lancet. 1990 Mar 24;335(8691):685–688. doi: 10.1016/0140-6736(90)90804-e. [DOI] [PubMed] [Google Scholar]
- 71.Ebringer R, Cooke D, Cawdell DR, Cowling P, Ebringer A. Ankylosing spondylitis: klebsiella and HL-A B27. Rheumatology and rehabilitation. 1977 Aug;16(3):190–196. doi: 10.1093/rheumatology/16.3.190. [DOI] [PubMed] [Google Scholar]
- 72.Mielants H, Veys EM, Cuvelier C, De Vos M, Botelberghe L. HLA-B27 related arthritis and bowel inflammation. Part 2. Ileocolonoscopy and bowel histology in patients with HLA-B27 related arthritis. J Rheumatol. 1985 Apr;12(2):294–298. [PubMed] [Google Scholar]
- 73.Yeremenko N, Paramarta JE, Baeten D. The interleukin-23/interleukin-17 immune axis as a promising new target in the treatment of spondyloarthritis. Curr Opin Rheumatol. 2014 Jul;26(4):361–370. doi: 10.1097/BOR.0000000000000069. [DOI] [PubMed] [Google Scholar]
- 74.Becker C, Wirtz S, Blessing M, et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. The Journal of clinical investigation. 2003 Sep;112(5):693–706. doi: 10.1172/JCI17464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeng L, Lindstrom MJ, Smith JA. Ankylosing spondylitis macrophage production of higher levels of interleukin-23 in response to lipopolysaccharide without induction of a significant unfolded protein response. Arthritis Rheum. 2011 Dec;63(12):3807–3817. doi: 10.1002/art.30593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Appel H, Maier R, Bleil J, et al. In situ analysis of interleukin-23- and interleukin-12-positive cells in the spine of patients with ankylosing spondylitis. Arthritis Rheum. 2013 Jun;65(6):1522–1529. doi: 10.1002/art.37937. [DOI] [PubMed] [Google Scholar]
- 77.Ciccia F, Bombardieri M, Principato A, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009 Apr;60(4):955–965. doi: 10.1002/art.24389. [DOI] [PubMed] [Google Scholar]
- 78.Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. The Journal of experimental medicine. 2004 Jan 5;199(1):125–130. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kopp T, Lenz P, Bello-Fernandez C, Kastelein RA, Kupper TS, Stingl G. IL-23 production by cosecretion of endogenous p19 and transgenic p40 in keratin 14/p40 transgenic mice: evidence for enhanced cutaneous immunity. Journal of immunology (Baltimore, Md : 1950) 2003 Jun 1;170(11):5438–5444. doi: 10.4049/jimmunol.170.11.5438. [DOI] [PubMed] [Google Scholar]
- 80.Kenna TJ, Davidson SI, Duan R, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012 May;64(5):1420–1429. doi: 10.1002/art.33507. [DOI] [PubMed] [Google Scholar]
- 81.Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. 2008 Aug;58(8):2307–2317. doi: 10.1002/art.23655. [DOI] [PubMed] [Google Scholar]
- 82.Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. The Journal of Investigative Dermatology. 2008 May;128(5):1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 83.Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Molecular and cellular biochemistry. 2012 Jan;359(1–2):419–429. doi: 10.1007/s11010-011-1036-6. [DOI] [PubMed] [Google Scholar]
- 84.Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4−CD8− entheseal resident T cells. Nature medicine. 2012 Jul;18(7):1069–1076. doi: 10.1038/nm.2817. [DOI] [PubMed] [Google Scholar]
- 85.Ciccia F, Guggino G, Rizzo A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Annals of the rheumatic diseases. 2015 Sep;74(9):1739–1747. doi: 10.1136/annrheumdis-2014-206323. [DOI] [PubMed] [Google Scholar]
- 86.Wendling D, Dougados M, Berenbaum F, et al. Rituximab treatment for spondyloarthritis. A nationwide series: data from the AIR registry of the French Society of Rheumatology. J Rheumatol. 2012 Dec;39(12):2327–2331. doi: 10.3899/jrheum.120201. [DOI] [PubMed] [Google Scholar]
- 87.Song IH, Heldmann F, Rudwaleit M, et al. Different response to rituximab in tumor necrosis factor blocker-naive patients with active ankylosing spondylitis and in patients in whom tumor necrosis factor blockers have failed: a twenty-four-week clinical trial. Arthritis Rheum. 2010 May;62(5):1290–1297. doi: 10.1002/art.27383. [DOI] [PubMed] [Google Scholar]
- 88.Song IH, Heldmann F, Rudwaleit M, et al. Treatment of active ankylosing spondylitis with abatacept: an open-label, 24-week pilot study. Annals of the rheumatic diseases. 2011 Jun;70(6):1108–1110. doi: 10.1136/ard.2010.145946. [DOI] [PubMed] [Google Scholar]
- 89.Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med. 2014 Jul 24;371(4):326–338. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 90.Griffiths CE, Strober BE, van de Kerkhof P, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010 Jan 14;362(2):118–128. doi: 10.1056/NEJMoa0810652. [DOI] [PubMed] [Google Scholar]
- 91.McInnes IB, Kavanaugh A, Gottlieb AB, et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. 2013 Aug 31;382(9894):780–789. doi: 10.1016/S0140-6736(13)60594-2. [DOI] [PubMed] [Google Scholar]
- 92.Ritchlin C, Rahman P, Kavanaugh A, et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Annals of the rheumatic diseases. 2014 Jun;73(6):990–999. doi: 10.1136/annrheumdis-2013-204655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McInnes IB, Mease PJ, Kirkham B, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015 Jun 26; doi: 10.1016/S0140-6736(15)61134-5. [DOI] [PubMed] [Google Scholar]
- 94.Baeten D, Baraliakos X, Braun J, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013 Nov 23;382(9906):1705–1713. doi: 10.1016/S0140-6736(13)61134-4. [DOI] [PubMed] [Google Scholar]
- 95.Poddubnyy D, Hermann KG, Callhoff J, Listing J, Sieper J. Ustekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS) Annals of the rheumatic diseases. 2014 May;73(5):817–823. doi: 10.1136/annrheumdis-2013-204248. [DOI] [PubMed] [Google Scholar]
- 96.Dick AD, Tugal-Tutkun I, Foster S, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology. 2013 Apr;120(4):777–787. doi: 10.1016/j.ophtha.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 97.Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annual review of medicine. 2014;65:223–244. doi: 10.1146/annurev-med-061512-150641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haibel H, Rudwaleit M, Listing J, Sieper J. Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Annals of the rheumatic diseases. 2005 Feb;64(2):296–298. doi: 10.1136/ard.2004.023176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Planck SR, Woods A, Clowers JS, Nicklin MJ, Rosenbaum JT, Rosenzweig HL. Impact of IL-1 signalling on experimental uveitis and arthritis. Annals of the rheumatic diseases. 2012 May 1;71(5):753–760. doi: 10.1136/annrheumdis-2011-200556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maya JR, Sadiq MA, Zapata LJ, et al. Emerging therapies for noninfectious uveitis: what may be coming to the clinics. Journal of ophthalmology. 2014;2014:310329. doi: 10.1155/2014/310329. [DOI] [PMC free article] [PubMed] [Google Scholar]