

Abstract
Reproduction is thought to come at a cost to longevity. Based on the assumption that increased energy expenditure during reproduction is associated with increased free-radical production by mitochondria, oxidative damage has been suggested to drive this trade-off. We examined the impact of reproduction on liver mitochondrial function by utilizing post-reproductive and non-reproductive house mice (Mus musculus) living under semi-natural conditions. The age-matched post-reproductive and non-reproductive groups were compared after the reproductive females returned to a non-reproductive state, so that both groups were in the same physiological state at the time the liver was collected. Despite increased oxidative damage (p = 0.05) and elevated CuZnSOD (p = 0.002) and catalase (p = 0.04) protein levels, reproduction had no negative impacts on the respiratory function of liver mitochondria. Specifically, in a post-reproductive, maintenance state the mitochondrial coupling (i.e., respiratory control ratio) of mouse livers show no negative impacts of reproduction. In fact, there was a trend (p = 0.059) to suggest increased maximal oxygen consumption by liver mitochondria during the ADP stimulated state (i.e., state 3) in post-reproduction. These findings suggest that oxidative damage may not impair mitochondrial respiratory function and question the role of mitochondria in the trade-off between reproduction and longevity. In addition, the findings highlight the importance of quantifying the respiratory function of mitochondria in addition to measuring oxidative damage.
Introduction
The notion that there is a tradeoff between reproductive investment and longevity is a central tenet of biology. The disposable soma theory of aging posits that this relationship arises because the allocation of energy to reproduction detracts from allocation to tissue maintenance [1]. Reproduction increases the demand for ATP produced by mitochondria, organelles that are also responsible for the production of reactive oxygen species (ROS) that damage intracellular lipids, proteins, and DNA [2]. This damage is thought to contribute to the decline in cell and tissue function that occurs with senescence (mitochondrial free radical theory of aging [2]). In this regard, it has been proposed that reproduction increases oxidative damage that may reduce the ability of cells to maintain function, which can negatively affect longevity [3–5]. But support for this assumption is equivocal. Recent reviews found no consistent relationship between reproduction and oxidative damage [6, 7].
Recently, research has shown that the assumption of a linear relationship between ROS production and ATP production was incorrect [7]. Indeed, the speed of electron movement through the electron transport chain (ETC) is tied to the redox state of the complexes. When the rate of oxygen consumption and ATP production slow, the rate of electron flow through the ETC also slows, and the ETC complexes become reduced. In this state, the probability that electrons escape the ETC and be converted to ROS is enhanced [8–10]. Moreover, studies also question whether oxidative damage is responsible for mitochondrial DNA mutations that lead to a decline in respiratory function [11, 12]. Few ecological or evolutionary studies have directly tested the effect of a trait assumed or known to induce oxidative damage, such as reproduction, on mitochondrial respiratory function. Without these data, it should not be assumed that oxidative damage is correlated with a negative impact on the ability of mitochondria to meet the respiratory demands of the cell or tissue being evaluated [13].
During pregnancy and lactation, the body undergoes dramatic physiological changes that are reversed after reproduction. Most investigators compare oxidative damage in reproductive versus non-reproductive animals [as reviewed by [6, 7]], but this approach makes it impossible to distinguish any irreversible costs of reproduction on the ability of tissues to support basic function (maintenance) from the reversible changes that enable reproduction. A more appropriate time to evaluate the impact of reproduction on maintenance is after reproduction, once the body returns to allocating resources to maintenance. While oxidative damage and antioxidants provide a measure of damage and maintenance, measuring mitochondrial respiratory function is critical to understanding the long-term functional impact of reproduction on longevity.
The aim of this study was to examine the impact of reproduction on liver mitochondrial respiratory function and oxidative stress, which is hypothesized to fuel the trade-off between reproduction and longevity. If oxidative damage is responsible for the reproduction-longevity trade-off, post-reproductive animals would display reduced mitochondrial respiratory function associated with accumulated oxidative stress.
Materials and Methods
Animals
We evaluated mitochondrial function in outbred lines of house mice, Mus musculus, founded by WKP [14]. Mice included in this study were 14 generations removed from the wild and housed in enclosures designed to mimic the conditions of mice living in a barn. Each enclosure was designed to match the natural home range (5 m2) and social group size (~ 10 adults) of wild mice [15]. The enclosures were divided between 2 wood buildings that each sit on a concrete slap; the buildings are covered and have hardware-cloth windows that excluded predators but exposed the mice to ambient temperatures.
Experimental design
At sexual maturity (~2 months of age), each population was founded with 3 male and 5–7 female mice. Within the enclosures, mice were allowed to establish a natural social structure and breed at their own pace. Mice were offered ad libitum access to a 10% or 20% protein, isocaloric diet following the design of another project. We found no effect of diet on mitochondrial quality (t17 = 0.08, p = 0.94) and thus excluded diet from further consideration. Due to postpartum estrus, females commonly nurse while gestating their next litter. Pups were removed at 4 week of age to maintain population densities. Males were kept in the enclosures with the females until the females were 10–11 months old. When the females were 10–11 months, the males were removed to ensure that all females were non-reproductive at the time of tissue collection. All females were then euthanized for tissue collection at approximately 1 year of age. This duration allowed females that had recently mated to complete pregnancy (3 weeks), lactation (3 weeks), and allowed reproductive tissues to regress after weaning. Reproductive females produced up to 7 litters.
After founding the populations, the number of mice in each enclosure was reduced over time due to death or injury before the study ended. At the time males were removed, there were 2:5, 3:5, 2:5, 2:5, 1:4,1:6 in the reproductive enclosures and 1:3 and 1:3 (M:F) in the non-reproductive (defined below) enclosures. In two enclosures, pups were never observed during daily checks; we refer to these females as non-reproductive. It is possible that some females experienced gestation and resorbed their fetuses or produced litters that were lost within the first 24 hours (between daily checks) due to cannibalism, but none were observed lactating. Because both of these enclosures ultimately had one male and several females, we speculate that the single dominant male in both enclosures was sterile. The body condition of these females was good and the condition of the non-reproductive females did not appear to differ from females in the other enclosures.
At 1 year of age, females were anesthetized with an overdose of isoflurane and then decapitated. Because mice were collected simultaneously from 1 or 2 enclosures and we could not measure respiratory function of mitochondria from more than 4 mice simultaneously, mitochondria were isolated from the livers of 2 to 4 randomly selected female mice per enclosure, resulting in a sample size of 15 post-reproductive and 4 non-reproductive females.
All animal procedures and housing conditions described herein were approved by the Auburn University IACUC (PRN# 2102–2104).
Isolated mitochondria isolation and measurements
Immediately following euthanasia, the liver was removed from each mouse and mitochondria were isolated as detailed in Sewell et al [16]. Briefly, livers were weighed and put into 10 volumes of a solution made up of 250 mM sucrose, 5 mM HEPES, and 1 mM EGTA and minced with scissors. This minced tissue was further homogenized with a Potter-Elvhjem PTFE pestle and glass tube. The resulting homogenate was centrifuged for 10 min at 500 g at 4°C, pelleting the cellular debris. The supernatant was then decanted through cheesecloth and then centrifuged for 10 min at 3,500 g at 4°C, pelleting the mitochondrial fraction. The supernatant was removed and the pellet resuspended in the sucrose solution. This solution was centrifuged for 10 min at 3,500 g at 4°C, the supernatant discarded and the final mitochondrial pellet suspended in 250 μl of a solution made up of 220 mM mannitol, 70 mM sucrose, 10 mM Tris+HCl, and 1 mM EGTA, at a pH of 7.4 [17]. Isolated mitochondria (20 μL) were incubated in 1 ml of respiration buffer at 37°C and respiration rates were determined polarographically (Oxytherm, Hansatech Instruments, UK) following Messer et al [18] using 2 mM pyruvate and 2 mM malate. The maximal respiration (state 3), defined as the rate of respiration in the presence of ADP was initiated by adding 0.25 mM ADP to the respiration chamber containing mitochondria and respiratory substrates. State 4 respiration was recorded following the phosphorylation of ADP. State 3 and state 4 respiration rates were normalized to mitochondrial protein concentration and expressed as O2/mg mitochondrial protein/min. The respiratory control ratio (RCR) was calculated by dividing state 3 by state 4 [13].
Western blotting
The protein levels of the antioxidants copper-zinc superoxide dismutase (CuZnSOD; GTX100554; GeneTex, Irvine, CA), manganese superoxide dismutase (MnSOD; GTX116093; GeneTex), glutathione peroxidase 1 (GPX-1; GTX116040; GeneTex), catalase (GTX110704; GeneTex), and the marker of lipid peroxidation (4-Hydroxynonenal; 4-HNE; ab46545; Abcam, Cambridge, MA) were measured by Western blotting [19] in isolated liver mitochondria [20, 21]. The protein content of these blots was normalized to α-tubulin (GTX112141; GeneTex) levels (the loading and transfer control) since alpha-tubulin is an inherent component of mitochondrial membranes [22]. This method does not address the purity of the mitochondrial isolation, as α-tubulin is not unique to mitochondria; however, centrifugation at 3,500 g during mitochondrial isolation has been shown to minimize contamination by peroxisomes and other organelles [23]. A chemiluminescent system was used to visualize marked proteins (GE Healthcare Life Sciences, Pittsburgh, PA). Images were taken with the ChemiDocIt Imaging System (UVP, LLC, Upland, CA).
Statistical analysis
Statistical analyses were completed using R version 3.0.3 [24]. Post-reproductive and non-reproductive groups were compared using two-sample t-test. Significance was established at p ≤0.05.
Results
Mitochondrial oxidative phosphorylation
To determine how reproduction affects mitochondrial efficiency, the respiratory control ratio of isolated mitochondria was measured. Specifically, we measured the respiratory function of the mitochondria by evaluating their respiratory control ratio (RCR), a measure of state 3 (maximum metabolic rate) to state 4 (basal metabolic rate) respiration. While alternative measures have been described [25], RCR is thought to be a particularly valuable measure of mitochondrial function because it is responsive essentially to all changes in the functionality of the ETC [13]. RCR of liver mitochondria was similar in post-reproductive compared to non-reproductive females, although there was a trend for higher RCR in post-reproductive mice (t17 = 1.80; p = 0.089; Fig 1A). When state 3 and state 4 respiration were considered independently, the difference in RCR between groups appears to be driven by state 3 respiration, or maximal respiration capacity, as the livers of females that reproduced display state 3 respiration that bordered on higher than non-reproductive controls (t17 = 2.02; p = 0.059; Fig 1B). Furthermore, state 4 respiration, i.e. basal respiratory rate, was similar between the two groups (t17 = 0.602; p = 0.555; Fig 1C).
Fig 1. Liver mitochondria respiratory function for non-reproductive (PL) and post-reproductive (PR) female house mice.
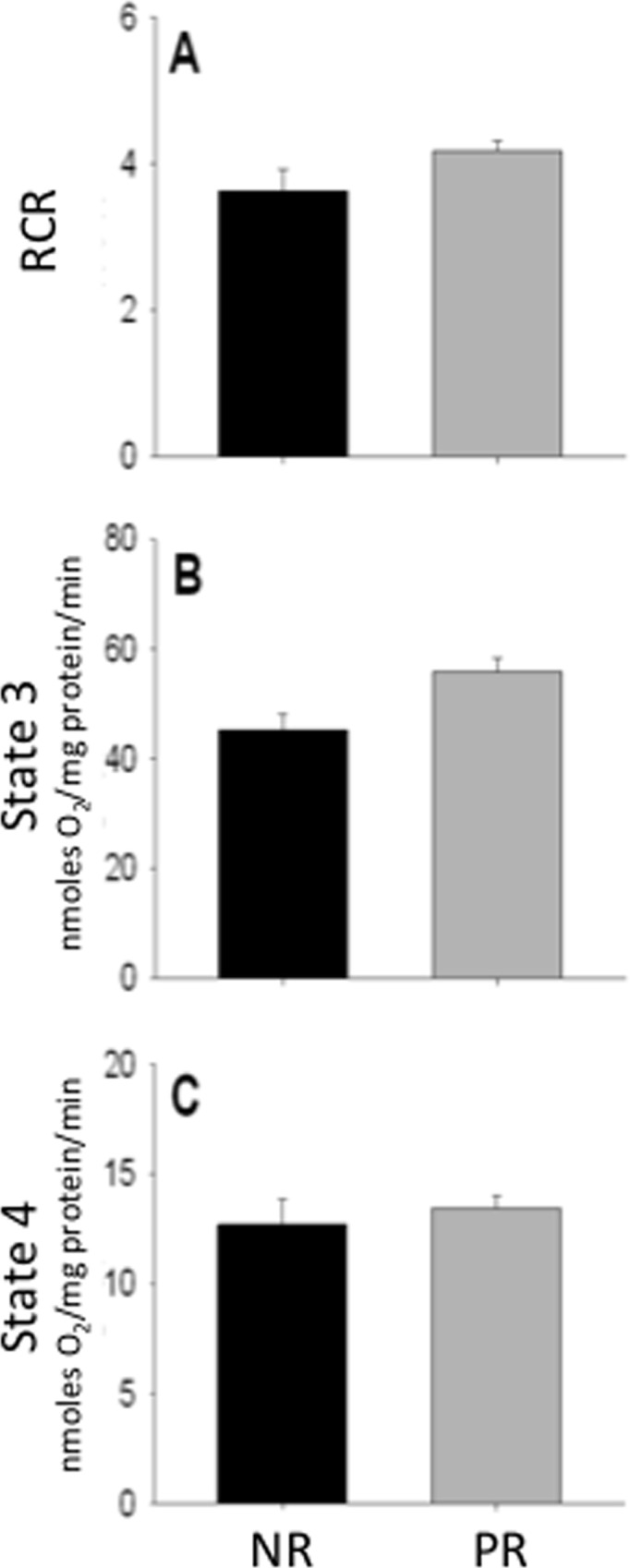
RCR (A), state 3 (B), and state 4 respiration (C) in liver mitochondria of non-reproductive and post-reproductive female mice are given. State 3 and state 4 respiration rates were measured polarographically and normalized to mitochondria content. RCR was calculated as the ratio of state3/state 4 respiration. Standard error bars are given and representative blots depicted for each graph. Non-reproductive, n = 4; Post-reproductive, n = 15. No significant differences were observed between groups.
Oxidative damage
A measure of lipid peroxidation, 4-HNE, was used to assess damage from oxidative stress [26]. The liver mitochondria of post-reproductive females exhibited significantly more lipid peroxidation than non-reproductive controls (t17 = 2.07; p = 0.050; Fig 2).
Fig 2. Liver peroxidation levels for non-reproductive (NR) and post-reproductive (PR) female house mice.

4-hydroxynonenal (4-HNE) levels (whole lane) from isolated liver mitochondria of non-reproductive and post-reproductive female mice are given. Each graph included standard error bars and representative blots. Concentrations were quantified by protein densities of Western blot bands and normalized by concentrations of α-tubulin. Non-reproductive, n = 4; Post-reproductive, n = 15; * p = 0.050, significantly different compared to non-reproductive group.
Antioxidants
The effects of reproduction on the relative level of several intra-mitochondrial antioxidants were quantified. Specifically, CuZnSOD and MnSOD dismutate superoxide to hydrogen peroxide and oxygen [20, 21]. In turn, intra-mitochondrial catalase and glutathione peroxidase 1 (GPX-1) convert hydrogen peroxide into water and oxygen [21]. Females that reproduced had significantly higher CuZnSOD (t17 = 3.73; p = 0.002; Fig 3) and catalase (t17 = 2.22; p = 0.040; Fig 3) than non-reproductive female, while MnSOD (t17 = 1.43; p = 0.169; Fig 3) and GPX-1 (t17 = 1.02; p = 0.320; Fig 3) were similar between groups.
Fig 3. Relative antioxidant levels for non-reproductive (NR) and post-reproductive (PR) female house mice.

Levels of Copper-zinc superoxide dismutase (CuZnSOD), manganese superoxide dismutase (MnSOD), catalase, and glutathione peroxidase 1 (Gpx-1) in isolated liver mitochondria of non-reproductive and post-reproductive female mice are given. Each graph included standard error bars and representative blots. Concentrations were quantified by protein densities of Western blot bands and normalized by concentrations of α-tubulin. Non-reproductive, n = 4; Post-reproductive, n = 15; * p<0.05, significantly different compared to non-reproductive group.
Discussion
The mitochondrial free radical theory of aging suggests that steady deterioration associated with aging is caused by the accumulation of ROS damage. Molecules that are damaged include mtDNA [27]. While most damaged mitochondria are destroyed, remaining mitochondria replicate [28, 29]. Turnover occurs every 3–4 days, contributing to an increasingly mutated mitochondrial population over a lifetime [29, 30]. Therefore, the energetic demands of reproduction are predicted to accelerate the decline in mitochondrial respiratory function that is associated with senescence.
Contrary to this prediction, we found that reproduction did not have a lasting effect on the respiratory capacity of liver mitochondria. Indeed, we found a trend, which warrants follow-up studies due to the sample size used, suggesting higher RCR in post-reproductive than non-reproductive females. This trend counters a previous report that livers from mice at peak-lactation do not have different RCR and state 3 respiration is lower (Clc+CII normalized to citrate synthase) than in virgin females [31]. Unlike the current study, Pichaud et al’s [31] wild-derived mice were housed in standard rodent boxes and were not re-mated immediately after birth. Thus, Pichaud et al’s mice did not experience concurrent gestation and lactation, as often occurs in mice in natural populations [31]. Furthermore, as Pichaud et al collected data at peak lactation and thus, their findings reflect the physiological changes that allow the liver to meet the energetic and fatty acid demands of milk production [31]. Changes that support reproductive demand cannot be uncoupled from the impact that reproduction has on future organ capacity during the reproductive event. Thus, organ specific mitochondrial function may display relatively high plasticity as females transition between reproductive and maintenance state. Our results show that post-reproductive females maintain the basal respiration rate of their mitochondria and show similar or perhaps even increased maximum mitochondrial respiratory capacity (Fig 1B), suggesting that there may be no negative impact of reproduction on mitochondrial function. It is important to recognize that these mitochondrial measurements were completed in vitro, and thus the relationship between mitochondrial respiratory function and reproduction may warrant further investigation in vivo.
Our results indicate that the membrane lipids of liver mitochondria in post- reproductive females sustained more oxidative damage than non-reproductive individuals (Fig 2). Due to rapid mitochondrial turnover, the liver mitochondria evaluated are the decedents of those mitochondria present during reproduction. While it is possible that the oxidative state of the animals was product of recent conditions, ROS induced damage to mtDNA over the life of an animal can alter mitochondrial proteins, reduce coupling, and as a result increase rate of ROS production [32]. While the mice in this study must be considered relatively old since they were maintained under naturalistic conditions, a significant accumulation of mtDNA mutations may be necessary before mitochondrial respiratory function is detected [33, 34]. It is also possible that the levels of ROS produced in both post-reproductive and non-reproductive mice were sufficient to upregulate antioxidants and repair mechanisms [35–37], but insufficient to increase rate of senescence, as is predicted by the theory of mitochondrial hormesis [35, 38].
Antioxidants neutralize free radicals to prevent damage to macromolecules [39]. Specifically, CuZnSOD and MnSOD convert superoxide to hydrogen peroxide and oxygen, and catalase and GPX-1 catalyze the conversion of hydrogen peroxide to water and oxygen [40–42]. We found that post-reproductive females displayed higher liver mitochondria protein levels of CuZnSOD and catalase and higher oxidative damage than non-reproductive controls. Assuming that an increase in protein abundance of these two antioxidant enzymes (i.e., CuZnSOD and catalase) translates into increased enzyme activity and detoxification of oxidants, it is predicted that liver mitochondria from post-reproductive females would eliminate more oxidants than non-reproductive females. This suggests that oxidant production (although not measured in this study) is higher in post-reproductive females, as high intra-mitochondrial antioxidants were insufficient to keep oxidative damage at the lower level observed in non-reproductive females. However, since the purity of the mitochondrial fraction was not measured, it is possible that antioxidants from other subcellular compartments could be contributing to these changes. Previous studies have reported higher antioxidant production, but lower oxidative damage at peak-lactation in female house mice compared to non-reproductive females [6, 43]. Our study housed mice in enclosures designed to mimic naturally occurring social groups. It is possible that the oxidative damage in post-reproductive females in this study is higher compared to reported oxidative damage in reproductive mice because our study houses mice in semi-natural enclosures that elicit stress from social interactions [43].
Finally, we note that associated with this being an opportunistic study, tissues could only be collected from one organ and thus, provides a limited snapshot of how the body responds to reproduction. The digestive track also displays dramatic hyperplasia in response to reproduction in rodents and thus, it is possible the reproduction could also have a significant effect on the function of this, and potentially other organs [44, 45]. Although the mammary gland typically displays dramatic changes in response to reproduction, its mass is minimal in non-reproductive female rodents and thus, the respiratory function of mammary mitochondria can’t be compared between reproductive and non-reproductive rodents using the methods described herein.
Conclusion
With our unique experimental design, comparing non-reproductive and experienced breeders in a post-reproductive state, we determined that reproduction does not adversely impact liver mitochondrial quality at the age of 1 year. Although this finding should be consider preliminary, these results question the nature of the trade-off between reproduction and longevity and highlight the importance of measuring mitochondrial respiratory function when measuring oxidative damage. The results of this study are consistent with previous findings suggesting that oxidative damage may not be associated with increased rate of senescence [11, 12, 46].
Acknowledgments
The authors thank the Hood lab undergraduates for assistance with husbandry and the Hood, Hill, and Wada labs and two anonymous reviewers for valuable comments that improved the manuscript. The authors acknowledge the Department of Biological Sciences, School of Kinesiology, College of Science and Mathematics and College of Education at Auburn University, American Society of Mammalogists, Alabama Academy of Sciences and National Science Foundation DBI-1453784 provided funding for this project.
Data Availability
All data are available from the Dryad database (data submitted 26 July 2016, accession number doi:10.5061/DRYAD.3BM04).
Funding Statement
This study was supported by Auburn University, Department of Biological Sciences and College of Science and Mathematics to WRH (http://www.auburn.edu/cosam/departments/biology/index.htm); Auburn University, School of Kinesiology and College of Education to ANK (http://www.education.auburn.edu/kinesiology); American Society of Mammalogist Grant-in-Aid to AVM (http://www.mammalsociety.org/grants); Alabama Academy of Sciences Research Grant to AVM (http://www.alabamaacademyofscience.org/students.php); and National Science Foundation DBI-1453784 to WRH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kirkwood TBL, Holliday R. Evolution of aging and longevity. Proceedings of the Royal Society B. 1979;205(1161):531–46. . [DOI] [PubMed] [Google Scholar]
- 2.Miquel J, Economos A, Fleming J, Johnson J Jr. Mitochondrial role in cell aging. Experimental Gerontology. 1980;15(6):575–91. [DOI] [PubMed] [Google Scholar]
- 3.Costantini D. Oxidative stress in ecology and evolution: lessons from avian studies. Ecology Letters. 2008;11(11):1238–51. 10.1111/j.1461-0248.2008.01246.x [DOI] [PubMed] [Google Scholar]
- 4.Dowling DK, Simmons LW. Reactive oxygen species as universal constraints in life-history evolution. Proceedings of the Royal Society B. 2009;276:1737–45. 10.1098/rspb.2008.1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monaghan P, Metcalfe NB, Torres R. Oxidative stress as a mediator of life history trade-offs: mechanisms, measurements and interpretation. Ecology Letters. 2009;12(1):75–92. 10.1111/j.1461-0248.2008.01258.x [DOI] [PubMed] [Google Scholar]
- 6.Blount JD, Vitikainen EI, Stott I, Cant MA. Oxidative shielding and the cost of reproduction. Biological Reviews. 2015. 10.1111/brv.12179 . [DOI] [PubMed] [Google Scholar]
- 7.Speakman JR, Garratt M. Oxidative stress as a cost of reproduction: Beyond the simplistic trade-off model. Bioessays. 2014;36(1):93–106. Epub 2013/11/29. 10.1002/bies.201300108 [DOI] [PubMed] [Google Scholar]
- 8.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proceedings of the National Academy of Sciences. 2006;103(20):7607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. Journal of bioenergetics and biomembranes. 1997;29(1):89–95. [DOI] [PubMed] [Google Scholar]
- 10.Lambert AJ, Brand MD. Superoxide production by NADH: ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochemical Journal. 2004;382(2):511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, et al. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. 2014. [DOI] [PMC free article] [PubMed]
- 12.Kujoth G, Hiona A, Pugh T, Someya S, Panzer K, Wohlgemuth S, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–4. [DOI] [PubMed] [Google Scholar]
- 13.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochemical Journal. 2011;435:297–312. 10.1042/BJ20110162 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meagher S, Penn DJ, Potts WK. Male-male competition magnifies inbreeding depression in wild house mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3324–9. 10.1073/pnas.060284797 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein J. Natural History of the Mouse Histocompatibility-2 Complex. New York: Springer-Verlag; 1975. [Google Scholar]
- 16.Sewell D, Wostmann B, Gairola C, Aleem M. Oxidative energy metabolism in germ-free and conventional rat liver mitochondria. American Journal of Physiology—Legacy Content. 1975;228(2):526–9. [DOI] [PubMed] [Google Scholar]
- 17.Makinen MW, Lee C-P. Biochemical studies of skeletal muscle mitochondria: I. Microanalysis of cytochrome content, oxidative and phosphorylative activities of mammalian skeletal muscle mitochondria. Arch Biochem Biophys. 1968;126(1):75–82. 10.1016/0003-9861(68)90561-4. [DOI] [PubMed] [Google Scholar]
- 18.Messer JI, Jackman MR, Willis WT. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. American Journal of Physiology—Cell Physiology. 2004;286(3):C565–72. Epub 2003/11/07. 10.1152/ajpcell.00146.2003 . [DOI] [PubMed] [Google Scholar]
- 19.Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radical Biology and Medicine. 2009;46(6):842–50. 10.1016/j.freeradbiomed.2009.01.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver Cu, Zn-SOD in mitochondria. Journal of Biological Chemistry. 2001;276(42):38388–93. [DOI] [PubMed] [Google Scholar]
- 21.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends in biochemical sciences. 2000;25(10):502–8. [DOI] [PubMed] [Google Scholar]
- 22.Carré M, André N, Carles G, Borghi H, Brichese L, Briand C, et al. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. Journal of Biological Chemistry. 2002;277(37):33664–9. [DOI] [PubMed] [Google Scholar]
- 23.Dumas J-F, Simard G, Roussel D, Douay O, Foussard F, Malthiery Y, Ritz P. Mitochondrial energy metabolism in a model of undernutrition induced by dexamethasone. British Journal of Nutrition. 2002;90(5);969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.RCoreTeam. R: A language and environment for statistical computing. Vienna, Austria2012.
- 25.Salin K, Auer SK, Rey B, Selman C, Metcalfe NB. Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance. Proceedings Biological sciences / The Royal Society. 2015;282(1812). 10.1098/rspb.2015.1028 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Powers SK, Smuder AJ, Kavazis AN, Hudson MB. Experimental guidelines for studies designed to investigate the impact of antioxidant supplementation on exercise performance. International journal of sport nutrition and exercise metabolism. 2010;20(1):2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loeb LA, Wallace DC, Martin GM. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proceedings of the National Academy of Sciences. 2005;102(52):18769–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galloway CA, Yoon Y. What comes first, misshape or dysfunction? The view from metabolic excess. The Journal of General Physiology. 2012;139(6):455–63. 10.1085/jgp.201210771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo AY, Joseph A-M, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123(15):2533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipsky NG, Pedersen PL. Mitochondrial turnover in animal cells—half-lives of mitochondria and mitochondrial subfractions of the rat liver based on biocarbonate C-14 incorporation. Journal of Biological Chemistry. 1981;256(16):8652–7. . [PubMed] [Google Scholar]
- 31.Pichaud N, Garratt M, Ballard JWO, Brooks RC. Physiological adaptations to reproduction. II. Mitochondrial adjustments in livers of lactating mice. Journal of Experimental Biology. 2013;216(15):2889–95. 10.1242/jeb.082685 . [DOI] [PubMed] [Google Scholar]
- 32.Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. 10.1146/annurev-biochem-060408-093701 . [DOI] [PubMed] [Google Scholar]
- 33.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat J-P, Letellier T. Mitochondrial threshold effects. Biochemical Journal. 2003;370(3):751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radical Biology and Medicine. 2015;85:250–8. 10.1016/j.freeradbiomed.2015.05.005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radical Biology and Medicine. 2011;51(2):327–36. 10.1016/j.freeradbiomed.2011.05.010 [DOI] [PubMed] [Google Scholar]
- 36.D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature reviews Molecular cell biology. 2007;8(10):813–24. [DOI] [PubMed] [Google Scholar]
- 37.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–R62. 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ristow M, Schmeisser K. Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-Response. 2014;12(2):288–341. 10.2203/dose-response.13-035.Ristow . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bast A, Haenen GR, Doelman CJ. Oxidants and antioxidants: state of the art. The American Journal of Medicine. 1991;91(3):S2–S13. [DOI] [PubMed] [Google Scholar]
- 40.Higashi T, Peters T. Studies on rat liver catalase I. Combined immunochemical and enzymatic determination of catalase in liver cell fractions. Journal of Biological Chemistry. 1963;238(12):3945–51. [PubMed] [Google Scholar]
- 41.Esworthy RS, Ho Y-S, Chu F-F. TheGpx1Gene encodes mitochondrial glutathione peroxidase in the mouse liver. Arch Biochem Biophys. 1997;340(1):59–63. [DOI] [PubMed] [Google Scholar]
- 42.de Haan JB, Cristiano F, Iannello R, Bladier C, Kelner MJ, Kola I. Elevation in the ratio of Cu/Zn-superoxide dismutase to glutathione peroxidase activity induces features of cellular senescence and this effect is mediated by hydrogen peroxide. Human molecular genetics. 1996;5(2):283–92. [DOI] [PubMed] [Google Scholar]
- 43.Garratt M, Vasilaki A, Stockley P, McArdle F, Jackson M, Hurst JL. Is oxidative stress a physiological cost of reproduction? An experimental test in house mice. Proceedings of the Royal Society B. 2011;278(1708):1098–106. 10.1098/rspb.2010.1818 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speakman JR, McQueenie J. Limits to sustained metabolic rate: The link between food intake, basal metabolic rate, and morphology in reproducing mice, Mus musculus. Physiological Zoology. 1996;69(4):746–69. . [Google Scholar]
- 45.Hammond KA. Adaptation of the Maternal Intestine During Lactation. Journal of Mammary Gland Biology and Neoplasia. 1997;2(3):243–52. [DOI] [PubMed] [Google Scholar]
- 46.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radical Biology and Medicine. 2007;43(4):477–503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available from the Dryad database (data submitted 26 July 2016, accession number doi:10.5061/DRYAD.3BM04).