

Abstract
β-Sheet forming peptides have attracted significant interest for the design of hydrogels for biomedical applications. One of the main challenges is the control and understanding of the correlations between peptide molecular structure, the morphology, and topology of the fiber and network formed as well as the macroscopic properties of the hydrogel obtained. In this work, we have investigated the effect that functionalizing these peptides through their hydrophobic face has on their self-assembly and gelation. Our results show that the modification of the hydrophobic face results in a partial loss of the extended β-sheet conformation of the peptide and a significant change in fiber morphology from straight to kinked. As a consequence, the ability of these fibers to associate along their length and form large bundles is reduced. These structural changes (fiber structure and network topology) significantly affect the mechanical properties of the hydrogels (shear modulus and elasticity).
Introduction
The use of noncovalent molecular self-assembly to construct materials has become a prominent strategy offering practical routes for the construction of increasingly functional materials. A variety of molecular building blocks can be used for this purpose; one such block that has attracted considerable attention in the past two decades is de novo designed peptides. The exploitation of peptides and their self-assembling properties to design hydrogels in particular has been the focus of significant efforts due to their potential for use in a variety of biomedical applications such as cell therapy,1,2 tissue regeneration,3−8 and drug delivery.9−11 A number of molecular designs have been developed for the synthesis of self-assembling peptides with the four main families being amphiphilic peptides,12 short peptide derivatives, α-helix/coiled-coil peptides,13,14 and β-sheet peptides.15−17 All these designs allow the synthesis of peptides that under appropriate conditions self-assemble to form elongated fibers. Above a critical gelation concentration (CGC) these fibers entangle and/or associate to form three-dimensional networks that have the ability to trap water, i.e., form hydrogels.
β-Sheet peptides are of particular interest as these peptides allow the fabrication of very stable hydrogels with properties that can be tailored through peptide design, media properties, and processing. We have recently investigated the self-assembly and gelation properties of a family of β-sheet peptides18−20 based on the design developed by Zhang and co-workers.21−23 This design, which is based on the alternation of hydrophilic and hydrophobic residues, allows the synthesis of peptides that self-assemble into antiparallel β-sheet fibrils. Because of the design used, these antiparallel β-sheet fibrils have a hydrophobic and a hydrophilic face as schematically shown in Figure 1. It is thought that in order to minimize contact between water and the hydrophobic faces, these β-sheet fibrils associate by pairing to form β-sheet fibers with the hydrophobic residue side chains buried in the fiber core (Figure 1).
Figure 1.

Top: schematic representation of the self-assembling and gelation processes of β-sheet forming peptides. Bottom left: schematic representation of an extended β-sheet fiber. Bottom right: molecular structures of F9 and FC9-BM.
One of the most attractive properties of these systems is their ease of functionalization. Usually this is achieved by covalently linking the desired functional group to the peptide to create a functional peptide which then can be dosed as required into the system to create functional hydrogels through self-assembly. In order to avoid affecting the self-assembling properties of the peptides, their functionalization is usually achieved by linking the functionality to one of the peptide’s chain termini.3,24−28 Here we are interested in looking at the possibility to functionalize the peptide fibrils through its hydrophobic face in order to bury the functionality in the hydrophobic core of the peptide fiber.
For this study we have chosen the self-assembling peptide FEFKFEFKK (F9) (F: phenylalanine; K: lysine; E: glutamic acid) which forms stable β-sheet-rich fibers and hydrogels at pH 5.5.29−31 The peptide was functionalized by replacing the third phenylalanine residue with cysteine. The nucleophilic thiol group of cysteine was then reacted with 2,3-dibromomaleimide to generate the peptide–bromomaleimide conjugate FEFKC(BM)EFKK (FC9-BM) through a straightforward nucleophilic substitution (Figure 2). The introduction of this modification does not affect the isoelectric point of the peptide and therefore does not affect the pH dependency of the peptide self-assembly and gelation properties. A pH of 5.5 was chosen for this study as it corresponds to the optimal pH (stability and homogeneity) for the gelation of these peptides, which at this pH carry a theoretical charge of +2. Hybrid hydrogels were prepared by mixing F9 and FC9-BM in the desired proportions while keeping the overall molar concentration of peptide constant. Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) and circular dichroism (CD) were used to investigate how the BM modification affected the conformation adopted by the peptide, while transmission electron microscopy (TEM), atomic force microscopy (AFM), and small-angle X-ray scattering (SAXS) were used to investigate the effect on fiber morphology and network topology. Finally, shear rheometry was used to investigate the effect on hydrogel mechanical properties.
Figure 2.

Scheme of the synthetic route used to produce FC9-BM peptide.
Materials and Methods
Materials
The peptides used for this study FEFKFEFKK and FEFKCEFKK were purchased from Biomatik Corporation (Wilmington, DE) with a purity >95% (confirmed in house by ESI-MS and RP-HPLC). All solvents and reagents were purchased from Sigma-Aldrich and used as received.
Synthesis of FEFKC(BM)EFKK (FC9-BM)
FC9-BM was synthesized according to the scheme shown in Figure 2. Peptide FEFKCEFKK (FC9) (75 mg, 55 μmol) was first dissolved in 4.5 mL of ddH2O. A solution of 2,3-dibromomaleimide (DBM) (14 mg, 55 μmol) in 500 μL of dimethylformamide (DMF) was added dropwise to the FC9 solution under vigorous stirring, and then the solution was left stirring for 24 h at room temperature. The reaction mixture was then lyophilized to give 79 mg (94% yield) of FC9-BM as a yellow residue. The molecular weight and purity of the product were confirmed by ESI-MS (Figure ESI 1; m/z: 1379.5 [M + H]+) and RP-HPLC (Figure ESI 2; purity 95%) (see Supporting Information for detailed methodology). The FC9-BM peptide was thus directly used for hydrogels preparation without further purification.
Sample Preparation
Peptide powders were dissolved in HPLC grade water by sonication at 80 kHz for 30 min, and the solution pH was adjusted to pH 5.5 by the stepwise addition of 0.5 M NaOH solution. The final sample concentration was 30 mM. The hydrogel was left overnight in a fridge at 4 °C to equilibrate and used the following day.
Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy (ATR-FTIR)
Hydrogels were spread as prepared onto the crystal surface of a Bruker ALPHA-P FT-IR spectrometer equipped with a multibounce attenuated total reflectance (ATR) plate. The transmittance spectra were recorded (128 scans) between 4000 and 400 cm–1 with a resolution of 4 cm–1. HPLC grade water was used as background and was automatically subtracted from the recorded spectra using the OPUS software provided with the instrument. Peak area relative intensity was evaluated by fitting the 1624 cm–1 peak using a GassAmp function (Origin 9.0) after subtraction of a straight baseline in the amide I region (1600–1700 cm–1). The total intensity of the amide I region was obtained by integration.
Circular Dichroism Spectroscopy (CD)
Circular dichroism measurements were performed on a ChiraScan, Applied Photophysics. Samples were prepared by diluting the hydrogels 20-fold in HPLC grade water. Samples were placed in a 0.1 mm quartz cell (Hellma) and spectra recorded at wavelengths from 200 to 280 nm, with a 1 nm step size and response time of 0.5 s. The ellipticity data acquired in mdeg were converted to mean residue molar ellipticity ε in deg cm2 dmol–1 residue–1 through
| 1 |
where θ is the ellipticity in mdeg, C the sample molar concentration, N the number of backbone amide bonds, and L the cell optical path length in cm.
Transmission Electron Microscopy (TEM)
Hydrogels were first diluted 10-fold using ddH2O and then negatively stained. A carbon-coated copper grid (400 mesh from Electron Microscopy Sciences) was placed on a 10 μL droplet of sample for 1 min and excess liquid drained off using lint-free tissue. The grid was then placed on a 10 μL droplet of ddH2O for 10 s before excess liquid was drained off. The grid was then transferred to a 10 μL droplet of a 1% uranyl acetate solution for 30 s, and again excess liquid was drained off. Finally, the grid was transferred to a 10 μL droplet of ddH2O for 10 s before excess liquid was drained off for a final time. The sample was then left to dry before imaging using a FEI Tecnai12 BioTwin TEM at 100 keV.
Atomic Force Microscopy (AFM)
Hydrogels were first diluted 10-fold using ddH2O. 50 μL of this solution was pipetted onto freshly cleaved mica. After 1 min excess solution was removed and the surface washed twice with 1 mL of ddH2O. Excess water was then removed once again by wicking using Whatman No.1 filter paper. The samples were allowed to air-dry prior to imaging. Samples were imaged by intermittent contact (“tapping”) mode in air using a Bruker Multimode AFM with a Nanoscope V controller and a “J” scanner. Imaging was performed using Olympus high aspect ratio etched silicon probes (OTESPA) with nominal spring constant of 42 N m–1 (Bruker AXS S.A.S, France). Drive amplitude and cantilever oscillation, which varied between 300 and 350 kHz, were determined by the Nanoscope (v8.15) software. The amplitude set point was adjusted to just below the point at which tip–sample interaction was lost. Height, phase, and amplitude images with scan sizes of either 2 or 5 μm2 were captured at a scan rate of 1.5 Hz and at a relative humidity of <40%. Data were first-order flattened using the Nanoscope Analysis (v1.4) software prior to image export.
Oscillatory Rheology
Rheological studies were carried out on a stress-controlled rheometer (Discovery HR-2, TA Instruments) equipped with a solvent trap to minimize evaporation, using a 20 mm parallel plate geometry. 500 μL of peptide hydrogel was loaded onto the stage, and the gap between the stage and the upper plate set to 250 μm. The loaded sample was then left for 2 min to equilibrate at 25 °C before measurement. Excess sample was carefully removed with a spatula from around the plate. Strain sweeps were measured between 0.04 and 40% strain at an oscillation frequency of 1 Hz. Radial frequency sweeps were undertaken between 0.01 and 15 Hz, using a strain of 0.2%, which is within the linear viscoelastic regime (LVR) of all samples. All measurements were repeated at least three times.
Small-Angle X-ray Scattering
SAXS measurements were performed at Diamond Light Source beamline B21 equipped with a using a BIOSAXS robot for sample loading and a PILATUS 2M (Dectris, Switzerland) detector. The X-ray wavelength used was 0.1 nm corresponding to an energy of 12.4 keV, and the sample–detector distance was 4.018 m giving an accessible q-range of 0.05–4.0 nm–1. Data were reduced, and solvent and capillary contributions were subtracted using the DawnDiamond software suite. Samples were prepared by directly at 3 mM concentration following the methodology described above. The SAXS patterns were collected 3 days after sample preparation.
Results and Discussion
A series of F9/FC9-BM hybrid hydrogels were prepared at constant overall peptide molar concentration of 30 mM by varying the ratio of FC9-BM added from 0% (pure F9) to 100% (pure FC9-BM). All samples formed hydrogels at pH 5.5 suggesting the formation, in all cases, of extended fibrillar networks.
First ATR-FTIR and CD were used to investigate the conformation adopted by the peptides. In Figure 3, the ATR-FTIR spectra obtained for all the samples as prepared are presented.
Figure 3.

Left: ATR-FTIR spectra obtained for F9/FC9-BM hybrid hydrogels with an overall peptide molar concentration of 30 mM. Right: 1624 cm–1 β-sheet peak relative intensity vs FC9-BM content.
As expected, the pure F9 hydrogel spectrum shows two strong absorption peaks at 1624 and 1524 cm–1 corresponding to the adoption by this peptide of an extended β-sheet conformation. For the pure FC9-BM hydrogel in addition to the peak at 1624 cm–1 a strong broad peak is also observed at 1651 cm–1 assigned to random coil conformation.32,33 The presence of both peaks suggests that the presence of the BM side group is frustrating the adoption by this peptide of a fully extended β-sheet conformation. As can be seen from the structures of the two peptides presented in Figure 1, the BM side group likely disrupts the overall ordering/packing of the phenylalanine rings, resulting in a loss of the fully extended β-sheet conformation. When the amount of FC9-BM in the hydrogels is increased, the relative intensity (in relation to the overall intensity of the amide I region) of the peak at 1624 cm–1 decreases (Figure 3), suggesting a decrease in β-sheet content. Interestingly, although the relative intensity decreases linearly for samples with FC9-BM content higher than 25%, the sample with an overall fraction of FC9-BM of 12.5% exhibits a lower than expected relative intensity, pointing toward a proportionally stronger disruptive effect of introducing small amounts of FC9-BM on the overall content of peptide adopting an extended β-sheet conformation. This would suggest that in order to accommodate FC9-BM in the structure the adjacent F9 peptides also lose some of their extended conformation.
CD confirmed these results. As can be seen from Figure 4 for the F9 sample a strong negative band is observed at 218 nm with a strong positive signal being observed below 205 nm. These CD spectra features are typical of β-sheet secondary structure in proteins.34 When FC9-BM is introduced into the system, although for 12.5% and 25% samples the peak at 218 nm is unchanged, the strong positive signal below 205 nm is lost, suggesting a decrease in β-sheet content. Indeed, the CD signal associated with random coil secondary structure in proteins presents a small positive band in the 205–230 nm region but a strong negative band in the 185–205 region.34 In agreement with the ATR-FTIR data, similar CD signals were obtained for the 12.5% and 25% samples, suggesting a similar β-sheet content. For samples with a FC9-BM content higher than 25% a significant decrease in the 218 nm band is observed, indicating a further decrease in β-sheet content. An additional band is observed at 222 nm, which in the literature has been assigned to aromatic residues, in our case phenylalanine. Its exact origin is still matter of debate and it is thought that it is related to the ordering of the aromatic rings.34
Figure 4.

CD spectra obtained for F9/FC9-BM solutions prepared by 20-fold dilution of hybrid hydrogels with overall peptide molar concentrations of 30 mM.
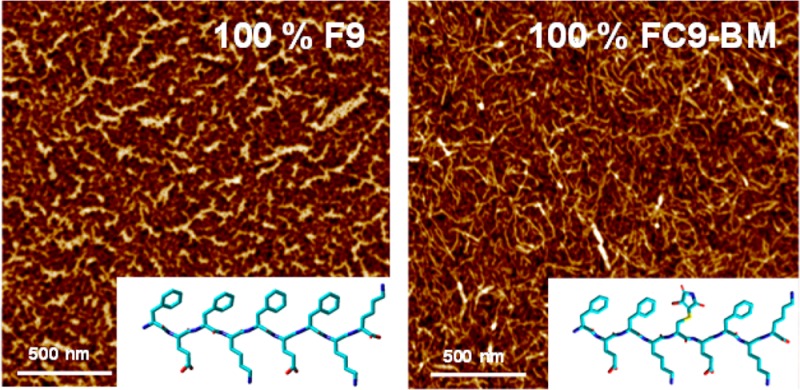
The ability of both peptides to form extended fibrillar structures was confirmed by TEM and AFM (Figure 5). Clear differences in fiber morphologies can be observed via TEM. For the F9 sample thin relatively straight fibers that associate along their length to form thicker bundles were observed. The width of the thinnest fibers observed was ∼3–4 nm, in good agreement with the adoption by this peptide of an extended β-sheet conformation while fiber bundle sizes varied from ∼5 up to ∼20 nm. For the FC9-BM sample thin kinked fibers were observed. In this case, too, the smallest fibers observed were found to have a width of ∼3–4 nm, but no extensive fiber association was observed resulting in very few large fiber bundles being observed. For the 50% F9/50% FC9-BM sample an intermediate morphology is observed. The TEM images suggest that the introduction of the BM modification results in a change in fiber morphology from straight to kinked correlating well with the loss of extended β-sheet conformation revealed by ATR-FTIR and CD. This change in fiber morphology is thought to result in a decrease in the ability of the fibers to associate along their length and form larger bundles. Close inspection of the FC9-BM TEM micrograph clearly shows fibers close to each other, but due to their kinked nature, they are unable to align and associate along their length and form large bundles (arrows in Figure 5) as observed for the F9 sample.
Figure 5.

TEM (top) and AFM height (bottom) images obtained for the different samples. Arrows in the TEM of 100% FC9-BM show example of kinks in fibers (see text for further details).
The reduced tendency for FC9-BM system to form extended fiber bundles was confirmed by AFM. It should be noted that TEM and AFM sample preparation methods are very different. While for TEM only few fibers/fiber aggregates are collected on the grids and imaged, in AFM significantly higher density of fibers/fiber aggregates are deposited on the mica surface, allowing us to observe a morphology which is closer to the overall network topology expected for these systems. As can be seen from Figure 5 much thinner features/fibers are observed for FC9-BM sample compared to F9 sample, confirming the TEM results.
TEM and AFM require significant sample preparation and give two-dimensional images of three-dimensional structures; therefore, SAXS was used to confirm the increased propensity for F9 to form fiber aggregates compared to FC9-BM. In this case the samples were prepared direct at the required concentration with no further sample manipulation. In Figure 6 the scattering patterns obtained for the three samples F9, 50% F9/50% FC9-BM, and FC9-BM are presented. As can be seen from the log vs log plots, at low q a clear q–1 behavior typical of the scattering of fibers can be observed for the FC9-BM sample. For the 50% F9/50% FC9-BM and F9 samples as the amount of FC9-BM decreases, the scattering pattern deviates increasingly from the expected q–1 behavior. This deviation from the expected scattering pattern of infinitely long thin fibers is even more evident in the ln qI(q) vs q2 representations. Indeed, it has been shown that for thin rod-like structures (i.e., fibers) for qRσ < 1 (Rσ: cross-section radius of gyration) the scattering intensity can be written as35,36
| 2 |
As can be seen from Figure 6, a linear behavior is observed at low q only for FC9-BM, suggesting that for this sample the fibers can indeed be considered as infinitely long thin rod-like structures. From the fitting of the linear region Rσ can be estimated as 1.7 ± 0.2 nm, which corresponds to a fiber diameter of 4–6 nm, in good agreement with the TEM results. For the other two samples no linear region is observed, indicating that in this case the scattering objects cannot be considered as thin infinitely long rods. Instead, a “roll-over” is present, characteristic of asymmetric objects,37,38 which is in good agreement with the interpretation made above of F9 favoring the formation of fiber bundles/aggregates. The scattering observed for these samples becomes complex and will be a mixture of scattering from the single fibers and fiber bundles/aggregates of different sizes.
Figure 6.

Log vs log (left) and ln qI(q) vs q2 representation of the SAXS scattering pattern obtained for the 100% F9, 50% F9 + 50% FC9-BM, and 100% FC9-BM samples prepared at 3 mM concentration.
FTIR and CD suggest that the introduction of FC9-BM results in a loss of β-sheet conformation, which is expected to affect the fiber intrinsic properties (e.g., flexibility). On the other hand, TEM, AFM, and SAXS suggest that the introduction of FC9-BM results in a change in the network topology, FC9-BM sample showing a significant lower level of fiber bundling/aggregation. This change in network topology is expected to affect the intrinsic properties of the network (e.g., elasticity). The bulk mechanical properties of the hydrogels will be a function of both the intrinsic properties of the fibers and the intrinsic properties of the fibrillar network.
We investigated how the introduction of FC9-BM affected the mechanical properties of the hydrogels using shear rheometry. In Figure 7 the shear moduli obtained for the hydrogels are presented as a function of FC9-BM content. As can be observed, there is a significant decrease in G′ (an order of magnitude) when 12.5% of FC9-BM is introduced in the system with an additional small decrease when the content of FC9-BM is increased further. The loss in β-sheet conformation as revealed by FTIR and CD translates at the macroscopic level into a significant decrease in G′. The TEM and AFM images suggest that the introduction of FC9-BM ultimately leads to a change in the network topology from a network formed by thick bundles of relatively straight fibers to a network formed by thin kinked fibers. One would expect indeed the latter topology to result in a hydrogel with a lower shear modulus but a higher elasticity. The increase in elasticity (in other words, a decrease in brittleness) is clearly shown by the strain sweep experiments that allow determination of the size of the linear viscoelastic regime (LVR). As can be seen from Figure 7, the LVR size increases significantly upon introduction of FC9-BM in the system, pointing toward the formation of a more elastic, less brittle, network.
Figure 7.

Left: shear modulus (G′) of F9/FC9-BM composite hydrogels at 15 Hz obtained from the frequency sweep experiments performed at 0.2% strain vs FC9-BM content. Right: strain sweep experiments for the 100% F9 (●, ○), 75% F9/25% FC9-BM (⧫, ◊), and 100% FC9-BM sample (■, □) (close symbols: G′; open symbols: G″). All samples had an overall peptide molar concentration of 30 mM.
A number of theories that relate topology of networks formed to moduli of hydrogels can be found in the literature. One such theory was developed by Jones and Marques39 for rigid polymer networks and relates the shear modulus to the fractal dimension DF of the objects forming the network through the power law
| 3 |
where C is the concentration of objects contributing to the network elasticity.
For a network formed by straight (between junctions) thin fibers DF is expected to be 1 corresponding to a power law of C2. Good agreement with the Jones and Marques theory was indeed obtained for the FC9-BM system for which a power law of C1.8±0.2 was obtained (Figure 8). For this system as our SAXS result show the system can indeed be considered as being formed by thin straight fibers.
Figure 8.

Log–log plot of shear moduli (G′) vs molar concentrations obtained for F9 (●) and FC9-BM (○) hydrogels.
For the F9 system a power law of C3.7±0.3 was obtained. As discussed by Ramzi et al., a higher exponent can originate from the presence of a high number of pendent fibers (not connected to the network) as the effective concentration (concentration of objects actively participating to the elasticity of the network) is lower than the nominal concentration of objects.40 From the TEM pictures obtained in our case, it is unlikely that a significant number of pendant fibers are present in our systems. On the other hand, for the F9 system the formation of large bundles of fibers is observed. In this case each bundle will act as a single element contributing to the elasticity of the network. As a result, the actual effective concentration of object participating to the elasticity of the network is reduced and leads probably to a higher exponent. This interpretation would imply that the difference in measured and expected exponent is an indirect measure of the level of fiber association and bundling in these systems and therefore that Jones and Marques theory could be used to characterize the association and bundling tendency of β-sheet forming peptide through shear moduli measurements.
Conclusions
We have investigated the effect of functionalizing the β-sheet forming peptide FEFKFEFKK through the hydrophobic face by replacing one of the phenyl ring groups with a bromomaleimide group. Our results show that the introduction of this modification results in a loss of the extended β-sheet conformation adopted by the peptide. This leads to a change in the fiber morphology from straight to kinked leading to a reduced ability to associate and bundle. As a result of these morphological changes, the macroscopic mechanical properties, in this case shear modulus, of the hydrogel decrease while its elasticity increases. The characterization of the level of association and bundling in these systems is notoriously difficult, and here we suggest an indirect characterization of this phenomenon through the use of the theory developed by Jones and Marques.
Acknowledgments
The authors are grateful to the Engineering and Physical Sciences Research Council (EPSRC Fellowship Grant No. EP/K016210/1) for funding this work. The authors thank the staff in the EM facility in the Faculty of Life Sciences, of the University of Manchester, for their assistance, and the Wellcome Trust for equipment grant support to the EM facility. The authors are also grateful to Diamond for beam time award and to Dr. K. Inoue and all the staff on beamline B21 at Diamond for their support with the SAXS experiments. All research data supporting this publication are directly available within this publication.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.5b03841.
RP-HPLC and ESI-MS graphs are presented confirming the molecular weight and purity of the FC9-BM peptide (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Haines-Butterick L.; Rajagopal K.; Branco M.; Salick D.; Rughani R.; Pilarz M.; Lamm M. S.; Pochan D. J.; Schneider J. P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (19), 7791–7796. 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.; Mackay M. E.; Czymmek K.; Nagarkar R. P.; Schneider J. P.; Pochan D. J. Injectable Solid Peptide Hydrogel as a Cell Carrier: Effects of Shear Flow on Hydrogels and Cell Payload. Langmuir 2012, 28 (14), 6076–6087. 10.1021/la2041746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier J. H.; Rudra J. S.; Gasiorowski J. Z.; Jung J. P. Multi-component extracellular matrices based on peptide self-assembly. Chem. Soc. Rev. 2010, 39 (9), 3413–3424. 10.1039/b914337h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkolar L.; Guilbaud J. B.; Miller A. F.; Gough J. E.; Saiani A. Enzymatically triggered peptide hydrogels for 3D cell encapsulation and culture. J. Pept. Sci. 2014, 20 (7), 578–584. 10.1002/psc.2666. [DOI] [PubMed] [Google Scholar]
- Matson J. B.; Stupp S. I. Self-assembling peptide scaffolds for regenerative medicine. Chem. Commun. 2012, 48 (1), 26–33. 10.1039/C1CC15551B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo Diaz L. A.; Saiani A.; Gough J. E.; Miller A. F., Human osteoblasts within soft peptide hydrogels promote mineralisation in vitro. J. Tissue Eng. 2014, 10.1177/2041731414539344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujeeb A.; Miller A. F.; Saiani A.; Gough J. E. Self-assembled octapeptide scaffolds for in vitro chondrocyte culture. Acta Biomater. 2013, 9 (1), 4609–4617. 10.1016/j.actbio.2012.08.044. [DOI] [PubMed] [Google Scholar]
- Liyanage W.; Vats K.; Rajbhandary A.; Benoit D. S. W.; Nilsson B. L. Multicomponent dipeptide hydrogels as extracellular matrix-mimetic scaffolds for cell culture applications. Chem. Commun. 2015, 51 (56), 11260–11263. 10.1039/C5CC03162A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts D.; Rochas C.; Saiani A.; Miller A. F. Effect of Peptide and Guest Charge on the Structural, Mechanical and Release Properties of beta-Sheet Forming Peptides. Langmuir 2012, 28 (46), 16196–16206. 10.1021/la303328p. [DOI] [PubMed] [Google Scholar]
- Tang C.; Miller A. F.; Saiani A. Peptide hydrogels as mucoadhesives for local drug delivery. Int. J. Pharm. 2014, 465, 427–435. 10.1016/j.ijpharm.2014.02.039. [DOI] [PubMed] [Google Scholar]
- Altunbas A.; Pochan D. J. Peptide-Based and Polypeptide-Based Hydrogels for Drug Delivery and Tissue Engineering. Top. Curr. Chem. 2011, 310, 135–167. 10.1007/128_2011_206. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Self-assembly of amphiphilic peptides. Soft Matter 2011, 7 (9), 4122–4138. 10.1039/c0sm01218a. [DOI] [Google Scholar]
- Dong H.; Paramonov S. E.; Hartgerink J. D. Self-Assembly of Alpha-Helical Coiled Coil Nanofibers. J. Am. Chem. Soc. 2008, 130 (41), 13691–13695. 10.1021/ja8037323. [DOI] [PubMed] [Google Scholar]
- Banwell E. F.; Abelardo E. S.; Adams D. J.; Birchall M. A.; Corrigan A.; Donald A. M.; Kirkland M.; Serpell L. C.; Butler M. F.; Woolfson D. N. Rational design and application of responsive alpha-helical peptide hydrogels. Nat. Mater. 2009, 8 (7), 596–600. 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman C. J.; Nilsson B. L. Review self-assembly of amphipathic beta-sheet peptides: Insights and applications. Biopolymers 2012, 98 (3), 169–184. 10.1002/bip.22058. [DOI] [PubMed] [Google Scholar]
- Maude S.; Tai L. R.; Davies R. P. W.; Liu B.; Harris S. A.; Kocienski P. J.; Aggeli A. Peptide Synthesis and Self-Assembly. Top. Curr. Chem. 2011, 310, 27–69. 10.1007/128_2011_234. [DOI] [PubMed] [Google Scholar]
- Kim S.; Kim J. H.; Lee J. S.; Park C. B. Beta-Sheet-Forming, Self-Assembled Peptide Nanomaterials towards Optical, Energy, and Healthcare Applications. Small 2015, 11 (30), 3623–3640. 10.1002/smll.201500169. [DOI] [PubMed] [Google Scholar]
- Boothroyd S.; Miller A. F.; Saiani A. From fibres to networks using self-assembling peptides. Faraday Discuss. 2013, 166, 195–207. 10.1039/c3fd00097d. [DOI] [PubMed] [Google Scholar]
- Saiani A.; Mohammed A.; Frielinghaus H.; Collins R.; Hodson N.; Kielty C. M.; Sherratt M. J.; Miller A. F. Self-assembly and gelation properties of alpha-helix versus beta-sheet forming peptides. Soft Matter 2009, 5 (1), 193–202. 10.1039/B811288F. [DOI] [Google Scholar]
- Boothroyd S.; Saiani A.; Miller A. F. Controlling network topology and mechanical properties of co-assembling peptide hydrogels. Biopolymers 2014, 101 (6), 669–680. 10.1002/bip.22435. [DOI] [PubMed] [Google Scholar]
- Zhang S. G.; Altman M. Peptide self-assembly in functional polymer science and engineering. React. Funct. Polym. 1999, 41 (1–3), 91–102. 10.1016/S1381-5148(99)00031-0. [DOI] [Google Scholar]
- Zhang S. G.; Holmes T.; Lockshin C.; Rich A. Spontaneous Assembly of a Self-Complementary Oligopeptide to Form a Stable Macroscopic Membrane. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (8), 3334–3338. 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. G.; Marini D. M.; Hwang W.; Santoso S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr. Opin. Chem. Biol. 2002, 6 (6), 865–871. 10.1016/S1367-5931(02)00391-5. [DOI] [PubMed] [Google Scholar]
- Maslovskis A.; Guilbaud J. B.; Grillo I.; Hodson N.; Miller A. F.; Saiani A. Self-Assembling Peptide/Thermoresponsive Polymer Composite Hydrogels: Effect of Peptide–Polymer Interactions on Hydrogel Properties. Langmuir 2014, 30 (34), 10471–10480. 10.1021/la502358b. [DOI] [PubMed] [Google Scholar]
- Radu L. C.; Yang J.; Kopecek J. Self-Assembling Diblock Copolymers of Poly[N-(2-hydroxypropyl)methacrylamide] and a beta-Sheet Peptide. Macromol. Biosci. 2009, 9 (1), 36–44. 10.1002/mabi.200800193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J. P.; Moyano J. V.; Collier J. H. Multifactorial optimization of endothelial cell growth using modular synthetic extracellular matrices. Integrative Biology 2011, 3 (3), 185–196. 10.1039/c0ib00112k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica F.; Alexander C.; Tirelli N.; Miller A. F.; Saiani A. Selective synthesis of double temperature-sensitive polymer-peptide conjugates. Chem. Commun. 2008, 37, 4433–4435. 10.1039/b806782a. [DOI] [PubMed] [Google Scholar]
- Hickling C.; Toogood H. S.; Saiani A.; Scrutton N. S.; Miller A. F. Nanofibrillar Peptide Hydrogels for the Immobilization of Biocatalysts for Chemical Transformations. Macromol. Rapid Commun. 2014, 35 (9), 868–874. 10.1002/marc.201400027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan M. R.; Moore P. N.; Zhang S. G.; Kamm R. D.; Lauffenburger D. A. Self-assembly of a beta-sheet protein governed by relief of electrostatic repulsion relative to van der Waals attraction. Biomacromolecules 2000, 1 (4), 627–631. 10.1021/bm005586w. [DOI] [PubMed] [Google Scholar]
- Caplan M. R.; Schwartzfarb E. M.; Zhang S. G.; Kamm R. D.; Lauffenburger D. A. Effects of systematic variation of amino acid sequence on the mechanical properties of a self-assembling, oligopeptide biomaterial. J. Biomater. Sci., Polym. Ed. 2002, 13 (3), 225–236. 10.1163/156856202320176493. [DOI] [PubMed] [Google Scholar]
- Caplan M. R.; Schwartzfarb E. M.; Zhang S. G.; Kamm R. D.; Lauffenburger D. A. Control of self-assembling oligopeptide matrix formation through systematic variation of amino acid sequence. Biomaterials 2002, 23 (1), 219–227. 10.1016/S0142-9612(01)00099-0. [DOI] [PubMed] [Google Scholar]
- Barth A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta, Bioenerg. 2007, 1767 (9), 1073–1101. 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35 (4), 369–430. 10.1017/S0033583502003815. [DOI] [PubMed] [Google Scholar]
- Kelly S. M.; Jess T. J.; Price N. C. How to study proteins by circular dichroism. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1751 (2), 119–139. 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Guenet J.-M.Thermoreversible Gelation of Polymers and Biopolymers; Academic Press: London, 1992. [Google Scholar]
- Guilbaud J.-B.; Saiani A. Using small angle scattering (SAS) to structurally characterise peptide and protein self-assembled materials. Chem. Soc. Rev. 2011, 40 (3), 1200–1210. 10.1039/C0CS00105H. [DOI] [PubMed] [Google Scholar]
- Ramachandran S.; Trewhella J.; Tseng Y.; Yu Y. B. Coassembling peptide-based biomaterials: Effects of pairing equal and unequal chain length oligopeptides. Chem. Mater. 2006, 18 (26), 6157–6162. 10.1021/cm061071l. [DOI] [Google Scholar]
- Higgins J. S.; Benoit H. C.. Polymer and Neutron Scattering; Clarendon Press: Oxford, 1994. [Google Scholar]
- Jones J. L.; Marques C. M. Rigid Polymer Network Models. J. Phys. 1990, 51 (11), 1113–1127. 10.1051/jphys:0199000510110111300. [DOI] [Google Scholar]
- Ramzi M.; Rochas C.; Guenet J. M. Structure-properties relation for agarose thermoreversible gels in binary solvents. Macromolecules 1998, 31 (18), 6106–6111. 10.1021/ma9801220. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.