

Abstract
Human deoxyhypusine hydroxylase (hDOHH) is an enzyme that is involved in the critical post-translational modification of the eukaryotic translation initiation factor 5A (eIF5A). Following the conversion of a lysine residue on eIF5A to deoxyhypusine (Dhp) by deoxyhypusine synthase, hDOHH hydroxylates Dhp to yield the unusual amino acid residue hypusine (Hpu), a modification that is essential for eIF5A to promote peptide synthesis at the ribosome, among other functions. Purification of hDOHH overexpressed in E. coli affords enzyme that is blue in color, a feature that has been associated with the presence of a peroxo-bridged diiron(III) active site. To gain further insight into the nature of the diiron site and how it may change as hDOHH goes through the catalytic cycle, we have conducted X-ray absorption spectroscopic studies of hDOHH on five samples that represent different species along its reaction pathway. Structural analysis of each species has been carried out, starting with the reduced diferrous state, proceeding through its O2 adduct, and ending with a diferric decay product. Our results show that the Fe•••Fe distances found for the five samples fall within a narrow range of 3.4–3.5 Å, suggesting that hDOHH has a fairly constrained active site. This pattern differs significantly from what has been associated with canonical dioxygen activating nonheme diiron enzymes such as soluble methane monooxygenase and Class 1A ribonucleotide reductases, for which the Fe•••Fe distance can change by as much as 1 Å during the redox cycle. These results suggest that the O2 activation mechanism for hDOHH deviates somewhat from that associated with the canonical nonheme diiron enzymes, opening the door to new mechanistic possibilities for this intriguing family of enzymes.
Introduction
Members of the ferritin-like protein superfamily are characterized by a four-helix bundle structural motif that generally bind pairs of redox-active metals in a 2-His-4-carboxylate coordination environment [1, 2]. Nonheme diiron members of this family activate O2 and facilitate a wide variety of reactions, including the biomineralization of iron by ferritins [3], the biosynthesis of DNA precursors from ribonucleotides by ribonucleotide reductase (RNR) [4], hydroxylation of C–H bonds by the hydroxylase components of bacterial multicomponent monooxygenases such as soluble methane monooxygenase (sMMO) [5, 6] and toluene/o-xylene monooxygenase (ToMO) [7], fatty acid desaturation by Δ9 stearoyl-acyl carrier protein desaturase (Δ9D) [8], arylamine N-oxygenation by AurF [9] and CmlI [10], and formation of alkanes from fatty aldehydes by aldehyde deformylating oxygenase (ADO) [11, 12]. Among the best studied members of this protein family is sMMO, which has been subjected to intensive crystallographic, spectroscopic and mechanistic investigations.
In 2006, a new nonheme diiron enzyme human deoxyhypusine hydroxylase (hDOHH) was characterized and found to be responsible for the post-translational modification of the eukaryotic translational initiation factor 5A (eIF5A) [13, 14]. eIF5A plays an essential role in the regulation of cell proliferation by facilitating peptide synthesis at the ribosome [15, 16]. Inactivation of the enzymes responsible for the post-translational modification of eIF5A results in cell death [17], making this pathway a potential therapeutic target for the treatment of diseases like malaria and certain cancers [18]. The substrate eIF5A utilizes a unique amino acid residue, hypusine (Hpu), to perform a critically important function [15] and is in fact the only known protein that harbors a Hpu residue [19]. The hypusine is derived from a lysine residue that is first converted to deoxyhypusine (Dhp) by deoxyhypusine synthase (Scheme 1). This modified eIF5A(Dhp) is the substrate for hDOHH, which activates O2 and hydroxylates the strong Cζ–H bond to generate the final eIF5A(Hpu) product [14]. As preparations of this enzyme from heterologous expression cells were reported to be blue in color [13], we carried out spectroscopic investigations that identified the blue chromophore as the diferric peroxo intermediate of hDOHH (hDOHH-P) [20]. Importantly, hDOHH-P exhibits spectroscopic properties similar to, yet distinct from, those of the peroxo intermediates associated with the canonical nonheme diiron enzymes [21–30].
Scheme 1.

Biosynthetic pathway of hypusine on eIF5A. Reproduced with permission from ref 20.
O2 activation by the majority of diiron enzymes is initiated by dioxygen binding to a diferrous center, generating a diferric peroxo species [31]. Subsequent O–O bond cleavage generates a high-valent diiron-oxo center (Fe(III)Fe(IV) or Fe(IV)2) that effects substrate oxidation [5, 32], but the detailed steps in the mechanisms by which the O–O unit is converted to the active oxidizing species remains unclear. Most of the enzymatic diferric peroxo species identified to date have fleeting half-lives on the order of seconds to a few minutes [30, 33–35], making characterization of these intermediates quite challenging. Additionally, most of the peroxo species described to date can only be accumulated through mutagenesis of the wild type (WT) enzyme [22, 24] or the use of reaction conditions that do not produce product [27]. In contrast, the hDOHH-P intermediate can persist for days. However, the binding of the eIF5A(Dhp) substrate to hDOHH-P significantly accelerates the decay of the peroxo intermediate and concomitantly generates eIF5A(Hpu) product [20]. Unlike members of the ferritin-like protein superfamily, hDOHH does not have a four-helix-bundle structural motif that provides the two histidine and four carboxylate residues that comprise the diiron active site. Instead, hDOHH utilizes HEAT repeat motifs to support a 4-His-2-carboxylate diiron active site, as established by site-directed mutagenesis studies [13] and the recent crystal structure of hDOHH [36].
Our initial study of hDOHH-P by X-ray absorption spectroscopy (XAS) revealed an Fe•••Fe distance of 3.44 Å, but the quality of the data limited the information that could be extracted about the active site [20]. In this follow-up of our earlier effort, we have obtained better XAS data on hDOHH, focusing on five different samples along the reaction pathway, starting with the reduced diferrous state and ending with a diferric product. XAS analysis of these samples reveals that the diiron center of hDOHH maintains a relatively invariant Fe•••Fe distance throughout its redox cycle despite changes in oxidation state, in contrast to the larger variations in Fe•••Fe distance observed for sMMO and the R2 subunit of RNR [23, 37–41], the canonical members of the family of nonheme diiron enzymes. This physical constraint imposed on the diiron center of hDOHH has implications on how the peroxo O–O bond can be cleaved and leads us to propose an O2 activation mechanism for hDOHH that is distinct from that for sMMO and RNR R2.
Results
We have used Fe-K-edge X-ray absorption spectroscopy (XAS) to gain insight into the the diiron active site structures of a series of hDOHH samples: chemically reduced diferrous hDOHH (hDOHH-R), diferric peroxo hDOHH (hDOHH-P), hDOHH-P bound with substrate, eIF5A(Dhp) (hDOHH-P•S) and the diferric species following decay of hDOHH-P, both in the presence and absence of substrate (hDOHH-D•S and hDOHH-D, respectively). hDOHH-P is the as-isolated form of the enzyme, hDOHH-R was generated by reacting hDOHH-P with dithionite until fully reduced, and hDOHH-P•S was prepared by adding eIF5A(Dhp) to a solution of the peroxo species. The peroxo-to-Fe(III) LMCT transition is maintained in both hDOHH-P and hDOHH-P•S (Figure S1). hDOHH-D was generated by allowing hDOHH-P to decay at room temperature in an XAS cup until the feature at A630 had reached a minimum value, resulting in a yellow colored species. Similarly, hDOHH-D•S was prepared by thawing the hDOHH-P•S sample and allowing the hydroxylation reaction to run for 72 hours at room temperature in an XAS cup. The previously reported XAS data for hDOHH-P was recorded with a k range of only 2–11.8 Å−1 and the sample showed evidence of significant photoreduction [20]. Here we present new hDOHH-P data with an improved k range of 2–13.5 Å−1 and a minimal degree of X-ray photoreduction.
X-ray absorption near edge structure (XANES) analysis provides information regarding the oxidation state (via the K-edge energy) and symmetry of the diiron centers (via the pre-edge area) in an enzyme active site. The five hDOHH species exhibit Fe K-edge energies consistent with oxidation state assignments that we had previously made by Mössbauer spectroscopy or deduced by UV-vis spectroscopy. The Fe K-edge energy for hDOHH-R was found to be 7122.7 eV (Figure 1, Table 1), which is similar to the K-edge energies reported for the diferrous forms of an R2-like ligand-binding oxidase (7121.4 eV) [42] and the ferroxidase site of frog M ferritin (7122.0 eV) [29]. In addition, previous Mössbauer analysis of hDOHH-R characterized the metal sites as high spin (S = 2) ferrous iron [20]. The Fe K-edge energy of 7125.6 eV for hDOHH-P is consistent with our previous analysis [20], and is approximately 3 eV higher than that of hDOHH-R. Mössbauer spectroscopy of hDOHH-P established that the metal centers are in the high spin (S = 5/2) ferric state and are antiferromagnetically coupled [20]. Assuming that the Kedge energy correlates to oxidation state, only a 1 eV jump would be anticipated in going from Fe(II)2 in hDOHH-R to Fe(III)2 in hDOHH-P. However, in transition metal complexes the ligand identity and hardness [43], effective nuclear charge [44], metal-ligand bond length [45] and spin state of the metal [46] all affect the K-edge energy. Given this and the lack of a systematic study of K-edge energies in diiron enzyme systems, the source of this 3-eV difference is not clear. hDOHH-P•S, hDOHH-D, and hDOHH-D•S all have K-edge energies similar to hDOHH-P (Table 1), consistent with a diferric center in the peroxo and the decayed species in the presence and the absence of substrate.
Figure 1.

XANES region of hDOHH-R (black dotted), hDOHH-P (blue solid), hDOHH-P•S (green dot dash) and hDOHH-D (red dash). Inset: zoom in of the pre-edge region.
Table 1.
XANES Analysis
| Species | K-edge energy (eV) | Peak Position (eV) | Peak Area (units) |
|---|---|---|---|
| hDOHH-R | 7122.7 | 7111.7 | 8.6 |
| hDOHH-P | 7125.6 | 7113.8 | 12.4 |
| hDOHH-P•S | 7124.9 | 7114.1 | 16.2 |
| hDOHH-D | 7125.2 | 7114.7 | 7.8 |
| hDOHH-D•S | 7125.6 | 7114.7 | 8.6 |
The pre-edge peak in the XANES region corresponds to forbidden 1s → 3d transitions in transition metal complexes [47]. The intensity of this transition is dependent on the degree of metal 4p mixing into 3d states, and increases as the metal center is distorted from centrosymmetry [48]. By comparing the area under the pre-edge peak, information about the symmetry and general coordination environment of the diiron site can be inferred. The pre-edge feature for hDOHH-R is fit by two pseudo-Voigt functions and is centered at 7111.7 eV with an area of 8.6 units (Table 1). This area falls between values typical of 6-coordinate (~5 units) and 5-coordinate diferrous species (~11 units) [49]. The pre-edge feature of hDOHH-P is centered at 7113.8 eV with an area of 12.4 units. This value is consistent with our previously published data [20] and higher than observed for hDOHH-R. hDOHH-P is likely 6-coordinate by comparison to synthetic 6-coordinate diferric peroxo complexes, which have pre-edge values that range from 13 – 16 units [50–52]. In contrast, hDOHH-P•S has a pre-edge feature centered higher in energy at 7114.1 eV with an area of 16.2 units, increased from hDOHH-P, indicating that the addition of substrate has decreased the symmetry around the diiron center. hDOHH-D and hDOHH-D•S have a feature centered around 7114.7 eV with areas of 7.8 and 8.6 units, respectively. These values are lower than observed for hDOHH-P and are consistent with those of synthetic 6-coordinate (μ-hydroxo)diferric centers, with reported areas of 5 – 9 units [53].
Additional structural information can be determined from Extended X-ray Absorption Fine Structure (EXAFS) analysis, which provides scattering distances for the ligands and close contacts near the Fe centers. The final fits for each species are presented in Table 2, and fit tables for individual complexes can be found in Supplementary Information (Tables S1 – S6).
Table 2.
EXAFS Fit Distances (Å) of hDOHH Speciesa
| Species | N | hDOHH-R | N | hDOHH-P | N | hDOHH-P•S | N | hDOHH-D/D•S | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe•••Fe (Å) | 1 | 3.47 | (5.18) | 1 | 3.41 | (1.86) | 1 | 3.41 | (5.44) | 1 | 3.42 | (5.09)/(3.51) |
| Fe-N/O (Å) | 4 | 2.18 | (2.78) | 3 | 2.15 | (1.95) | 4 | 2.11 | (5.61) | 4 | 2.09/2.08 | (4.45)/(4.40) |
| Fe-O/N (Å) | 2 | 2.07 | (3.39) | 3 | 1.98 | (4.57) | 1 | 1.98 | (4.47) | 2 | 1.95 | (2.88) |
| Fe•••C (Å) | 3 | 3.10 | (6.20) | - | - | 3 | 3.09 | (6.50) | 3 | 3.08 | (4.90)/(6.29) | |
| 3 | 3.68 | (4.05) | 3 | 3.58 | (2.93) | 3 | 3.56 | (3.51) | 3 | − /3.59 | − /(3.30) | |
| 3 | 4.35 | (2.51) | 4 | 4.29 | (1.55) | 4 | 4.30 | (1.72) | ||||
Numbers in parenthesis are σ2 values in units of 10−3 Å2. See SI for individual fitting tables.
The primary coordination sphere of hDOHH-P consists of 3 Fe-N/O scatterers at 2.15 Å with a relatively low Debye-Waller factor (σ2) of 1.95 × 10−3 Å2 (Table 2) and 3 Fe-O/N at 1.98 Å with higher σ2 values (4.45 × 10−3 Å2). The higher σ2 value for the latter Fe-O/N shell suggests a broader range of distances for the scatterers comprising the 1.98-Å shell. Second and third sphere Fe•••C are fit at 3.58 Å and 4.29 Å, respectively, with reasonable σ2 values. The Fe•••Fe distance fit for hDOHH-P is 3.41 Å, which agrees quite well with the previously reported value of 3.44 Å [20].
The primary coordination sphere of hDOHH-R consists of two shells, both with reasonable σ2 values (Table 2). The first shell contains 4 Fe-N/O scatterers at 2.18 Å, and the second shell contains 2 Fe-O/N scatterers at 2.07 Å. However these two shells are within 0.11 Å of each other, which are just slightly outside of the resolution of the data. The resolution to distinguish one shell from another in a given fit for hDOHH-R is determined by the equation:
where ΔR is the resolution and Δk is the difference in the k-space range used. For hDOHH-R, ΔR = 0.12 Å. Although these shells are outside the ability to resolve the two shells (0.11 Å), there is an improvement in fit when the 2.07 Å shell is included (Table S1, Fit 12 vs Fit 18). Consequently, this shell was included in the best fit for hDOHH-R. The Fe•••Fe distance for this species was fit at 3.47 Å, which is only slightly longer than the Fe•••Fe distance of 3.41 Å observed in hDOHH-P. Three additional carbon shells were found at 3.10 Å, 3.68 Å and 4.35 Å.
Fitting of hDOHH-D resulted in two acceptable fits of the data (Fits A and B, Table S4 and S5). Both fits A and B have similar primary shells and reasonable σ2 values with 4 Fe-N/O at 2.07 Å and 2.09 Å, respectively. An additional Fe-O/N shell with two scattering atoms is needed for hDOHH-D, at similar distances of 1.93 Å for fit A and 1.95 Å for fit B. Significant differences between fits A and B are evident in the Fe•••Fe distances as well as the carbon scattering shells. Fit A has a shorter Fe•••Fe distance at 3.07 Å with a σ2 of 9.87 × 10−3 Å2 and 5 Fe•••C at 3.41 Å, while fit B has a longer Fe•••Fe distance at 3.42 Å with a σ2 of 5.09 × 10−3 Å2 and 3 Fe•••C at 3.08 Å. Fit A resembles a bis-hydroxo “diamond-core”-like species, if the two Fe-O bonds at 1.93 Å were assigned to μ-hydroxo ligands with a metal-metal separation of 3.07 Å (synthetic models have Fe•••Fe distances between 2.8 Å and 3.1 Å [54–60]). However, the σ2 for the Fe scatterer at this short distance is unreasonably high. Moreover, a carbon scatterer at 3.41 Å seems unlikely as this has not, to our knowledge, been observed in previously reported species and would require significant movement of the histidine rings (vide infra). Fit B differs from fit A by switching the assignments for the scatterers at ~3.1 Å and ~3.4 Å. This model structure would accommodate the longer Fe•••Fe distance of 3.42 Å and has a more reasonable σ2 value than fit A. Additionally, the carbon scatterer at 3.08 Å is more clearly assigned to the ligands bound to the iron centers (see Discussion). For these reasons, fit B is favored over fit A for hDOHH-D, with fit B being more consistent with a single-atom-bridged “open core” species (Scheme 2), with the single atom bridge falling into the shell at 1.95 Å. Our preference of fit B for hDOHH-D is supported by the best fit to hDOHH-D•S, which consists of four Fe-N/O scatterers at 2.08 Å, two Fe-O/N at 1.95 Å, one Fe•••Fe at 3.42 Å, and three Fe•••C at both 3.08 Å and 3.59 Å (Table 2). This congruence in the fits of hDOHH-D and hDOHH-D•S strengthens our argument for a 3.42 Å Fe•••Fe distance in the decayed species.
Scheme 2.

Models derived from the EXAFS fits from the samples shown above
The best fit for hDOHH-P•S consists of 1 Fe-O/N at 1.98 Å and 4 Fe-N/O at 2.11 Å (Table 2). The changes in distance and the number of scatterers for each shell in the first coordination sphere, together with the previously noted increase in pre-edge area, suggest that substrate binding causes a distortion of the diiron active site prior to O–O bond cleavage. The Fe•••Fe distance however remains unchanged at 3.41 Å. Additional Fe•••C shells at 3.09 Å, 3.56 Å and 4.30 Å are required to achieve a reasonable fit, similar to hDOHH-R.
Discussion
The importance of peroxo intermediates in the dioxygen activation chemistry of diiron enzymes is underscored by their prominent role in the catalytic cycles proposed for these enzymes. However, detailed study of the chemistry of O–O activation is challenging due to the general instability of these peroxo species and the protein modifications required to observe them [22, 24, 27, 30, 33, 35]. Here, we report a detailed XAS analysis of the diferric-peroxo intermediate from the human hydroxylase, hDOHH. This enzyme is unique within its class as it is purified as a long-lived diferric-peroxo species, thereby facilitating its characterization.
Despite its stability, hDOHH-P has been shown to carry out substrate hydroxylation [20], making it the first catalytically competent peroxo intermediate to be characterized from a native enzyme. Our XAS analysis has provided structural information on five species in the reaction cycle. EXAFS analysis of the five hDOHH samples we have studied show two prominent features in the Fourier-transformed data (see Figures S3, S5, S7, S10, S12). The taller feature between R+Δ 1.5 – 1.7 Å is assigned to scatterers in the primary coordination sphere of the iron atoms of the diiron active site. The second, less intense feature at R+Δ ~ 3.0 Å is comprised of contributions arising mainly from the other iron atom. Included in the first shell scatterers are the ligating atoms of one glutamate and two histidine residues on each metal center. This ligation scheme was initially deduced from sequence comparisons and site-directed mutagenesis [13] and recently identified from the crystal structure of the enzyme [36]. Crystallographically, imidazole ligands typically give rise to Fe–N bond distances between 2.0–2.5 Å that average to 2.2 Å irrespective of whether the iron is in the +2 or +3 oxidation state [61–69]. This is consistent with the distances observed in all five hDOHH species studied here, with distances ranging from 2.08 – 2.18 Å. Typical Fe–O bond distances found for carboxylate ligands span a similar range of values (1.9 – 2.6 Å) [61–69] but can vary depending on the binding mode of the carboxylate, i.e. terminal vs bridging or monodentate vs bidentate. In addition to the protein-derived ligands, the diiron center is likely to have solvent-derived ligands with distances that vary among the five hDOHH species, suggesting some variation in the identities and binding modes of these solvent derived ligands but not in the protein derived ligands. Finally, hDOHH-P and hDOHH-P•S contain one additional ligand unique to these species – a bound O2 molecule. Our data suggests that dioxygen is likely bound to the diiron(III) center as a 1,2-peroxo bridge [20]. Based on the available structures for synthetic peroxo-bridged diiron(III) complexes [50, 51, 70–72], such a ligand would be expected to have Fe–O bond distances of 1.87 – 1.98 Å, consistent with our observed distance of 1.98 Å.
Interestingly, the Fe•••Fe distances found for the five hDOHH complexes vary within a narrow range of 3.41 – 3.47 Å, suggesting that the diiron core dimensions do not change significantly as it undergoes redox transformations. Similar Fe•••Fe distances have been observed in other diiron proteins such as deoxyhemerythrin and reduced sMMO, where the diiron unit has a single-atom bridge and additional bidentate carboxylate bridges [9, 23, 29, 38, 62, 63, 66]. However the hDOHH crystal structure [36] suggests that the two carboxylate ligands of hDOHH are not well positioned to bridge the diiron center but instead bind as terminal ligands. Both the core architecture and the invariant Fe•••Fe distance are features that differ significantly from the approximately 1-Å change in Fe•••Fe distance observed as the diiron active sites of sMMO and E. coli RNR convert from the diferrous forms to their respective high-valent intermediates [38, 41, 73]. Presumably, these differences reflect changes in the redox chemistry associated with the diiron centers of each enzyme.
Analysis of fits of the EXAFS data for the five samples leads to the proposed diiron site structures shown in Scheme 2. Based on the iron-ligand bond length comparisons presented above, the six scatterers observed for the first coordination sphere of each Fe atom in the EXAFS fit of hDOHH-R can be reasonably assigned in the following manner. The carboxylate and two histidine ligands on each Fe would give rise to one of the two 2.07-Å scatterers and two of the four 2.18-Å scatterers. The three remaining scatterers on each Fe most likely derive from solvent derived exogenous ligands. The second scatterer at 2.07 Å would correspond to a hydroxo bridge between the two ferrous ions, as this distance matches the average FeII-(μ-OH) distance (2.072 Å) found for a number of (μ-hydroxo)diferrous complexes, which have FeII–OH distances ranging from 1.99 to 2.21 Å (Table S8) [74–76]. On the other hand, corresponding complexes with bridging aqua ligands typically have longer FeII-μ-OH2 distances and exhibit a wider range of values (2.13 – 2.40 Å, Table S8) [74, 77, 78]. A particularly useful diiron(II) complex for comparison is [FeII2(μ-OH)(μ-OH2)(TPA)2]3+ (TPA = tris(2-pyridylmethyl)amine) [74], which was found to have respective FeII-μ-OH and FeII-μ-OH2 bonds averaging 2.07 and 2.17 Å, values that support our proposed assignment of the 2.07-Å scatterer in hDOHH-R as a hydroxo bridge. The remaining two scatterers at 2.18 Å have Fe–O distances consistent with either terminal (2.04 – 2.16 Å, Table S8) [51, 77, 79–83] or bridging water ligands (2.13 – 2.40 Å) [77, 78], but the Fe•••Fe distance of 3.22 Å observed for [FeII2(μ-OH)(μ-OH2)(TPA)2]3+ is too short to match the distance deduced for hDOHH-R.
hDOHH-D and hDOHH-D•S are diferric species that form upon decay of hDOHH-P respectively in the absence and the presence of the eIF5A substrate, which is indicated by the loss of the visible chromophore associated with the peroxo intermediate. As reported previously, eIF5A binding to hDOHH-P significantly destabilizes this intermediate and results in its immediate decay [20]. These two decayed samples give rise to very similar EXAFS spectra and fits. Like hDOHH-R, the first coordination spheres of hDOHH-D and hDOHH-D•S are best fit with two shells of N/O scatterers, two at 1.95 Å and four at 2.11 Å. The decrease in the Fe-ligand distance for each shell relative to those of hDOHH-R is rationalized by the change in iron oxidation state from +2 to +3. We propose that the 2.11-Å shell consists of the three protein-derived ligands as well as a terminal solvent ligand, while the 1.95-Å shell can be reasonably assigned to hydroxo ligands. One of the latter scatterers very likely corresponds to the μ-OH bridge found in hDOHH-R, as analogous bridges in synthetic diferric complexes have FeIII-(μ-OH) distances between 1.94 Å and 2.02 Å [55, 60, 84–87]. The other scatterer could, in principle, be assigned to a second hydroxo bridge, but such an FeIII2(μ-OH)2 core should give rise to an Fe•••Fe distance much shorter than the 3.41-Å separation found for hDOHH-D and hDOHH-D•S. Alternatively, the other 1.95-Å scatterer could arise from a terminal hydroxo ligand on each Fe, as synthetic high-spin FeIII–OH units have bond distances that fall within the range of 1.82 – 1.93 Å [88–96]. This formulation would also maintain the +1 charge associated with the diiron sites in hDOHH-R and hDOHH-P (see below).
The best fit for hDOHH-P shows N/O scatterers at 2.15 Å and 3 O/N scatterers at 1.98 Å. The 2.15-Å shell likely consists of two histidine ligands and a terminal water ligand based on bond metrics discussed earlier. The 1.98-Å shell would comprise a terminal carboxylate ligand, the hydroxo bridge (1.94 – 2.02 Å) [55, 60, 85–87, 97], and the proximal oxygen of a μ-1,2-peroxo ligand (1.86 – 1.94 Å) [52, 70–72]. These results compare well to the parameters found by Suzuki and co-workers in the crystal structure of [FeIII2(μ-OH)(μ-1,2-O2)(L)2]+ (L = bis(6-methylpyridyl-2-methyl)-3-aminopropionate), which has an Fe•••Fe distance of 3.396 Å and average Fe–O and Fe–N distances of 1.95 Å and 2.21 Å, respectively [85].
The complex of hDOHH-P with its substrate eIF5A(Dhp), which we refer to as hDOHH-P•S, was also prepared and analyzed by EXAFS. Figure 2 shows an overlay of the Fourier transformed (FT) data for hDOHH-P and hDOHH-P•S, revealing obvious differences between these two species. The feature at R+Δ ~1.5 Å representing the first coordination sphere is shifted to a slightly longer distance in hDOHH-P•S compared to hDOHH-P. Also, there is a notable feature at ~1.8 Å in hDOHH-P that is absent in hDOHH-P•S. Lastly, the feature at R+Δ ~ 3 Å, assigned to contributions mainly from the second iron atom, becomes less intense and shifted to a slightly lower R value relative to that in hDOHH-P. As hDOHH-P and hDOHH-P•S have identical Fe•••Fe distances based on their respective EXAFS fits, the differences in the feature at R+Δ ~ 3 Å may arise from changes in the multiple scattering pathways that contribute to this peak. This shows that the hDOHH-P active site changes upon substrate binding to hDOHH-P, demonstrating that eIF5A binds to the hDOHH enzyme.
Figure 2.

Fit (solid line) of the unfiltered (dotted) EXAFS data (inset) and corresponding Fourier transform. k = 2 – 13.5 Å−1; hDOHH-P (blue, Figure S5) hDOHH-P•S (red, Figure S7).
Intriguingly, the Fe•••Fe distance of hDOHH-P is not significantly perturbed by the addition of substrate. However, the changes in the first coordination sphere noted above upon substrate binding result in a larger pre-edge area for hDOHH-P•S (16.2 units vs 12.4 for hDOHH-P), which is consistent with a larger distortion from octahedral symmetry engendered by a decrease in iron coordination number from 6 to 5. This change is accompanied by alterations in the composition of the first shell of scatterers in the best fit found for hDOHH-P•S. Although the average Fe–N/O bond distances for the two hDOHH complexes differ by just 0.01 Å, the shorter 1.98-Å shell now has only one scatterer, while the other shell now consists of four Fe–N/O scatterers at 2.11 Å. Attempts to increase the number of scattering atoms in either the 1.98 Å or 2.11 Å shells resulted in substantial increases in the σ2 values for the shell of interest (Table S3, Fits 19 and 20). The shortening of a three-scatterer shell at 2.15 Å in hDOHH-P to a four-scatterer shell at 2.11 Å corresponds to the shift of a 1.98-Å scatterer into the shell at longer distance. Clearly, there is some rearrangement in the iron coordination spheres upon substrate binding to hDOHH-P.
In the fit of hDOHH-P, the 1.98-Å subshell consists of three O scatterers, which we have assigned to the μ-OH bridge, the 1,2-bridging peroxo, and a terminal carboxylate ligand. In hDOHH-P•S, this shell consists of only one scatterer. As there is no change in the peroxo charge transfer band at 630 nm (Figure S1), it would seem likely that the peroxo ligand remains as the only scatterer at 1.98 Å. The carboxylate ligand and the hydroxo bridge would presumably shift into the 2.11-Å shell together with the two His residues concomitant with the loss of the terminal aqua ligand, but we have insufficient data to establish the fates of these ligands.
For all five of the hDOHH samples in this study, there are scatterers included in the fits at distances ranging from 3.1 – 4.3 Å that are likely derived from the imidazole rings of the histidine ligands, as observed for the diiron sites in sMMO and ToMO [37, 98], methemerythrin and RNR R2 [99], and CmlA [100]. Histidine ligands typically give rise to scatterers at 3.1 and 4.3 Å, which are respectively associated with the C atoms adjacent to the coordinated N atom and the C and N atoms further away (Figure 3). Interestingly, no scatterer at ~3.1 Å is required in the fit for hDOHH-P. The absence of this carbon shell in hDOHH-P could be due to slightly different orientations of the imidazole ring relative to the Fe-NHis bond for the four histidines bound to the diiron center, which lead to destructive interference that decreases the contributions of these scatterers to the EXAFS spectrum of hDOHH-P. This is visually apparent in the relative intensities of the second-sphere peaks at ~ 3 Å in the Fourier transforms of hDOHH-P and hDOHH-P•S (Figure 2). Similarly, the fits for hDOHH-D and hDOHH-D•S do not require scatterers at ~4.3 Å. A similar phenomenon was reported for reduced MMOH with the appearance of a new light atom scatterer between 3.2 Å and 4.0 Å upon binding of the accessory protein MMOB, [37] which was rationalized by the contributing amino acid residues becoming more ordered in the MMOH:MMOB complex. We propose that a different ordering of the active site affects the outer-sphere contributions of ligands in hDOHH, particularly the imidazole rings of histidine, which results in the disappearance of light atom scatterers from the EXAFS fit.
Figure 3.
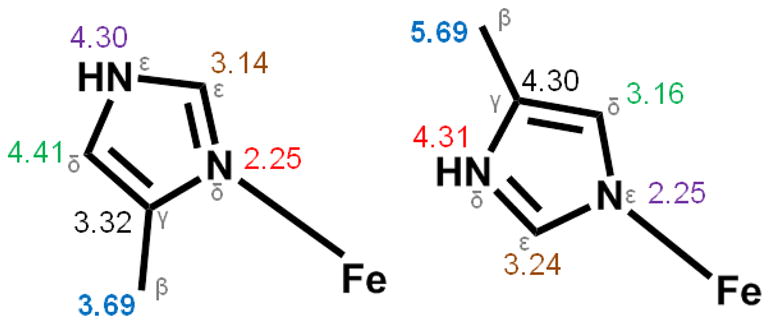
Comparison of average Fe•••C/N distances of His ligands bound to Fe at Nδ (left) and Nε (right). Color corresponds to the same atom position in the imidazole ring of His.
Interestingly, there is an additional scattering interaction at ~3.6 Å observed in most of the species studied here. This feature does not arise from an iron scatterer, as our attempts to introduce an iron atom at 3.6 Å always refined to a distance of 3.4 Å. Moreover, the inclusion of a light scattering atom at this distance improves the fit (See Tables S1 – S6). A scatterer at a similar distance has been found in EXAFS studies of sMMO and assigned to multiple scattering pathways involving the β carbon of an Nδ-bound His [39]. To find additional support for this assignment, Fe•••C distances were collected from available diiron protein crystal structures with Nδ-bound histidine ligands [61–67] or Nε-bound histidine ligands [61, 62, 68, 69] (Figure 3). His ligands bound to Fe through Nδ have Fe•••Cβ distances ranging from 3.5 Å to 3.9 Å and Fe•••Cγ distances ranging from 3.1 – 3.6 Å, while those bound to Fe through the Nε position have Fe•••Cε distances of 3.1–3.5 Å. The 3.58 Å scatterer fits within all of these observed ranges. However, the Fe-N bond lengths that relate to the respective Fe•••C distances can suggest which options best agree with the 3.58 Å scatterer. With a Nε-bound His ligand and an Fe•••Cε interaction at ~3.6 Å, the Fe-N distance is around 2.3 Å. The same is true for an Nδ-bound His with an Fe•••Cγ distance of ~3.6 Å. These Fe–N distances are longer than our experimentally determined Fe–N range of 2.1–2.2 Å, and so do not support assignment of the 3.58 Å scatterer. A Nδ-bound His with an Fe•••Cβ interaction has an Fe–N distance of ~2.2 Å, which agrees with both the multiple scattering and Fe-N observations. We thus propose that the available crystallographic data supports the assignment of the ~3.6 Å scatterer as arising from the Cβ-atom of an Nδ-bound His ligand in the XAS samples analyzed herein.
A 1.7-Å resolution crystal structure of a truncated form of hDOHH-P (hDOHH-PT) was recently reported by Han et al. [36] (PDB ID 4D50), providing the first crystallographic information about this interesting enzyme (Figure 4). This structure confirmed the HEAT repeat protein motif predicted by Park and coworkers [13] that distinguishes this enzyme from most diiron enzymes, which are typically found to use a 4-helix bundle structural motif [1, 2, 11]. Additionally, the histidine-rich coordination environment for the diiron center predicted by site-directed mutagenesis experiments [13] was supported by this structure. Lastly, the active site was deduced to have a (μ-solvento)(μ-1,2-peroxo)diiron core structure as suggested by our earlier spectroscopic analysis [20]. However, the metrical information provided by the crystal structure about the diiron site differs considerably from what we have extracted from our XAS analysis. On average, the Fe-ligand distances appear to be 0.1–0.2 Å longer in the crystallographically derived data compared to our EXAFS measurements (Table 3), but the latter are likely to be of greater precision (± 0.02 Å) [101].
Figure 4.

Diiron site of hDOHH-P generated by PyMOL using PDB ID 4D50. Fe atoms are shown as brown spheres, while peroxo O-atoms are shown as red spheres. Residues from the N-terminal domain are in blue, while residues from the C-terminal domain are in green. Amino acid residues coordinating the diiron site are shown in stick representation. The left panel shows the diiron site within the HEAT repeat motif, while the right panel shows a zoomed-in view showing the peroxodiiron unit within a hydrophobic cleft, with access for the substrate presumably via the less restricted approach.
Table 3.
Comparison of Fe–X distances found in hDOHH-P and hDOHH-PT
| Ligand type | hDOHH-P (Å) | hDOHH-PT (Å) |
|---|---|---|
| Fe•••Fe | 3.41 | 3.7 |
| Fe-N(His) | 2.15 | 2.3 |
| Fe-O(Glu) | 1.98 | 2.1 |
| Fe-OH2 | 2.15 | 2.2 |
| Fe-μ-OH | 1.98 | 2.2 |
| Fe-O(peroxo) | 1.98 | 2.2 |
Even more significantly, the diiron core dimensions from the XRD and EXAFS analyses are distinct. The Fe•••Fe distance found in the crystal structure of hDOHH-PT is 0.3 Å longer than the value found by EXAFS (Table 3). The authors of the protein structure paper suggest that the EXAFS-deduced μ-hydroxo bridge may have become protonated during the 48-hour period needed for crystallization, resulting in longer Fe–Obridge bonds and, consequently, a larger Fe•••Fe distance. Alternatively, we suggest that the observed elongation of the Fe•••Fe distance may result from photoreduction of the diiron(III) center during the XRD experiment, which is a well-recognized problem in metalloenzyme crystallography [102–104]. Photoreduction would also rationalize the elongation of the Fe-ligand bonds observed in the XRD study. Indeed, we have previously noted that hDOHH-P is sensitive to photoreduction even under the comparatively mild XAS conditions, as evidenced by an observed downshift in the K-edge energy with increasing exposure to the synchrotron beam [20]. In the XAS experiments reported in this paper, we minimized the effect of photoreduction on our analysis by moving the X-ray beam during the XAS experiments to a fresh spot on the sample surface after each scan.
There are also spectroscopic differences between hDOHH-PT used in crystallography experiments and the hDOHH samples prepared for the XAS studies. Han et al. note that the visible absorption maximum of hDOHH-PT is blue-shifted from ~630 nm to 600 nm, suggesting some perturbation of the peroxo-to-iron(III) charge transfer chromophore. Moreover, while the Mössbauer spectrum collected for the hDOHH-PT solution sample showed quadrupole doublets with parameters (δ1/2 = 0.57/0.55 mm s−1, ΔEQ1/2 = 0.85/1.17 mm s−1) similar to those previously reported for hDOHH-P (δ1/2 = 0.58/0.55 mm s−1, ΔEQ1/2 = 0.88/1.16 mm s−1) [20], the crystalline material exhibited somewhat different parameters (δ1/2 = 0.62/0.62 mm s−1, ΔEQ1/2 = 0.72/1.25 mm s−1), hinting at a possible structural change.
There may be additional differences in how the His residues are bound to the diiron center. In the crystal structure, they are shown to bind through the Nε atom, whereas XAS analysis suggests binding via Nδ, due to the presence of a ~3.6 Å carbon scattering shell that is assigned to the His β carbon (vide supra). The His ligands are typically Nδ-bound in a number of other crystallographically characterized diiron enzymes [61, 63–67], with the notable exceptions of hemerythrin [68] and mammalian desaturase [105] where all iron-bound His residues are Nε-bound. At the present time, we cannot rationalize this difference between the XRD and XAS results.
An important generalization for the hDOHH series is that the Fe•••Fe distance determined from the XAS studies remains relatively invariant as hDOHH-R (3.47 Å) progresses through hDOHH-P and hDOHH-P•S (3.41 Å) and eventually to hDOHH-D (3.42 Å) following decay of the peroxo intermediate. In contrast, larger changes in the Fe•••Fe distances are observed for several diiron enzymes upon oxidation of diiron(II) forms to diiron(III) (Table 4). In the case of sMMO, the Fe•••Fe distance contracts by 0.3–0.4 Å by substitution of the monodentate carboxylate bridge in the (μ-1,1-carboxylato)(μ-1,3-carboxylato)diiron(II) core by two hydroxo bridges in the diiron(III) core. For RNR R2 and the fatty acid desaturases, the change in distance upon oxidation of the diferrous form to the diferric form can be even more dramatic from ~4.0 Å to ~3.1 Å in the conversion of the bis(μ-1,3-carboxylato)diiron(II) core to a (μ-oxo)(μ-1,3-carboxylato)diiron(III) core. In these examples, the binding mode of a bridging carboxylate plays a key role in the changes in Fe•••Fe distance due to their versatility in binding a metal center, namely monodentate or bidentate as well as terminal or bridging [110]. However, such a role for carboxylates in hDOHH appears unlikely, as there are only two carboxylate ligands, both of which are positioned within the active site such that only a terminal binding mode is plausible. To maintain the 3.4–3.5 Å Fe•••Fe distance in hDOHH throughout its catalytic cycle, we propose that the Fe–OH–Fe unit remains intact throughout the series. Further constraints to the Fe•••Fe distance may also be imposed by the unique HEAT repeat protein fold that houses the diiron center in hDOHH, which significantly differs from the 4-helix bundle motif that supports the diiron centers of many nonheme diiron enzymes. In the crystal structure of hDOHH-PT [36], each iron is ligated by a histidine and a glutamate from one protein domain and by a second histidine that derives from the second domain (Figure 4, left panel), creating a cross-domain interaction that may rigidly position the bound diiron center and could be responsible for the limited change in the Fe•••Fe distance.
Table 4.
Fe•••Fe Distances (Å) of Related Diiron Cores in Enzymesa
| Species | FeIIFeII | FeIIIFeIII | Peroxo | FeIII/IVFeIV |
|---|---|---|---|---|
| sMMO | 3.4 [106] | 3.0 [64] | - | 2.46 [38] |
| 3.43 [38], 3.29 [37] | 3.02 [40], 3.03 [39] | |||
| Ec RNR R2 | 3.9 [65] | 3.3 [107] | 2.50 [23] | 2.79 [41] |
| 3.41 [23] | 3.22 [41] | |||
| St RNR R2 | 3.7 [66] | 3.3 [66] | - | - |
| Ferroxidase center of frog M ferritin | 3.43 [29] | 2.99 [29] | 2.53 [29] | - |
| Fatty acid desaturases | 4.1 [67] | 3.2 [108] | - | - |
| 3.13 [109] | ||||
| hDOHH | 3.47 b | 3.42 b | 3.41 b | - |
Distances shown in italics derived from X-ray crystallography; all other distances obtained from XAS studies.
This work.
Closer scrutiny of the hDOHH-PT structure [36] shows the diiron unit to be buried in a narrow hydrophobic wedge-like pocket, with the peroxo moiety pointing away from the wider mouth of the pocket where eIF5A is proposed to bind hDOHH (Figure 4, right panel). The location and the hydrophobic environment of the peroxo pocket may account for the impressive stability of the peroxo intermediate. However, with this orientation for the intermediate as a starting point, the peroxo ligand would have to undergo rearrangement for it to be effective in substrate hydroxylation. We thus propose a mechanism shown in Scheme 3 that begins with the (μ-hydroxo)(μ-1,2-peroxo)diiron(III) active site as described by our EXAFS analysis (Scheme 3 A), with substrate binding initiating a series of steps (Scheme 3 B-F) that lead to formation of the high-valent diiron oxidant that cleaves the substrate C–H bond. Substrate binding causes the loss of the terminal water ligand on the left Fe followed by the shift of the hydroxo bridge to become a terminal ligand on that Fe (Scheme 3 B). These changes result in the decrease in coordination number for both Fe’s, allowing the μ-1,2-peroxo bridge to twist and isomerize into a side-on bound μ-η2:η2-peroxo moiety (Scheme 3 C). This conversion activates the O–O bond for cleavage to generate a diiron(IV) intermediate (Scheme 3 D) that carries out substrate hydroxylation (Scheme 3 E & F).
Scheme 3.

Proposed mechanism of O–O bond scission and substrate hydroxylation by hDOHH. A is hDOHH-P, B is hDOHH-P•S, and F is hDOHH-D•S. C, D, and E correspond to yet unobserved intermediates on the reaction pathway.
The high-valent diiron intermediate proposed for hDOHH has a core structure distinct from that associated with sMMO intermediate Q. The latter has been shown by EXAFS analysis and resonance Raman spectroscopy to have a bis(μ-oxo)diiron(IV) diamond core [38, 111]. The Raman evidence that both oxygen atoms bridging the two Fe’s in Q derive from one O2 molecule supports the hypothesis of Banerjee et al. that a (μ-η2:η2-peroxo)diiron(III) moiety serves as the precursor to Q [111]. Our mechanistic proposal relies on this precedent. The more compact core structure established for Q with an EXAFS-deduced Fe•••Fe distance of 2.46 Å may not be able to form within the constraints of the hDOHH active site, for which a relatively invariant Fe•••Fe distance (3.4–3.5 Å) is found for all five hDOHH complexes described in this study. Importantly, both the (μ-η2:η2-peroxo)diiron(III) moiety and the open-core isomer of the bis(μ-oxo)diiron(IV) diamond core, respectively proposed in Scheme 3 C & D, would fit within the Fe•••Fe distance constraints of the hDOHH active site. Although synthetic complexes with (μ-η2:η2-peroxo)diiron(III) cores have yet to be described, corresponding dicopper complexes are well known and exhibit Cu•••Cu distances of 3.6 Å for those with planar Cu2O2 units [112–114]; in one example, the Cu•••Cu distance can be decreased by conversion to a nonplanar butterfly configuration [115]. On the other hand, there are two precedents for the open-core formulation among synthetic high-valent diiron complexes which have been shown to have respective Fe•••Fe distances of 3.3 and 3.6 Å [116, 117]. In addition, reactivity comparisons among this series of diiron complexes show that the subset with terminal Fe=O units exhibits significantly higher reactivity with respect to H-atom abstraction than those with only bridging oxo units [118], which led us to speculate that the sMMO-Q diamond core may isomerize to an open core in order to be able to cleave the 105-kcal/mol C–H bond of methane. This notion has also been proposed independently on the basis of DFT calculations [119, 120]. In any case, the terminal Fe=O unit in the open core configuration shown in Scheme 3 D may be well set up to attack the target C–H bond on eIF5A and effect its hydroxylation.
In summary, we have used X-ray absorption spectroscopy to characterize five different species in the catalytic cycle of hDOHH, including its O2 adduct hDOHH-P, which we propose to have a (μ-hydroxo)(cis-μ-1,2-peroxo)diiron(III) center that gives rise to its characteristic blue color and an Fe•••Fe distance of 3.4 Å. These results are in good agreement with the X-ray structure reported for hDOHH-PT, making this peroxo intermediate the best characterized to date within the dioxygen activating nonheme diiron enzyme family. hDOHH-P resembles peroxo intermediates found for other diiron enzymes in this family such as mutant RNR R2 [24, 25], Δ9D [28], and the ferroxidase center of ferritin [121] in exhibiting a ν(O–O) vibration within the 850–900 cm−1 range, which has been associated with synthetic (μ-1,2-peroxo)diiron(III) complexes [50–52]. However, unlike for hDOHH-P, available EXAFS data for RNR W48A/D84E R2 [23] and the ferroxidase center of ferritin [29] implicate a much shorter Fe•••Fe distance of 2.5 Å, an observation that is difficult to reconcile with the peroxo binding mode derived from the Raman data [25]. A major difference between hDOHH and most of the other diiron enzymes characterized thus far is the ligand combination that supports the diiron center. Most enzymes in this family utilize a 2-His-4-carboxylate ligand set that is neutralized upon binding of the diiron(II) center. In contrast, hDOHH employs a 4-His-2-carboxylate combination and requires one solvent-derived ligand to be ionized in order to mitigate the excess positive charge engendered by the binding of the diiron(II) center. The same argument may also be applied to justify the presence of a hydroxo bridge in deoxyhemerythrin, which has a 5-His-2-carboxylate binding site [68]. We suggest that the presence of the hydroxo bridge in hDOHH-P enhances the structural stability of the peroxo intermediate to allow the Fe scatterer to be observed unequivocally in the EXAFS data.
The differences noted above between hDOHH-P and the peroxo intermediates of canonical nonheme diiron enzymes suggest a greater diversity in peroxo binding modes in nonheme diiron active sites than initially imagined. Two other examples support this notion. ToMO gives rise to a peroxo intermediate with no visible chromophore [122] but nevertheless gives rise to a hydroxylated product, while CmlI generates a peroxo intermediate with a visible chromophore at ~500 nm and a much lower ν(O-O) at 791 cm−1, which are inconsistent with a (cis-μ-1,2-peroxo)diiron(III) center [10]. Clearly additional work is called for to clarify the questions raised by this work. Despite the structural differences, Nature has selected for these diiron enzymes to catalyze fundamentally similar reactions. In widening the scope of the diiron family of proteins, we can learn more about design principles and structural motifs that are used to control the reactivity in biological systems.
Experimental Procedures
Overexpression and Purification of hDOHH
The overexpression and purification were modified from a previously reported procedure [20]. hDOHH was overexpressed using the pGEX-4T-3_hdohh plasmid containing the hdohh gene in BL21(DE3) pLysS E. coli cells. Cells were grown in 4 L of LB growth media using a fermentation flask (New Brunswick) containing 100 μg/mL ampicillin at 37 °C with air bubbling continuously throughout the growth. Protein expression was induced at an OD600 of 0.6 by the addition of 1 mM IPTG for 4 hours at 30 °C. The cells were harvested by centrifugation (8000 rpm, 10 min, 4 °C) and the cell pellet resuspended in ice cold 30 mL Buffer A (50 mM Tris-HCl, pH 7.5) supplemented with 1 mM PMSF. The resuspended cell pellet was stored at −80 °C. For protein purification, cells were lysed by sonication and cellular debris removed by centrifugation (25,000 rpm, 30 minutes, 4°C). The clarified supernatant was rotated with 1 mL GSH-Sepharose resin (GE Healthcare) for four hours at 4 °C. The protein-bound resin was then poured into two empty PD-10 columns and washed with 50 mL Wash Buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl) followed by 25 mL Thrombin Cleavage Buffer (20 mM Tris-HCl, pH 8, 150 mM NaCl, 2.5 mM CaCl2). Thrombin (10 U) was added to the protein-bound resin and incubated overnight at 4 °C to cleave the GSH affinity tag from hDOHH. Free hDOHH was eluted from the column by the addition of 3 mL Thrombin Cleavage Buffer. Subsequent purification was carried out using a Superdex 200 size-exclusion column (GE Healthcare) and buffer. Blue fractions containing hDOHH-P were pooled and concentrated using a Vivaspin concentrator with 10 000 MWCO (Millipore) and stored at −80 °C.
Overexpression and Purification of eIF5A(Dhp)
The overexpression and purification of modified eIF5A(Dhp) was modified from a previously reported procedure [123]. eIF5A(Dhp) was overexpressed using the polycistronic plasmid pST39_eIF5A/DHS containing both eIF5A(Lys) and DHS genes in BL21(DE3)pLysS E. coli cells.
Sample Preparation
hDOHH-P was prepared from as-isolated enzyme following purification. The blue species was concentrated using a Vivaspin concentrator with 10 000 MWCO at 4 °C in 50 mM Tris-HCl, pH 8, 125 mM NaCl. Glycerol was added for a final concentration of 20% and 4 mM [Fe]T. To make the hDOHH-P•S sample, eIF5A(Dhp) was concentrated in Tris-HCl, pH 7.5, 125 mM NaCl and 20% glycerol at 4 °C using a Vivaspin concentrator. Concentrated hDOHH-P (in 50 mM Tris-HCl, 125 mM NaCl) was added to eIF5A(Dhp) on ice at a final ratio of 1.2 eq substrate. Enzyme and substrate were mixed and frozen for a final [Fe]T of 3.2 mM. The decayed species (hDOHH-D and hDOHH-D•S) were prepared by thawing the hDOHH-P and hDOHH-P•S XAS samples. The samples were incubated at room temperature until the peroxo absorption feature had reached a minimum value (approximately 48 hours). The samples were then frozen in liquid nitrogen. hDOHH-R was prepared by chemically reducing hDOHH-P using 7 equivalents dithionite and 0.1 equivalents of methyl viologen at room temperature. The sample was then concentrated, and glycerol was added for a final concentration of 3 mM [Fe]T in Tris-HCl, pH 8 with 20 % glycerol. All samples were frozen in liquid nitrogen in a small Mössbauer/XAS cup.
X-ray Absorption Spectroscopy
Iron K-edge X-ray absorption spectra of hDOHH-R, hDOHH-P, hDOHH-D, and hDOHH-D•S were collected on SSRL beam line 7-3 using a 30 element solid state Ge detector (Canberra) with a SPEAR storage ring current of ~500 mA at a power of 3.0 GeV. The incoming X-rays were unfocused using a Si(220) double crystal monochromator, which was detuned by 40% from the maximal flux to attenuate harmonic X-rays. For hDOHH-R, hDOHH-P, hDOHH-D and hDOHH-D•S 14, 12, 12 and 10 scans (respectively) were collected from 6882 eV to 8000 eV at a temperature (~10 K) that was controlled by an Oxford Instruments CF1208 continuous flow liquid helium cryostat. Iron K-edge X-ray absorption spectra of hDOHH-P•S were collected on NSLS beam line X3B using a 30 element solid state Ge detector (Canberra) with a storage ring current of ~100–300 mA at a power of 2.8 GeV. A sagittally focused Si (111) double crystal monochromator was used for energy selection, with a downstream nickel-coated mirror providing vertical focusing and rejection of higher harmonics. Nine scans were collected from 6882 eV to 8000 eV at a temperature (~20 K) that was controlled by closed-system liquid helium Displex cyrostat. An iron foil was placed in the beam pathway prior to I0 and scanned concomitantly for an energy calibration, with the first inflection point of the edge assigned to 7112.0 eV. A 3 μm Mn filter and additionally on 7-3, a Soller slit, were used to increase the signal to noise ratio of the spectra. Photoreduction was monitored by scanning the same spot on the sample twice and comparing the first derivative peaks associated with the edge energy during collection.
The detector channels from the scans were examined, calibrated, averaged, and processed for EXAFS analysis using EXAFSPAK [124] to extract χ(k). Theoretical phase and amplitude parameters for a given absorber-scatterer pair were calculated using FEFF 8.40 [125] and were utilized by the “opt” program of the EXAFSPAK package during curve fitting. Parameters for each species were calculated using a model derived from the crystal structure (PDB code 4D50). In all analyses, the coordination number of a given shell was a fixed parameter and was varied iteratively in integer steps, while the bond lengths (R) and mean-square deviation (σ2) were allowed to freely float. The amplitude reduction factor S0 was fixed at 0.9, while the edge-shift parameter E0 was allowed to float as a single value for all shells. Thus, in any given fit, the number of floating parameters was typically equal to (2 × num shells) + 1. hDOHH-P, hDOHH-P•S, hDOHH-D, and hDOHH-D•S all have a k range of 2 – 13.5 Å−1, and hDOHH-R has a range of 2 – 15 Å−1
Pre-edge analysis was performed on data normalized in the “process” program of the EXAFSPAK package, and pre-edge features of the samples were fit between 7108 eV to 7118 eV (except for hDOHH-R which was fit between 7108 eV to 7115 eV) using the Fityk [126] program with pseudo-Voigt functions composed of 50:50 Gaussian/Lorentzian functions.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grant GM-38767 to L.Q. and postdoctoral fellowship 5F32GM106612-02 to L.M.E. and by the Intramural Research Program of the National Institute of Dental and Craniofacial Research. V.V.V. is grateful to the Vietnam Education Foundation for providing him with a predoctoral fellowship. XAS data were collected on Beamline 7-3 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory and at Beamline X3B of the National Synchrotron Light Source (NSLS). SLAC is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Use of Beamline 7-3 is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). NSLS is a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-AC02-98CH10886. We thank Dr. Caleb Allpress for helpful discussions.
References
- 1.Lundin D, Poole AM, Sjöberg B-M, Högbom M. J Biol Chem. 2012;287:20565–20575. doi: 10.1074/jbc.M112.367458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nordlund P, Eklund H. Curr Opin Struct Biol. 1995;5:758–766. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Theil EC. Acc Chem Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 4.Nordlund P, Reichard P. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 5.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 6.Tinberg CE, Lippard SJ. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bochevarov AD, Li J, Song WJ, Friesner RA, Lippard SJ. J Am Chem Soc. 2011;133:7384–7397. doi: 10.1021/ja110287y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox BG, Lyle KS, Rogge CE. Acc Chem Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 9.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci U S A. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Lipscomb JD. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krebs C, Bollinger JM, Booker SJ. Curr Opin Chem Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das D, Eser BE, Han J, Sciore A, Marsh ENG. Angew Chem Int Ed. 2011;50:7148–7152. doi: 10.1002/anie.201101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YS, Kang KR, Wolff EC, Bell JK, McPhie P, Park MH. J Biol Chem. 2006;281:13217–13225. doi: 10.1074/jbc.M601081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park MH. J Biochem. 2006;139:161–169. doi: 10.1093/jb/mvj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saini P, Eyler DE, Green R, Dever TE. Nature. 2009;459:118–121. doi: 10.1038/nature08034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutierrez E, Shin B-S, Woolstenhulme CJ, Kim J-R, Saini P, Buskirk AR, Dever TE. Mol Cell. 2013;51:35–45. doi: 10.1016/j.molcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park MH, Nishimura K, Zanelli CF, Valentini SR. Amino Acids. 2010;38:491–500. doi: 10.1007/s00726-009-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaiser A. Amino Acids. 2012;42:679–684. doi: 10.1007/s00726-011-1042-8. [DOI] [PubMed] [Google Scholar]
- 19.Greganova E, Altmann M, Bütikofer P. FEBS J. 2011;278:2613–2624. doi: 10.1111/j.1742-4658.2011.08199.x. [DOI] [PubMed] [Google Scholar]
- 20.Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que L. Proc Natl Acad Sci U S A. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 22.Bollinger JM, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. J Am Chem Soc. 1998;120:1094–1095. [Google Scholar]
- 23.Baldwin J, Krebs C, Saleh L, Stelling M, Huynh BH, Bollinger JM, Riggs-Gelasco P. Biochemistry. 2003;42:13269–13279. doi: 10.1021/bi035198p. [DOI] [PubMed] [Google Scholar]
- 24.Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM. Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 25.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Solomon EI. J Am Chem Soc. 2004;126:8842–8855. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 26.Krebs C, Bollinger JM, Theil EC, Huynh BH. J Biol Inorg Chem. 2002;7:863–869. doi: 10.1007/s00775-002-0371-1. [DOI] [PubMed] [Google Scholar]
- 27.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 28.Broadwater JA, Achim C, Münck E, Fox BG. Biochemistry. 1999;38:12197–12204. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 29.Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 30.Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871–9876. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 31.Solomon EI, Park K. J Biol Inorg Chem. 2016;21 doi: 10.1007/s00775-016-1372-9. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 33.Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1994;116:7465–7466. [Google Scholar]
- 34.Korboukh VK, Li N, Barr EW, Bollinger JM, Krebs C. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray LJ, Naik SG, Ortillo DO, García-Serres R, Lee JK, Huynh BH, Lippard SJ. J Am Chem Soc. 2007;129:14500–14510. doi: 10.1021/ja076121h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Z, Sakai N, Böttger LH, Klinke S, Hauber J, Trautwein AX, Hilgenfeld R. Structure. 2015;23:882–892. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Rudd DJ, Sazinsky MH, Lippard SJ, Hedman B, Hodgson KO. Inorg Chem. 2005;44:4546–4554. doi: 10.1021/ic048794w. [DOI] [PubMed] [Google Scholar]
- 38.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 39.Rudd DJ, Sazinsky MH, Merkx M, Lippard SJ, Hedman B, Hodgson KO. Inorg Chem. 2004;43:4579–4589. doi: 10.1021/ic049716b. [DOI] [PubMed] [Google Scholar]
- 40.Shu L, Liu Y, Lipscomb JD, Que L., Jr J Biol Inorg Chem. 1996;1:297–304. [Google Scholar]
- 41.Dassama LMK, Silakov A, Krest CM, Calixto JC, Krebs C, Bollinger JM, Green MT. J Am Chem Soc. 2013;135:16758–16761. doi: 10.1021/ja407438p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griese JJ, Kositzki R, Schrapers P, Branca RMM, Nordström A, Lehtiö J, Haumann M, Högbom M. J Biol Chem. 2015;290:25254–25272. doi: 10.1074/jbc.M115.675223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Mascharak PK, Baidya N. Inorg Chem. 1991;30:920–928. [Google Scholar]
- 44.Wirt MD, Sagi I, Chen E, Frisbie SM, Lee R, Chance MR. J Am Chem Soc. 1991;113:5299–5304. [Google Scholar]
- 45.Penner-Hahn JE, Fronko RM, Pecoraro VL, Yocum CF, Betts SD, Bowlby NR. J Am Chem Soc. 1990;112:2549–2557. [Google Scholar]
- 46.Rudd DJ, Goldsmith CR, Cole AP, Stack TDP, Hodgson KO, Hedman B. Inorg Chem. 2005;44:1221–1229. doi: 10.1021/ic048765l. [DOI] [PubMed] [Google Scholar]
- 47.de Groot F. Chem Rev. 2001;101:1779–1808. doi: 10.1021/cr9900681. [DOI] [PubMed] [Google Scholar]
- 48.Sarangi R. Coord Chem Rev. 2013;257:459–472. doi: 10.1016/j.ccr.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Randall CR, Shu L, Chiou Y-M, Hagen KS, Ito M, Kitajima N, Lachicotte RJ, Zang Y, Que L. Inorg Chem. 1995;34:1036–1039. [Google Scholar]
- 50.Cranswick MA, Meier KK, Shan X, Stubna A, Kaizer J, Mehn MP, Münck E, Que L. Inorg Chem. 2012;51:10417–10426. doi: 10.1021/ic301642w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pap JS, Cranswick MA, Balogh-Hergovich E, Baráth G, Giorgi M, Rohde GT, Kaizer J, Speier G, Que L. Eur J Inorg Chem. 2013:3858–3866. doi: 10.1002/ejic.201300162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frisch JR, Vu VV, Martinho M, Münck E, Que L. Inorg Chem. 2009;48:8325–8336. doi: 10.1021/ic900961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 54.Carson EC, Lippard SJ. Inorg Chem. 2006;45:837–848. doi: 10.1021/ic051476s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ou C-C, Lalancette RA, Potenza JA, Schugar HJ. J Am Chem Soc. 1978;100:2053–2057. doi: 10.1021/ja00426a078. [DOI] [PubMed] [Google Scholar]
- 56.Schmitt W, Murugesu M, Goodwin JC, Hill JP, Mandel A, Bhalla R, Anson CE, Heath SL, Powell AK. Polyhedron. 2001;20:1687–1697. [Google Scholar]
- 57.Thich JA, Ou CC, Powers D, Vasiliou B, Mastropaolo D, Potenza JA, Schugar HJ. J Am Chem Soc. 1976;98:1425–1433. [Google Scholar]
- 58.Yoon S, Lippard SJ. J Am Chem Soc. 2004;126:2666–2667. doi: 10.1021/ja031603o. [DOI] [PubMed] [Google Scholar]
- 59.Makhlynets OV, Oloo WN, Moroz YS, Belaya IG, Palluccio TD, Filatov AS, Müller P, Cranswick MA, Que L, Rybak-Akimova EV. Chem Commun. 2014;50:645–648. doi: 10.1039/c3cc47632d. [DOI] [PubMed] [Google Scholar]
- 60.Yoon S, Lippard SJ. J Am Chem Soc. 2005;127:8386–8397. doi: 10.1021/ja0512531. [DOI] [PubMed] [Google Scholar]
- 61.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci U S A. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makris TM, Knoot CJ, Wilmot CM, Lipscomb JD. Biochemistry. 2013;52:6662–6671. doi: 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whittington DA, Lippard SJ. J Am Chem Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 64.Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 66.Eriksson M, Jordan A, Eklund H. Biochemistry. 1998;37:13359–13369. doi: 10.1021/bi981380s. [DOI] [PubMed] [Google Scholar]
- 67.Lindqvist Y, Huang W, Schneider G, Shanklin J. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 68.Holmes MA, Le Trong I, Turley S, Sieker LC, Stenkamp RE. J Mol Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 69.Holmes MA, Stenkamp RE. J Mol Biol. 1991;220:723–737. doi: 10.1016/0022-2836(91)90113-k. [DOI] [PubMed] [Google Scholar]
- 70.Dong Y, Yan S, Young VG, Que L. Angew Chem Int Ed. 1996;35:618–620. [Google Scholar]
- 71.Ookubo T, Sugimoto H, Nagayama T, Masuda H, Sato T, Tanaka K, Maeda Y, Ōkawa H, Hayashi Y, Uehara A, Suzuki M. J Am Chem Soc. 1996;118:701–702. [Google Scholar]
- 72.Kim K, Lippard SJ. J Am Chem Soc. 1996;118:4914–4915. [Google Scholar]
- 73.Riggs-Gelasco PJ, Shu L, Chen S, Burdi D, Huynh BH, Que L, Stubbe J. J Am Chem Soc. 1998;120:849–860. [Google Scholar]
- 74.Korendovych IV, Kryatov SV, Reiff WM, Rybak-Akimova EV. Inorg Chem. 2005;44:8656–8658. doi: 10.1021/ic051739i. [DOI] [PubMed] [Google Scholar]
- 75.Kryatov SV, Taktak S, Korendovych IV, Rybak-Akimova EV, Kaizer J, Torelli S, Shan X, Mandal S, MacMurdo VL, Mairata i Payeras A, Que L. Inorg Chem. 2005;44:85–99. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]
- 76.Chaudhury P, Wieghardt K, Nuber B, Weiss J. Angew Chem Int Ed. 1985;24:778–779. [Google Scholar]
- 77.Yoon S, Lippard SJ. J Am Chem Soc. 2004;126:16692–16693. doi: 10.1021/ja044489y. [DOI] [PubMed] [Google Scholar]
- 78.Yoon S, Kelly AE, Lippard SJ. Polyhedron. 2004;23:2805–2812. [Google Scholar]
- 79.Mitra M, Lloret-Fillol J, Haukka M, Costas M, Nordlander E. Chem Commun. 2014;50:1408–1410. doi: 10.1039/c3cc47830k. [DOI] [PubMed] [Google Scholar]
- 80.Widger LR, Siegler MA, Goldberg DP. Polyhedron. 2013;58:179–189. doi: 10.1016/j.poly.2013.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Z-T, Cheng X-L. Acta Crystallogr C. 2005;61:m529–m531. doi: 10.1107/S0108270105036218. [DOI] [PubMed] [Google Scholar]
- 82.Liang Y, Li W, Guo B-J. Acta Crystallogr Sect E Struct Reports Online. 2005;61:m1782–m1784. [Google Scholar]
- 83.Galet A, Muñoz MC, Agustí G, Martínez V, Gaspar AB, Real JA. Z Anorg Allg Chem. 2005;631:2092–2095. [Google Scholar]
- 84.Turowski PN, Armstrong WH, Liu S, Brown SN, Lippard SJ. Inorg Chem. 1994;33:636–645. [Google Scholar]
- 85.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2005;127:826–827. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 86.Shakya R, Powell DR, Houser RP. Eur J Inorg Chem. 2009;2009:5319–5327. [Google Scholar]
- 87.Jozwiuk A, Ingram AL, Powell DR, Moubaraki B, Chilton NF, Murray KS, Houser RP. Dalton Trans. 2014;43:9740–9753. doi: 10.1039/c4dt00047a. [DOI] [PubMed] [Google Scholar]
- 88.Hazell A, Jensen KB, McKenzie CJ, Toftlund H. Inorg Chem. 1994;33:3127–3134. [Google Scholar]
- 89.Dong Y, Fujii H, Hendrich MP, Leising RA, Pan G, Randall CR, Wilkinson EC, Zang Y, Que L. J Am Chem Soc. 1995;117:2778–2792. [Google Scholar]
- 90.Ogo S, Wada S, Watanabe Y, Iwase M, Wada A, Harata M, Jitsukawa K, Masuda H, Einaga H. Angew Chem Int Ed. 1998;37:2102–2104. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2102::AID-ANIE2102>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 91.MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J Am Chem Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- 92.Yamashita M, Furutachi H, Tosha T, Fujinami S, Saito W, Maeda Y, Takahashi K, Tanaka K, Kitagawa T, Suzuki M. J Am Chem Soc. 2007;129:2–3. doi: 10.1021/ja063987z. [DOI] [PubMed] [Google Scholar]
- 93.Celenligil-Cetin R, Paraskevopoulou P, Dinda R, Staples RJ, Sinn E, Rath NP, Stavropoulos P. Inorg Chem. 2008;47:1165–1172. doi: 10.1021/ic702154z. [DOI] [PubMed] [Google Scholar]
- 94.Mukherjee J, Lucas RL, Zart MK, Powell DR, Day VW, Borovik AS. Inorg Chem. 2008;47:5780–5786. doi: 10.1021/ic800048e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Majumdar A, Lippard SJ. Inorg Chem. 2013;52:13292–13294. doi: 10.1021/ic4019508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Majumdar A, Apfel U-P, Jiang Y, Moënne-Loccoz P, Lippard SJ. Inorg Chem. 2014;53:167–181. doi: 10.1021/ic4019585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Armstrong WH, Lippard SJ. J Am Chem Soc. 1984;106:4632–4633. [Google Scholar]
- 98.DeWitt JG, Bentsen JG, Rosenzweig AC, Hedman B, Green J, Pilkington S, Papaefthymiou GC, Dalton H, Hodgson KO, Lippard SJ. J Am Chem Soc. 1991;113:9219–9235. [Google Scholar]
- 99.Scarrow RC, Maroney MJ, Palmer SM, Que L, Roe AL, Salowe SP, Stubbe J. J Am Chem Soc. 1987;109:7857–7864. [Google Scholar]
- 100.Vu VV, Makris TM, Lipscomb JD, Que L. J Am Chem Soc. 2011;133:6938–6941. doi: 10.1021/ja201822v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cramer SP, Hodgson KO, Stiefel EI, Newton WE. J Am Chem Soc. 1978;100:2748–2761. [Google Scholar]
- 102.Sousa CM, Carpentier P, Matias PM, Testa F, Pinho F, Sarti P, Giuffrè A, Bandeiras TM, Romão CV. Acta Crystallogr D Biol Crystallogr. 2015;71:2236–2247. doi: 10.1107/S1399004715015825. [DOI] [PubMed] [Google Scholar]
- 103.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Ståhlberg J, Eijsink VGH, Beckham GT, Sandgren M. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sigfridsson KGV, Chernev P, Leidel N, Popovic-Bijelic A, Gräslund A, Haumann M. J Biol Chem. 2013;288:9648–9661. doi: 10.1074/jbc.M112.438796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bai Y, McCoy JG, Levin EJ, Sobrado P, Rajashankar KR, Fox BG, Zhou M. Nature. 2015;524:252–256. doi: 10.1038/nature14549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 107.Nordlund P, Eklund H. J Mol Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 108.Guy JE, Whittle E, Kumaran D, Lindqvist Y, Shanklin J. J Biol Chem. 2007;282:19863–19871. doi: 10.1074/jbc.M702520200. [DOI] [PubMed] [Google Scholar]
- 109.Shu L, Broadwater JA, Achim C, Fox BG, Münck E, Que L., Jr J Biol Inorg Chem. 1998;3:392–400. [Google Scholar]
- 110.Rardin RL, Tolman WB, Lippard SJ. New J Chem. 1991;15:417–430. [Google Scholar]
- 111.Banerjee R, Proshlyakov Y, Lipscomb JD, Proshlyakov DA. Nature. 2015;518:431–434. doi: 10.1038/nature14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Magnus KA, Hazes B, Ton-That H, Bonaventura C, Bonaventura J, Hol WG. Proteins. 1994;19:302–309. doi: 10.1002/prot.340190405. [DOI] [PubMed] [Google Scholar]
- 113.Kitajima N, Fujisawa K, Fujimoto C, Morooka Y, Hashimoto S, Kitagawa T, Toriumi K, Tatsumi K, Nakamura A. J Am Chem Soc. 1992;114:1277–1291. [Google Scholar]
- 114.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 115.Funahashi Y, Nishikawa T, Wasada-Tsutsui Y, Kajita Y, Yamaguchi S, Arii H, Ozawa T, Jitsukawa K, Tosha T, Hirota S, Kitagawa T, Masuda H. J Am Chem Soc. 2008;130:16444–16445. doi: 10.1021/ja804201z. [DOI] [PubMed] [Google Scholar]
- 116.Xue G, Fiedler AT, Martinho M, Munck E, Que L. Proc Natl Acad Sci. 2008;105:20615–20620. [Google Scholar]
- 117.Xue G, Geng C, Ye S, Fiedler AT, Neese F, Que L. Inorg Chem. 2013;52:3976–3984. doi: 10.1021/ic3027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xue G, De Hont R, Münck E, Que L. Nat Chem. 2010;2:400–405. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Siegbahn PEM, Crabtree RH. J Am Chem Soc. 1997;119:3103–3113. [Google Scholar]
- 120.Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. J Am Chem Soc. 2007;129:3135–3147. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Moënne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM. Biochemistry. 1999;38:5290–5295. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- 122.Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. J Am Chem Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Park JH, Dias CAO, Lee SB, Valentini SR, Sokabe M, Fraser CS, Park MH. Protein Eng Des Sel. 2011;24:301–309. doi: 10.1093/protein/gzq110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.George GN. EXAFSPAK: A Suite of Computer Programs for Analysis of X-ray Absorption Spectra 1990 [Google Scholar]
- 125.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Phys Rev B. 1998;58:7565–7576. [Google Scholar]
- 126.Wojdyr M. J Appl Crystallogr. 2010;43:1126–1128. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.