

Abstract
AIM
To identify the genetic defects of a Chinese patient with sporadic retinitis pigmentosa (RP).
METHODS
Ophthalmologic examinations were performed on the sporadic RP patient, 144 genes associated with retinal diseases were scanned with capture next generation sequencing (CNGS) approach. Two heterozygous mutations in PDE6B were confirmed in the pedigree by Sanger sequencing subsequently. The carrier frequency of PDE6B mutations of reported PDE6B mutations based on the available two public exome databases (1000 Genomes Project and ESP6500 Genomes Project) and one in-house exome database was investigated.
RESULTS
We identified compound heterozygosity of two novel nonsense mutations c.1133G>A (p.W378X) and c.2395C>T (p.R799X) in PDE6B, one reported causative gene for RP. Neither of the two mutations in our study was presented in three exome databases. Two mutations (p.R74C and p.T604I) in PDE6B have relatively high frequencies in the ESP6500 and in-house databases, respectively, while no common dominant mutation in each of the database or across all databases.
CONCLUSION
We demonstrates that compound heterozygosity of two novel nonsense mutations in PDE6B could lead to RP. These results collectively point to enormous potential of next-generation sequencing in determining the genetic etiology of RP and how various mutations in PDE6B contribute to the genetic heterogeneity of RP.
Keywords: compound heterozygosity, retinitis pigmentosa, mutation, capture next generation sequencing, PDE6B
INTRODUCTION
Retinitis pigmentosa (RP) is one of the leading causes of incurable blindness in the world and its prevalence is estimated at approximately 1 in 3000 to 1 in 5000 individuals [1]. It is typically characterized by initial symptoms of night blindness, with onset in adolescence or early adulthood, loss of peripheral vision and, as the disease progresses, loss of central vision leading to complete blindness or severe visual impairment. It is a clinically and genetically heterogeneous disorder[1]–[2]. Clinically, the age-at-onset of symptoms is highly variable and ranges from childhood to mid-adulthood. Genetically, so far, more than 190 genes have been identified as the cause of one or another form of inherited retinal disease. More than 60 genes were known as the cause of non-syndromic RP[1]–[3]. Syndromic forms of RP are heterogeneous: mutations in 13 genes cause Usher syndrome and 17 genes are associated with Bardet-Biedl syndrome. In addition to genetic heterogeneity, different diseases may be caused by mutations in the same gene, symptoms of different diseases may overlap, and there is extensive variation in clinical expression even among individuals sharing the same mutation in the same gene[1]. It makes RP as one of the most complicate single gene inherited diseases. It is inherited in autosomal [autosomal dominant (AD) and autosomal recessive (AR)], sex-linked (x-Link RP, XLRP) and mitochondrial modes of inheritance. Among the modes of inheritance in RP, AR inheritance appears to be more common than other form, accounting for approximately a third or more of all cases of RP[1].
The goal of the present study was to identify the causative mutations of the RP patient. We studied the using of Capture Next Generation Sequencing (CNGS) for genetic screening of a sporadic patient with RP, followed by Sanger sequencing and segregation analysis. Meanwhile, we sought to investigate the carrier frequency of the reported causative mutations based on the available two public exome databases (1000 Genomes Project and ESP6500 Genomes Project) and one in-house exome database.
SUBJECTS AND METHODS
Subjects
This study abode by the treaty of the Declaration of Helsinki and was ratified by the Ethics Committee of the Eye Hospital of Wenzhou Medical University. All participating individuals signed the written informed consent. A five milliliter venous blood sample of the patient and his parents was drawn into an ethylenediaminetetraaceticacid (EDTA) sample tube. Genomic DNA was extracted from peripheral blood leukocytes using standard phenol/chloroform extraction protocols.
Identification of the Disease Mutation
As previously described, 144 genes that associated with retinal diseases, such as PDE6B, were selected by a gene capture strategy by the GenCap custom enrichment kit (MyGenostics)[4]. In brief, taking advantage of the Solexa QA, cutadapt (http://code.google.com/p/cutadapt/), BWA (http://bio-bwa.sourceforge.net/bwa.shtml), SOAP aligner (http://soap.genomics.org.cn/) and GATK (https://www.broadinstitute.org/gatk/) programs to retrieve and align, identifying SNPs or insertions and deletions (InDels). We used the exome-assistant program (http://122.228.158.106/exomeassistant) to note SNPs and InDels. SIFT, PolyPhen2_HDIV, PolyPhen2_HVAR and likelihood ratio test (LRT)[5] were used to predict the impact of an amino acid substitution on protein function in order to evaluate and determine pathogenicity[4].
As for mutations, according to journal guidelines (www.hgvs.org/mutnomen), the number of nucleotide reflects the number of cDNA with +1 that corresponds to the A of the ATG initiation codon of translation in the reference sequence. And the initiation codon is codon 1.
Frequency and Evaluation of PDE6B Mutations
We searched the PubMed database with keywords: “PDE6B AND mutations” (http://www.ncbi.nlm.nih.gov/pubmed/?term=PDE6B+AND+Mutation), then collected all the reported causative mutations in the human PDE6B gene. The corresponding frequencies of DNA mutations identified in the 1000 Genomes Project, ESP6500 Genomes Project and the in-house databases with 1402 samples from Chinese population.
RESULTS
Clinical Data
The patient was a 32-year-old male. He provided a history of being suspect for RP in early childhood due to profound nyctalopia. When examined at age 32, his decimal best-corrected visual acuity (BCVA) was 0.4. Fundus photographs and optical coherence tomography (OCT) images revealed widespread macular atrophy, retinal vascular attenuation, optic disc pallor, intraretinal bone spicule pigmentation and overall thinning of the outer retinal layer (Figure 1). There is no family history. In summary, the diagnosis of this patient is sporadic RP according to the clinical manifestations.
Figure 1. Fundus photograph and OCT image of the patient (OD).
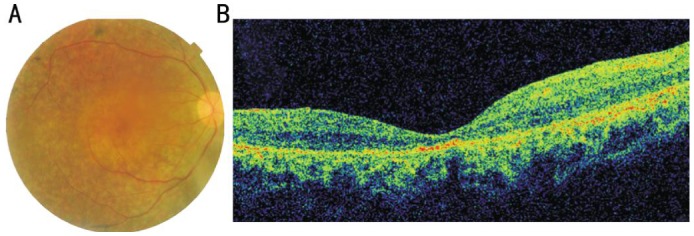
A: Color fundus photographs of the patient revealed widespread macular atrophy, narrow vessels, optic disc pallor and early bone spicule pigment; B: OCT of the patient showed that retinal thickness was significantly decreased, and inner segment/outer segment (IS/OS) are not visualized in some part of the retina.
Capture Next Generation Sequencing and Mutation Identification
We used a CNGS platform for the screening of all the 144 genes which associated with retinal diseases. We obtained a mean coverage of 164X of the sequencing depth by manual checking, and the depth is sufficient to ensure that all the exons of disease genes have been well covered. Here we showed one (PDE6B) of disease genes coverage. Results of primary bioinformatic analysis showed 13 heterozygous variants in 11 different genes, and 7 variants seemed to be deleterious proved by three prediction tools (SIFT, PolyPhen2_HDIV and PolyPhen2_HVAR). Then two novel compound nonsense mutations (p.W378X, p.R799X) were confirmed by Sanger sequencing, both of which encode a truncated PDE6B protein. The parents of the patient were non-symptomatic and were heterozygous carriers of the corresponding mutations. We performed multiple sequence alignments (Figure 2) and found that the nonsense mutations of PDE6B were located in a phylogenetically conserved region (Figure 3). Moreover, it has been predicted to be deleterious using prediction tools (Table 1).
Figure 2. DNA sequence chromatograms.

DNA sequence chromatograms of the patient with RP and non-related control. The heterozygous peaks of the mutations are pointed out by gray arrows.
Figure 3. Multiple-sequence alignment of p.W378X and p.R799X.

Structure-based sequence multiple-sequence alignment of PDE6B different species and human PDE6A, PDE6C revealed that two mutations (p.W378X and p.R799X) from RP patient were located within a conserved region. Secondary structure elements are indicated above.
Table 1. DNA variants of PDE6B in RP pateint.
| Exon | DNA change | Protein change | Mutation type | Novel | SIFT | Polyphen2_HDIV | PolyPhen2_HVAR | LRT |
| 9 | c.1133G>A | p.W378X | Nonsense | Yes | N/A | N/A | N/A | P |
| 21 | c.2395C>T | p.R799X | Nonsense | Yes | N/A | N/A | N/A | P |
N/A: No answer; P: Pathological.
Frequency and Evaluation of PDE6B Mutations
There were 28 reported mutations found in the literature. Among these mutations, only three (p.R74C, p.T604I and p.H620fs) of them were observed in one of the databases (Table 2). Mutations p.R74C and p.T604I have relatively high frequencies of 0.000385 and 0.0024, in the ESP6500 and in-house databases, respectively. We found that the mutational hot spot residue glycine 323 is critical because p.G323C and p.G323A were identified in two distinct families (Table 2). Four nonsense mutations (p.C270X, p.Q298X, p.Q534X, p.K706X) identified in the literature were not present in the databases. These data provides additional evidence to show that RP is a genetically heterogeneous disease, because there is no common dominant mutation in each of the populations or across all populations.
Table 2. Frequency of reported causative mutations in PDE6B.
| Exon | DNA change | Protein change | 1000G | ESP6500 | In-house |
| 1 | c.220C>T | p.R74C | <0.0004 | 0.000385 | <0.0007 |
| 2 | c.469-1G>T | Splicing | <0.0004 | <0.000005 | <0.0007 |
| 4 | c.810C>A | p.C270X | <0.0004 | <0.000005 | <0.0007 |
| 5 | c.892C>T | p.Q298X | <0.0004 | <0.000005 | <0.0007 |
| 5 | c.922G>A | p.G308S | <0.0004 | <0.000005 | <0.0007 |
| 6 | c.967G>T | p.G323C | <0.0004 | <0.000005 | <0.0007 |
| 6 | c.968G>C | p.G323A | <0.0004 | <0.000005 | <0.0007 |
| 9 | c.1160C>T | p.P387L | <0.0004 | <0.000005 | <0.0007 |
| 10 | c.1328A>C | p.N443T | <0.0004 | <0.000005 | <0.0007 |
| 11 | c.1417delC | p.L473fs | <0.0004 | <0.000005 | <0.0007 |
| 12 | c.1487delC | p.P496fs | <0.0004 | <0.000005 | <0.0007 |
| 12 | c.1580T>C | p.L527P | <0.0004 | <0.000005 | <0.0007 |
| 12 | c.1600C>T | p.Q534X | <0.0004 | <0.000005 | <0.0007 |
| 12 | c.1604T>A | p.I535N | <0.0004 | <0.000005 | <0.0007 |
| 13 | c.1655G>A | p.R552Q | <0.0004 | <0.000005 | <0.0007 |
| 13 | c.1669C>T | p.H557Y | <0.0004 | <0.000005 | <0.0007 |
| 13 | c.1688T>A | p.F563Y | <0.0004 | <0.000005 | <0.0007 |
| 14 | c.1727G>A | p.G576D | <0.0004 | <0.000005 | <0.0007 |
| 14 | c.1798G>A | p.D600N | <0.0004 | <0.000005 | <0.0007 |
| 14 | c.1811C>T | p.T604I | <0.0004 | <0.000005 | 0.0024 |
| 15 | c.1860delC | p.H620fs | <0.0004 | 0.00008 | <0.0007 |
| 16 | c.1976T>C | p.L659P | <0.0004 | <0.000005 | <0.0007 |
| 17 | c.2096T>G | p.L699R | <0.0004 | <0.000005 | <0.0007 |
| 17 | c.2116_2118del | p.706_706del | <0.0004 | <0.000005 | <0.0007 |
| 17 | c.2116A>T | p.K706X | <0.0004 | <0.000005 | <0.0007 |
| 18 | c.2131G>A | p.A711T | <0.0004 | <0.000005 | <0.0007 |
| 21 | c.2419T>A | p.W807R | <0.0004 | <0.000005 | <0.0007 |
| 22 | c.2560C>G | p.L854V | <0.0004 | <0.000005 | <0.0007 |
1000G: http://www.1000genomes.org/; ESP6500: http://evs.gs.washington.edu/EVS/; In-house: In-house exome database.
Reported PDE6B Mutations and Their Associated Phenotypes
Various PDE6B mutations have been reported to cause retinitis autosomal recessive retinitis pigmentosa (ARRP) and congenital stationary night blindness (CSNB), which include p.H258N, p.L228I, p.C270X, p.Q298X, p.P387L, p.L527P, p.R531X, p.I535N, p.R552Q, p.H557Y, p.G576D, p.E640fs, p.D600N, p.H620fs, p.L699R, p.K706X, p.W807R and two splicing mutations c.469-1G>T and c.2193+1G>T [6]–[20]. In this study, we summarized these mutations and their associated phenotypes from 18 families. Patients from 6 unrelated families were reported carrying two compound heterozygosity mutations. The clinical phenotypes in these cases with ARRP has early childhood onset of night vision loss, bone spicule pigment, attenuated retinal vessels, elevated dark adapted threshold, and generally well-preserved visual acuity until late stages. Forty-five point five percent (10/22) of these patients from 6 families had cataracts. The clinical manifestations of the present patients were generally consistent with the literatures. There appears to be no particularly unique phenotype associated with compound heterozygosity of nonsense mutations, or with mutations affecting the catalytic domain of PDE6B.
DISCUSSION
The comprehensive diagnosis of RP is made challenging by the large number of disease genes and alleles associated with RP. Significant progress has been made in determining the molecular causes of RP based on the next generation sequencing technique. In our study, we used CNGS method to scan 144 genes associated with retinal diseases. Genetic examination revealed two novel nonsense heterozygous mutations c.1133G>A (p.W378X) and c.2395C>T (p.R799X) in PDE6B. The two novel nonsense mutations in this study were predicted as likely to be disease-causing mutations as both of them will lead to truncated proteins.
Mutations in PDE6B (NM_000283) have been linked to humans autosomal recessive RP (arRP)[6]–[7]. The human PDE6B gene has been mapped to chromosome 4p16.3, spans approximately 45 kb, and has 22 exons, encoding a protein of 854 amino acid residues, named phosphodiesterase 6B. Which is the β-subunit of rod phosphodiesterase 6 (PDE6)[21]. PDE6 regulates the cytoplasmic level of cyclic guanosine monophosphate (cGMP) in the photoreceptors. Activated by light stimulation, PDE6 could reduce the level of cGMP, which leads to closure of the cGMP-gated Na+ and Ca++ channels then to hyperpolarize the rod plasma membrane [22]. A dysfunction of the β-subunit of PDE6 results in a high concentration of cGMP and Ca++ in rod photoreceptors, and it promote apoptosis of the rod photoreceptors[23].
PDE6B mutations are associated with one of the earliest onset and most aggressive forms of RP[6]–[9]. Mice homozygous for the rd mutation display hereditary retinal degeneration and have been considered a model for human retinitis pigmentosa. Adeno-associated virus (AAV) -mediated gene therapy had been reported available to prevent retinal degeneration in rd10 mice containing a recessive PDEbeta mutation[24]. Since there are no effective treatments for this disease, so far, it is important to identify the underlying genetic defects. It would be helpful for prenatal diagnosis, and the selection of patients for clinical trials, for example, virus-based gene therapy for patients with PDE6B mutations, to restore visual function[25]–[26].
The CNGS strategy has been reported to be a fast, effective, and reliable tool to detect known and novel mutations in AD RP patients[27]. Approximately 40% of ARRP families also could be successfully molecular diagnosed by CNGS[28]. Our data also clearly support that CNGS is a useful approach to identify the mutation of RP patients, even in sporadic cases because of its ability to detect variants in PDE6B with full coverage. However, for CNGS clinical screening to become a widely accepted stand-alone PDE6B mutation screening tool, more studies need to be done, especially, to assign pathogenicity status to newly discovered or rare variants.
In our recent study, we showed missing coverage of some exons in the PROM1 gene from CNGS based molecular diagnosis of putative Stargardt disease[4]. It has been reported that with deep sequencing, missing coverage of some regions is common[29]. So before searching for the disease-associated mutations, it is necessary to check the coverage of the targeted sequence, even if no mutations were found in the missing coverage region. We also showed, in the present study, that PDE6B has been well covered. Missing coverage issue may be treated case by case.
We noticed that two novel nonsense compound mutations in our study are still not present in databases. This highlights that the present mutational spectra from these databases may still need to be enlarged and also reveals that PDE6B gene contributes to the genetic heterogeneity of RP disease. In order to increase the rate of RP mutation detection and to correctly evaluate the residual risk of being a RP carrier after molecular analysis, it is essential that genetic tests are designed based on the characteristic mutation frequency profile for a given population and that the sensitivity of mutation detection assays is as high as possible [30]–[31]. However, so far, no data are available to define the carrier frequency of PDE6B mutations. To address this, one approach is to collect a large number of DNA samples in different populations and sequence the whole PDE6B coding sequence with classical Sanger sequencing. However, this is a labor intensive approach[30]. Public and in-house exome databases from large population samples would be expected to uncover many variants, some of them known to be benign, some rare, some not yet described[12]. Several studies have been performed to analyze carrier frequency in RP or even being into question the previously reported mutations[32]–[33]. Therefore, here we sought to take advantage of public and in-house exome databases to address it. We found 28 reported mutations in the literature. Only three (p.R74C, p.T604I and p.H620fs) reported mutations were observed in one of the databases (Table 2). This study provides additional evidence to show that RP is a genetically heterogeneous disease, because there is no common dominant mutation in present databases. While, we still can't clearly define the carrier frequency with each reported mutations due to relative small capacity of the exome databases even we have in-house databases with 1402 samples from Chinese population.
We took advantage of online prediction tools to predict the impact of amino acid substitutions on PDE6B protein function. The prediction results strongly suggested that all the reported mutations except p.L854V are potentially pathogenic mutations, although p.L854V is found in patients[34]. Additional studies may be required before excluding it as a “disease-associated” mutation.
Token together, here we demonstrated a successful case of using next generation sequencing to identify compound heterozygosity of two novel nonsense PDE6B mutations in a Chinese patient with sporadic retinitis pigmentosa, which points to enormous potential of next-generation sequencing in determining the genetic etiology of RP, and also we showed how various mutations in PDE6B contribute to the genetic heterogeneity of RP.
Acknowledgments
We thank the patient for the participation in this project. We are indebted to Dr. Jin-Yu Wu (Institute of Genomic Medicine, Wenzhou Medical University) for bioinformatics analysis.
Foundations: Supported by the Chinese National Program on Key Basic Research Project (973 Program, No.2013CB967502); the Natural Science Foundation of China (No.81201181/H1818); Zhejiang Provincial & Ministry of Health Research Fund for Medical Sciences (No.2016KYA145); Wenzhou City Grant (No.Y20140633); Chinese National Training Programs of Innovation and Entrepreneurship for Undergraduates (No.20130343005).
Conflicts of Interest: Cheng LL, None; Han RY, None; Yang FY, None; Yu XP, None; Xu JL, None; Min QJ, None; Tian J, None; Ge XL, None; Zheng SS, None; Lin YW, None; Zheng YH, None; Qu J, None; Gu F, None.
REFERENCES
- 1.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roosing S, Lamers IJ, de Vrieze E, van den Born LI, Lambertus S, Arts HH, POC1B Study Group. Peters TA, Hoyng CB, Kremer H, Hetterschijt L, Letteboer SJ, van Wijk E, Roepman R, den Hollander AI, Cremers FP. Disruption of the basal body protein POC1B results in autosomal-recessive cone-rod dystrophy. Am J Hum Genet. 2014;95(2):131–142. doi: 10.1016/j.ajhg.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sergouniotis PI, Chakarova C, Murphy C, Becker M, Lenassi E, Arno G, Lek M, MacArthur DG, UCL-Exomes Consortium, Bhattacharya SS, Moore AT, Holder GE, Robson AG, Wolfrum U, Webster AR, Plagnol V. Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am J Hum Genet. 2014;94(5):760–769. doi: 10.1016/j.ajhg.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Ge X, Shi W, Huang P, Min Q, Li M, Yu X, Wu Y, Zhao G, Tong Y, Jin ZB, Qu J, Gu F. Molecular diagnosis of putative Stargardt disease by capture next generation sequencing. PLoS One. 2014;9(4):e95528. doi: 10.1371/journal.pone.0095528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J, Jiang R. Prediction of deleterious nonsynonymous single-nucleotide polymorphism for human diseases. Scientific World Journal. 2013;2013:675851. doi: 10.1155/2013/675851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet. 1994;7(4):551. doi: 10.1038/ng0894-551a. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4(2):130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 8.Bayés M, Giordano M, Balcells S, et al. Homozygous tandem duplication within the gene encoding the beta-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum Mutat. 1995;5(3):228–234. doi: 10.1002/humu.1380050307. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995;92(8):3249–3253. doi: 10.1073/pnas.92.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dryja TP, Hahn LB, Reboul T, Arnaud B. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet. 1996;13(3):358–360. doi: 10.1038/ng0796-358. [DOI] [PubMed] [Google Scholar]
- 11.Kuniyoshi K, Sakuramoto H, Yoshitake K, Ikeo K, Furuno M, Tsunoda K, Kusaka S, Shimomura Y, Iwata T. Reduced rod electroretinograms in carrier parents of two Japanese siblings with autosomal recessive retinitis pigmentosa associated with PDE6B gene mutations. Doc Ophthalmol. 2015;131(1):71–79. doi: 10.1007/s10633-015-9497-7. [DOI] [PubMed] [Google Scholar]
- 12.Shen S, Sujirakul T, Tsang SH. Next-generation sequencing revealed a novel mutation in the gene encoding the beta subunit of rod phosphodiesterase. Ophthalmic Genet. 2014;35(3):142–150. doi: 10.3109/13816810.2014.915328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valverde D, Baiget M, Seminago R, del Rio E, Garcia-Sandoval B, del Rio T, Bayés M, Balcells S, Martinez A, Grinberg D, Ayuso C. Identification of a novel R552O mutation in exon 13 of the beta-subunit of rod phosphodiesterase gene in a Spanish family with autosomal recessive retinitis pigmentosa. Hum Mutat. 1996;8(4):393–394. doi: 10.1002/humu.1380080403. [DOI] [PubMed] [Google Scholar]
- 14.Ali S, Riazuddin SA, Shahzadi A, Nasir IA, Khan SN, Husnain T, Akram J, Sieving PA, Hejtmancik JF, Riazuddin S. Mutations in the β-subunit of rod phosphodiesterase identified in consanguineous Pakistani families with autosomal recessive retinitis pigmentosa. Mol Vis. 2011;17:1373–1380. [PMC free article] [PubMed] [Google Scholar]
- 15.Danciger M, Heilbron V, Gao YQ, Zhao DY, Jacobson SG, Farber DB. A homozygous PDE6B mutation in a family with autosomal recessive retinitis pigmentosa. Mol Vis. 1996;2:10. [PubMed] [Google Scholar]
- 16.Danciger M, Blaney J, Gao YQ, Zhao DY, Heckenlively JR, Jacobson SG, Farber DB. Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa. Genomics. 1995;30(1):1–7. doi: 10.1006/geno.1995.0001. [DOI] [PubMed] [Google Scholar]
- 17.Valverde D, Solans T, Grinberg D, Balcells S, Vilageliu L, Bayés M, Chivelet P, Besmond C, Goossens M, González-Duarte R, Baiget M. A novel mutation in exon 17 of the beta-subunit of rod phosphodiesterase in two RP sisters of a consanguineous family. Hum Genet. 1996;97(1):35–38. doi: 10.1007/BF00218829. [DOI] [PubMed] [Google Scholar]
- 18.Hmani-Aifa M, Benzina Z, Zulfiqar F, Dhouib H, Shahzadi A, Ghorbel A, Rebaï A, Söderkvist P, Riazuddin S, Kimberling WJ, Ayadi H. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur J Hum Genet. 2009;17(4):474–482. doi: 10.1038/ejhg.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saga M, Mashima Y, Akeo K, Kudoh J, Oguchi Y, Shimizu N. A novel homozygous Ile535Asn mutation in the rod cGMP phosphodiesterase beta-subunit gene in two brothers of a Japanese family with autosomal recessive retinitis pigmentosa. Curr Eye Res. 1998;17(3):332–335. doi: 10.1076/ceyr.17.3.332.5214. [DOI] [PubMed] [Google Scholar]
- 20.Tsang SH, Tsui I, Chou CL, Zernant J, Haamer E, Iranmanesh R, Tosi J, Allikmets R. A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am J Ophthalmol. 2008;146(5):780–788. doi: 10.1016/j.ajo.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber B, Riess O, Hutchinson G, Collins C, Lin BY, Kowbel D, Andrew S, Schappert K, Hayden MR. Genomic organization and complete sequence of the human gene encoding the beta-subunit of the cGMP phosphodiesterase and its localisation to 4p 16.3. Nucleic Acids Res. 1991;19(22):6263–6268. doi: 10.1093/nar/19.22.6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfister C, Bennett N, Bruckert F, Catty P, Clerc A, Pagès F, Deterre P. Interactions of a G-protein with its effector: Transducin and cGMP phosphodiesterase in retinal rods. Cell Signal. 1993;5(3):235–241. doi: 10.1016/0898-6568(93)90015-e. [DOI] [PubMed] [Google Scholar]
- 23.Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347(6294):677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- 24.Pang JJ, Boye SL, Kumar A, Dinculescu A, Deng W, Li J, Li Q, Rani A, Foster TC, Chang B, Hawes NL, Boatright JH, Hauswirth WW. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008;49(10):4278–4283. doi: 10.1167/iovs.07-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estrada-Cuzcano A, Roepman R, Cremers FP, den Hollander AI, Mans DA. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum Mol Genet. 2012;21(R1):R111–R124. doi: 10.1093/hmg/dds298. [DOI] [PubMed] [Google Scholar]
- 26.Han J, Dinculescu A, Dai X, Du W, Smith WC, Pang J. Review: the history and role of naturally occurring mouse models with Pde6b mutations. Mol Vis. 2013;19:2579–2589. [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez-San Jose P, Corton M, Blanco-Kelly F, Avila-Fernandez A, Lopez-Martinez MA, Sanchez-Navarro I, Sanchez-Alcudia R, Perez-Carro R, Zurita O, Sanchez-Bolivar N, Lopez-Molina MI, Garcia-Sandoval B, Riveiro-Alvarez R, Ayuso C. Targeted next-generation sequencing improves the diagnosis of autosomal dominant retinitis pigmentosa in Spanish patients. Invest Ophthalmol Vis Sci. 2015;56(4):2173–2182. doi: 10.1167/iovs.14-16178. [DOI] [PubMed] [Google Scholar]
- 28.Fu Q, Wang F, Wang H, Xu F, Zaneveld JE, Ren H, Keser V, Lopez I, Tuan HF, Salvo JS, Wang X, Zhao L, Wang K, Li Y, Koenekoop RK, Chen R, Sui R. Next-generation sequencing-based molecular diagnosis of a Chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(6):4158–4166. doi: 10.1167/iovs.13-11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14(5):R51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arzimanoglou II, Tuchman A, Li Z, Gilbert F, Denning C, Valverde K, Zar H, Quittell L, Arzimanoglou I. Cystic fibrosis carrier screening in Hispanics. Am J Hum Genet. 1995;56(2):544–547. [PMC free article] [PubMed] [Google Scholar]
- 31.Hallam S, Nelson H, Greger V, Perreault-Micale C, Davie J, Faulkner N, Neitzel D, Casey K, Umbarger MA, Chennagiri N, Kramer AC, Porreca GJ, Kennedy CJ. Validation for clinical use of, and initial clinical experience with, a novel approach to population-based carrier screening using high-throughput, next-generation DNA sequencing. J Mol Diagn. 2014;16(2):180–189. doi: 10.1016/j.jmoldx.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Cross JL, Iben J, Simpson CL, Thurm A, Swedo S, Tierney E, Bailey-Wilson JE, Biesecker LG, Porter FD, Wassif CA. Determination of the allelic frequency in Smith-Lemli-Opitz syndrome by analysis of massively parallel sequencing data sets. Clin Genet. 2015;87(6):570–575. doi: 10.1111/cge.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jabbari J, Jabbari R, Nielsen MW, Holst AG, Nielsen JB, Haunso S, Tfelt-Hansen J, Svendsen JH, Olesen MS. New exome data question the pathogenicity of genetic variants previously associated with catecholaminergic polymorphic ventricular tachycardia. Circ Cardiovasc Genet. 2013;6(5):481–489. doi: 10.1161/CIRCGENETICS.113.000118. [DOI] [PubMed] [Google Scholar]
- 34.Kim C, Kim KJ, Bok J, Lee EJ, Kim DJ, Oh JH, Park SP, Shin JY, Lee JY, Yu HG. Microarray-based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Mol Vis. 2012;18:2398–2410. [PMC free article] [PubMed] [Google Scholar]