

Abstract
Cancer and stromal cell metabolism is important for understanding tumor development, which highly depends on the tumor microenvironment (TME). Cell or animal models cannot recapitulate the human TME. We have developed an ex vivo paired cancerous (CA) and noncancerous (NC) human lung tissue approach to explore cancer and stromal cell metabolism in the native human TME. This approach enabled full control of experimental parameters and acquisition of individual patient's target tissue response to therapeutic agents while eliminating interferences from genetic and physiological variations. In this two-case study of non-small-cell lung cancer, we performed stable isotope-resolved metabolomic (SIRM) experiments on paired CA and NC lung tissues treated with a macrophage activator β-glucan and 13C6-glucose, followed by ion chromatography–Fourier transform mass spectrometry (IC-FTMS) and nuclear magnetic resonance (NMR) analyses of 13C-labeling patterns of metabolites. We demonstrated that CA lung tissue slices were metabolically more active than their NC counterparts, which recapitulated the metabolic reprogramming in CA lung tissues observed in vivo. We showed β-glucan-enhanced glycolysis, Krebs cycle, pentose phosphate pathway, antioxidant production, and itaconate buildup in patient UK021 with chronic obstructive pulmonary disease (COPD) and an abundance of tumor-associated macrophages (TAMs) but not in UK049 with no COPD and much less macrophage infiltration. This metabolic response of UK021 tissues was accompanied by reduced mitotic index, increased necrosis, and enhaced inducible nitric oxide synthase (iNOS) expression. We surmise that the reprogrammed networks could reflect β-glucan M1 polarization of human macrophages. This case study presents a unique opportunity for investigating metabolic responses of human macrophages to immune modulators in their native microenvironment on an individual patient basis.
Keywords: immune dysregulation, increased inflammatory response, neoplasm of the lung, progressive pulmonary function impairment
INTRODUCTION
In the past several years, there has been rekindled interest in cancer metabolism as an important component of cancer progression and survival, which has led to the discovery of many novel aspects of metabolic reprogramming in cancer cells, far beyond the enhanced aerobic glycolysis originally described by Warburg (1956). Metabolic reprogramming is a hallmark of cancer (Hanahan and Weinberg 2011) and is important for promoting all stages of cancer cell development, such as growth, survival, and metastasis (Yuneva et al. 2007; Dang et al. 2009; Nomura et al. 2010; Cheng et al. 2011; Le et al. 2012; Mullen et al. 2012; Faubert et al. 2013; Son et al. 2013; Xie et al. 2014; Kim et al. 2015; Sellers et al. 2015). Altered cancer cell metabolism has also been implicated in modulating the cancer cell's interaction with tumor microenvironment (TME) to enable its escape from immune surveillance (Zamanakou et al. 2007; Ho et al. 2015) and to promote tumor progression, survival, metastasis, and angiogenesis (Stern et al. 2002; Walenta et al. 2004; Sonveaux et al. 2008; Wysoczynski and Ratajczak 2009; Bonuccelli et al. 2010; Hanahan and Weinberg 2011; Guillaumond et al. 2013; Caneba et al. 2014; Leithner et al. 2014; Thayanithy et al. 2014). However, with a few exceptions (Fan et al. 2009; Maher et al. 2012; Sellers et al. 2015), the majority of human cancer metabolic studies have been conducted in two-dimensional (2D) cell culture systems or in mouse xenografts, which lack the native 3D architecture of human TME, and therefore cannot recapitulate the altered metabolic networks that are induced by interactions of cancer cells with their stroma. Consideration of such interactions is essential to both fundamental understanding of tumor metabolism and clinical translation of such understanding into diagnostic and therapeutic benefits (Sellers et al. 2015).
We and others have recently demonstrated the feasibility of interrogating tumor metabolism in vivo in human patients using a combination of stable isotope tracers and metabolomic analyses (Fan et al. 2009; Maher et al. 2012; Sellers et al. 2015). Our studies uncovered novel metabolic reprogramming events and activation of anaplerotic pyruvate carboxylase, in particular, in human lung cancer, which is important for supporting lung cancer cell growth both in vitro and in vivo (Sellers et al. 2015). However, experimentation on human subjects is very limited in terms of experimental design and treatment options including tracer use or therapeutic agents.
To circumvent these issues, we have extended the ex vivo tissue slice approach pioneered by Warburg (1924) for interrogating large-scale metabolic responses of human non-small-cell lung cancer (NSCLC) tissues (Xie et al. 2014; Sellers et al. 2015; Fan et al. 2016) to an immune modulator, β-glucan. We have shown that this particulate yeast-derived polysaccharide can polarize alternatively activated (M2) or immunosuppressive macrophages to the classical M1 immunoactivating phenotype in mouse models, leading to reduced tumor burden (Liu et al. 2015). This approach has the advantages of (1) maintaining the native human tissue 3D architecture of TME; (2) flexibility of tracer use and treatment options; (3) delineation of target tissue responses without systemic influences; (4) complete experimental control of the environment including nutrient supply; (5) circumvention of genetic, physiologic, and environmental complications in elucidating treatment responses by adopting the paired cancerous (CA) and surrounding noncancerous (NC) tissue design; and (6) the ability to determine treatment effects on target tissues on an individual patient basis.
In this study, we describe ex vivo stable isotope resolved metabolomics (SIRM) studies utilizing 13C6-glucose as tracer and β-glucan as an anticancer treatment on fresh lung tissue slices from two NSCLC patients. We found opposite metabolic network responses to β-glucan in the CA lung tissue slices for these two patients, which were accompanied by distinct histologic and immunohistochemical changes in terms of necrosis, mitotic index, and the expression of inducible nitric oxide synthase (iNOS, marker of M1-like macrophages). In particular, the enhanced aerobic glycolysis, Krebs cycle, antioxidant production, and itaconate buildup is consistent with macrophage polarization toward the M1 type by β-glucan in reference to animal studies (Rodríguez-Prados et al. 2010; Tannahill et al. 2013) in one patient with chronic obstructive pulmonary disease (COPD) but not in the other without COPD.
RESULTS
Clinical Presentation, Patient History, and Molecular Pathology
The lung tumor characteristics, patient demographics, and smoking/health history are listed in Table 1. The histopathological properties of the CA and surrounding NC lung tissues (proximal and distal to the tumor margin) for two patients are illustrated in Supplemental Figure S1, where both tissues of patient UK021 were more fibrotic than those of patient UK049. Immunofluorescent staining for mitotic index (PCNA [proliferating cell nuclear antigen]) and necrosis (RIP-1) indicated that the UK021 tissues were less proliferative and more necrotic (Fig. 1A,B). Immunofluorescent staining for resident macrophages (CD68) indicated their higher abundance in UK021 than UK049 lung tissues (Fig. 1C,D), which confirmed the hematoxylin and eosin (H&E) staining data (Supplemental Fig. S1A). Double immunofluorescent staining of iNOS and CD68 provided a measure of iNOS expression (marker of M1-like macrophages) normalized to the abundance of CD68+ macrophages. This ratio was lower for UK021 than for UK049 CA lung tissues (Fig. 1C,D), which could reflect a less M1-like phenotype of tumor-associated macrophages (TAMs) in the former tissue. All of these histopathological differences could be related to the COPD that UK021 had suffered but not UK049.
Table 1.
Clinical attributes and demographics of the two non-small-cell lung cancer (NSCLC) patients who consented for the ex vivo studies
| Patient # | UK021 | UK049 |
|---|---|---|
| Tumor type | Poorly differentiated adenocarcinoma | Moderately differentiated adenocarcinoma |
| Tumor size | 3.8 × 2.6 × 2.5 cm | 9 × 8 × 5 cm |
| TNM staging (pathology) | T3N0M0 | T3N0M0 |
| Overall stage | IIB | IIB |
| Tumor grade | 3 | 2 |
| Tumor PET SUV | 15.4 | Not available |
| FEV1/FVC ratio | 86 | 72 |
| Gender | F | M |
| Age | 42 | 76 |
| Ethnicity | White | White |
| Other health issues | COPD, hypercholesterolemia | None |
| Smoking history (pack years) | 20 | 50 |
PET, positron emission tomography; SUV, standardized uptake value; FEV1/FVC ratio, forced expiratory volume 1/forced vital capacity; COPD, chronic obstructive pulmonary disease.
Figure 1.

UK021 cancerous (CA) lung tissue is less mitotic, has less nitric oxide (NO)-producing potential, and is more necrotic than UK049 CA lung tissues. UK021 and UK049 CA and noncancerous (NC) lung tissues were sampled in the operating room and immediately preserved in formalin for immunohistochemical analysis for PCNA (mitotic index marker), RIP-1 (necrosis marker), and iNOS (inducible nitric oxide synthase, marker for M1-like macrophages [Ohri et al. 2009, 2011]) as green fluorescence, CD68 (general macrophage marker) as red fluorescence, and DAPI (nuclear marker) as blue fluorescence. (A,C) An example frame of the PCNA, RIP-1 (60× magnification), and iNOS + CD68 fluorescence images (60× magnification), as well as their normalized image intensity averaged over three to seven image frames for UK021 tissues. The fluorescence intensity of PCNA/RIP-1 and iNOS was respectively normalized to that of DAPI and CD68. (B,D) The corresponding image frames and normalized image intensity for UK049 tissues are shown. The UK021 CA lung tissue displayed a relatively lower mitotic index, higher necrotic state, and lower iNOS to CD68 ratio than the UK049 CA tissue. For both patients, the CA lung tissues were more mitotic and necrotic with a higher iNOS/CD68 ratio than the NC lung tissues proximal and distal to the CA tissues.
UK021 but Not UK049 Tumor Tissue Slices Exhibited Accelerated Glycolysis and Glutathione Synthesis in Response to β-Glucan Treatment
When UK021 CA tissue slices were treated with or without WGP (whole glucan particle) β-glucan in the presence of 13C6-glucose (Glc), an enhanced level of 13C3-lactate was observed in both intracelluar (Fig. 2D) and extracellular compartments (Supplemental Fig. S2) for the WGP compared with the control treatments. This, together with the increased production of 13C6-fructose-1,6-bisphosphate (Fig. 2B) and 13C3-pyruvate (Fig. 2C), pointed to accelerated glycolysis under normoxia in UK021 CA tissue slices in response to WGP. In contrast, UK049 CA tissue slices responded to WGP with diminished aerobic glycolysis, as evidenced by the attenuated release of 13C3-lactate into the medium (Supplemental Fig. S2) and decreased production of 13C3-pyruvate (Fig. 3C). The decreased buildup of 13C-glucose in UK021 but not in UK049 CA tissue slices is further consistent with increased glucose metabolism into lactate in the former case (Figs. 2 and 3).
Figure 2.

β-Glucan enhances glycolysis, Krebs cycle, and glutathione synthesis in UK021 CA tissue slices. Slices of freshly resected UK021 CA and NC lung tissues were treated with 0 or 100 µg/mL whole glucan particle (WGP) for 24 h in the presence of 13C6-glucose (Glc) with n = 1 for each treatment because of limited tissue availability. Tissue slices were processed, extracted for polar metabolites, and analyzed by ion chromatography–Fourier transform mass spectrometry (IC-FTMS), as described in Methods. The 12C (0) and 13C (1–5) isotopologues of glycolytic, Krebs cycle, and glutathione (GSH) synthesis pathway metabolites are shown as bar graphs in A–I along with the atom-resolved tracing of these pathways. Not all possible 13C isotopologues are shown. PDH, pyruvate dehydrogenase; PCB, pyruvate carboxylase; ME, NADP+-dependent malic enzyme; F1,6P, fructose-1,6-bisphosphate; •, 12C; •,•,•, 13C derived from PDH-, PCB-, and ME-mediated Krebs cycle reactions, respectively. In the WGP-treated CA tissue slice, accelerated glycolysis, Krebs cycle, and GSH synthesis were evident, respectively, from the accumulation of 13C6-F1,6P (B)/13C3-pyruvate (C)/13C3-lactate (D)/medium 13C3-lactate (cf. Supplemental Fig. S2), 13C-citrate (E)/13C-αketoglutarate (αKG, F )/13C-succinate, and 13C-Glu (J)/13C-GSH (K), as compared with control (Ctl) CA tissues. In contrast, the response of these metabolites in the NC tissue slice to WGP was opposite to that in the CA tissue slice.
Figure 3.

β-Glucan does not alter Krebs cycle and glutathione synthesis while attenuating glycolysis in UK049 CA tissue slices. Tissue slice treatment, extraction, and analysis, as well as abbreviations and symbols used, were as described in Figure 2, except that n = 2 for each treatment (control vs. WGP). Each value was an average of two replicates with the error bars as standard errors of the mean. The decreased production of tissue 13C3-pyruvate (C) and rate of 13C3-lactate release into the medium (cf. Supplemental Fig. S2) in CA tissue slices in response to WGP is consistent with attenuated glycolysis. In addition, the lack of buildup in 13C-αKG (F)/ 13C-succinate/13C-fumarate/13C-malate/13C-Glu/13C-GSH indicates lack of stimulation of the Krebs cycle and GSH synthesis in CA tissue slices by WGP. However, WGP elicited 13C-citrate accumulation in CA tissue slices, similar to the UK021 case, which points to a specific effect on citrate metabolism via citrate synthase and/or aconitase/isocitrate dehydrogenase. The 13C enrichment of the corresponding metabolites responded negatively to WGP in NC tissue slices.
WGP-treated UK021 CA tissue slice also showed enhanced oxidation of 13C3-pyruvate via the first part of the Krebs cycle, as evidenced by the increased production of 13C-citrate (Fig. 2E) and α-ketoglutarate (αKG, Fig. 2F), relative to the control (Ctl) tissue slice. A major fraction of this enhanced Krebs cycle flow appeared to be diverted to the synthesis of 13C-Glu (Fig. 2J) and then 13C-glutathione (GSH) (Fig. 2K). This was not the case for the WGP-treated UK049 CA tissue slices. Although 13C-citrate synthesis was elevated, the subsequent flow through the Krebs cycle and the Glu-GSH pathway did not change in these tissue slices, relative to the Ctl tissue slices (Fig. 3E,F,J,K).
UK021 but Not UK049 Tumor Tissues Display Elevated PPP and Malic Enzyme Activity While Producing More NADPH and Macrophage Metabolite Itaconate in Response to β-Glucan Treatment
The two sets of CA tissue slices also showed opposite response to WGP in terms of the itaconate level (Figs. 2L, 3L) (i.e., itaconate accumulated in the UK021 but was depleted in the UK049 CA tissue slices). Itaconate synthesis from glucose via decarboxylation of the Krebs cycle intermediate cis-aconitate is known to be induced in murine macrophages to activate immunoactive M1 phenotypes in response to lipopolysaccharide and IFN-γ stimulation (Strelko et al. 2011; Michelucci et al. 2013). However, no 13C-labeled itaconate was evident in any of the tissue slices incubated in 13C6-glucose, despite the high level of 13C enrichment in the citrate precursor (Figs. 2E, 3E). As itaconate is reportedly not produced by lung cancer cells (Michelucci et al. 2013), changes in itaconate levels could reflect metabolism in resident lung macrophages in these tissue slices. If so, our results suggest that itaconate metabolism in human lung macrophages is distinct from that in murine macrophages. The data also support the notion that UK021 CA tissues respond to WGP by undergoing the M2 to M1 polarization of resident macrophages, whereas UK049 tissues fail to do so.
Because NADPH metabolism is crucially important to the differentiation and killing action of M1 macrophages via NADPH oxidase activity (Benoit et al. 2008; Michelucci et al. 2013; Graham et al. 2015), we also examined the total level and 13C-labeling pattern of NADPH in both UK021 and UK049 tissue slices. As shown in Figure 4C, the total level of NADPH was elevated in UK021 but not in UK049 CA tissue slices in response to WGP, which can be largely attributed to the enhanced synthesis of NADPH from glucose via the salvage pathway (Belenky et al. 2007; Muller and Prendergast 2007). This is evidenced by the predominance of 13C-ribose labeling in NADPH and the enhancement of this labeling in UK021 (Fig. 4C) but not UK049 CA tissue slices under WGP treatment (Fig. 5C) (cf. also Supplemental Figure S3 vs. S4 for 13C-N1′-ribose of NAD+). In addition, the hydride sources of NADPH for sustaining superoxide and NO production involved in the killing action of M1 macrophages include the malic enzyme (ME) reaction and the pentose phosphate pathway (PPP) (Ghesquiere et al. 2014). 13C6-glucose flow through the PPP was substantially enhanced in UK021 CA tissue slices by WGP, as evidenced by the increased production of many 13C-labeled PPP intermediates (Fig. 4D–G), particularly sedoheptulose-7-phosphate (Sed7P, Fig. 4E), which is unique to PPP. Although some of the PPP intermediates had a higher level of 13C enrichment in WGP-treated UK049 CA tissue slices (Fig. 5D,F), the 13C enrichment patterns in Sed7P in these slices did not respond to WGP (Fig. 5E). We also noted that other than the dominant 13C7-isotopologue of Sed7P, the 13C2- and 13C5-species were present in appreciable amounts in both sets of tissue slices, which presumably arose from reaction of 13C-enriched intermediates with unlabeled intermediates, either preexisting or derived from an alternative source such as glycogen. All three 13C-isotopologues of Sed7P can be produced via the oxidative and/or nonoxidative branches of PPP. However, the absence of 13C1- and 13C3-species, produced via the transaldolase (TA) reaction in the nonoxidative branch of PPP (denoted by green lines in Figs. 4 and 5), suggests that the oxidative PPP predominates in human lung CA tissue slices. Thus, the heightened PPP activity induced by WGP in UK021 CA tissue slices supplied, at least in part, the hydride for elevated NADPH production, whereas WGP had little effect on both PPP activity and NADPH generation in UK049 CA tissue slices.
Figure 4.

β-Glucan stimulates pentose phosphate pathway (PPP), NADPH production, and glycogen synthesis in UK021 CA tissue slices. PPP metabolite analysis by IC-FTMS is as described in Figure 2. NADPH was analyzed by liquid chromatography (LC)-FTMS, whereas glycogen was quantified by 1H NMR, as described in Methods. 13C-ribose enrichment in NADPH was estimated from the enrichment sum of 13C5- + 13C10-isotopologues, whereas 13C-ring (adenine) enrichment in NADPH was derived from the enrichment sum of the remaining 13C-isotopologues (Sellers et al. 2015). •, 12C; •, 13C; R5P, ribose-5-phosphate; X5P, xylulose-5-phosphate; Sed7P, sedoheptulose-7-phosphate; Gly3P, glyceraldehyde-3-phosphate; E4P, erythrose-4-phosphate; F6P, fructose-6-phosphate; G6P, glucose-6-phosphate; G1P, glucose-1-phosphate; Glyc, glycogen; G6PDH, glucose-6-phosphate dehydrogenase; TK, transketolase; TA, transaldolase. The green dashed curves denote C–C bond break in TA reaction, and the green arrows delineate the addition of the 13C3- and 13C1-3 carbon fragment to the unlabeled E4P to form 13C3- and 13C1-Sed7P, which represent contributions from the nonoxidative branch of PPP. This contribution was minimal because of the very low level of 13C3- and 13C1-Sed7P species in E. Comparing WGP- versus control-treated CA tissue slices, WGP enhanced buildup of PPP-specific intermediates 13C7-Sed7P (E)/13C4-E4P (F) and NADPH (product of the oxidative branch, C) was evident. This supports stimulation of the oxidative branch of PPP. Likewise, enhanced buildup of glycogen precursor 13C6-G1P (H) and 13C-glycogen (I) by WGP was observed in the CA tissue slice. In contrast, PPP metabolites, NADPH, and glycogen metabolites responded negatively to WGP in the NC tissue slice.
Figure 5.

β-Glucan has little effect on PPP, NADPH production, and glycogen synthesis in UK049 CA tissue slices. The same analyses as in Figure 4 were performed for UK049 tissue slices. Each value was an average of two replicates with the error bars as standard error of means. No significant buildup of 13C7-Sed7P (unique metabolite of PPP, E) or total and 13C-ribose containing NADPH (C) was evident in CA tissue slices in response to WGP. As with the UK021 case, there was no significant presence of 13C3- and 13C1-Sed7P in E. This is consistent with little effect of WGP on the oxidative branch of PPP in CA tissue slices. A small buildup of 13C6-G1P (H) was induced by WGP in CA tissue slices but no significant accumulation of the product 13C-glycogen (I) was evident. NC tissues showed little response to WGP in terms of all of these metabolites. All abbreviations are as in Figure 4. •, 12C; •, 13C.
NADPH production via the ME reaction appeared also to occur in both CA tissue slices, as evidenced by the presence of 13C1-malate (magenta 13C; Figs. 2I, 3I). The predominant and perhaps the only path for the synthesis of 13C1-malate from 13C6-glucose is via the reaction sequence of pyruvate dehydrogenase (PDH)-initiated (canonical) Krebs cycle, ME, and pyruvate carboxylase (PCB) reaction (denoted by dashed magenta arrow, Figs. 2 and 3). PCB is active in both UK021 and UK049 CA tissue slices, as evidenced by their significant buildup of 13C3- and 13C5-citrate (Figs. 2E and 3E). Alternatively, the 13C1-pyruvate product of ME can generate 13C1-acetyl CoA, which reacts with unlabeled oxaloacetate (OAA), and undergo the rest of the Krebs cycle again to produce 13C1-malate. Although the effect was small, WGP induced 13C1-malate production in UK021 CA tissue slices (Fig. 2I) but attenuated it in UK049 CA tissue slices (Fig. 3I), which suggests that the ME activity may also contribute to WGP-induced NADPH buildup in the former but not in the latter. As NADPH is the reductant for regenerating glutathione (GSH) from GSSG (the oxidized form of glutathione), the WGP effect on NADPH production can account for the buildup of unlabeled GSH in UK021 CA tissue slices (0 isotopologue, Fig. 2K) but not in UK049 CA tissue slices (Fig. 3K).
Glycogen Synthesis Is Activated by β-Glucan in UK021 but Not in UK049 CA Tissue Slices
WGP appeared to activate the glycogen synthesis pathway in the UK021 CA tissue slice, as evidenced by the excess buildup of the labeled glycogen precursor 13C6-glucose-1-phosphate (G1P, Fig. 4H) and the 13C-glycogen product when comparing WGP versus control treatments (Glyc, Fig. 4I). Although 13C6-G1P accumulated to a small extent in the UK049 CA tissue slices in response to WGP (Fig. 5H), the 13C-glycogen product did not build up significantly (Fig. 5I). Mammalian macrophages have been shown to synthesize glycogen, and the glycogen content depletes during phagocytotic activity (Gudewicz 1975). Recently, exogenous glycogen was shown to stimulate the production of nitric oxide (NO) and inflammatory cytokines by murine macrophage via the Toll-like receptor 2 pathway (Kakutani et al. 2012). These data point to a role of glycogen metabolism in macrophage-mediated immune stimulation, such as WGP-induced iNOS expression in UK021 CA lung tissue slice (cf. Fig. 7C).
Figure 7.
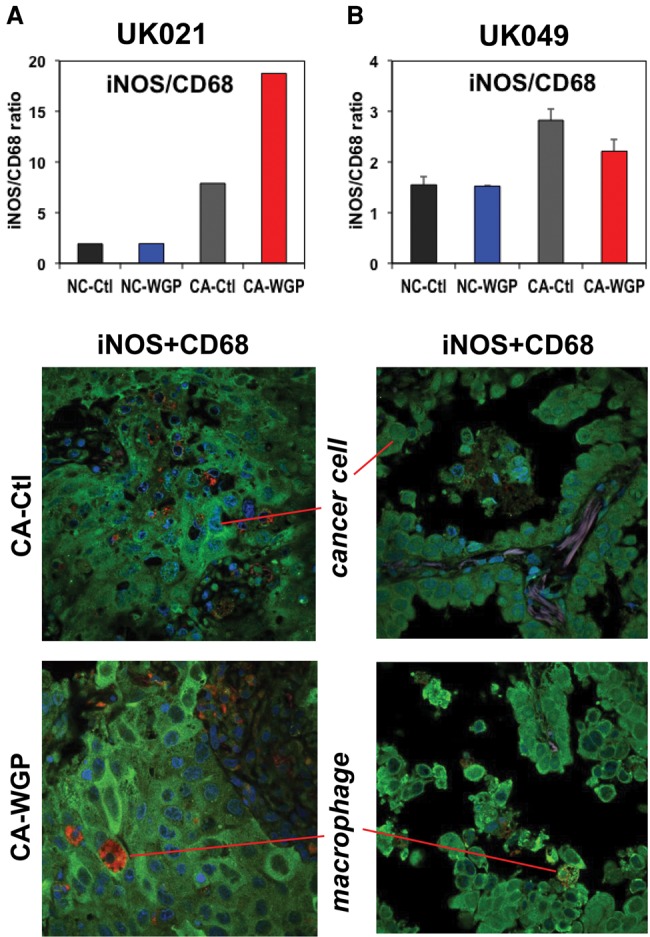
β-Glucan-enhanced iNOS expression in UK021 but not UK049 CA lung tissue slices. WGP treatment, tissue preservation, and immunohistochemical analysis are as in Figure 6. As in Figure 1, the green fluorescence (iNOS) was normalized to the red fluorescence (CD68) to reflect WGP-induced increase in iNOS expression relative to CD68 expression for UK021 (A) but not for UK049 (B) CA lung tissue slices.
We further note that 13C6-glucose metabolism via glycolysis, the Krebs cycle, glutathione synthesis, PPP, and/or nucleotide synthesis was enhanced in CA over NC tissue slices for both UK021 and UK049 patients, as evidenced by the excess buildup in the 13C-labeled metabolites of these pathways in the former tissue slices (Figs. 2–5; Supplemental Figs. S2 and S5–S6). These ex vivo metabolic differences recapitulate those of the CA versus NC tissues in vivo as we have previously reported (Sellers et al. 2015). Notably, the UK021 patient had a lower capacity for glycogen synthesis in the NC than CA tissue slices, whereas the opposite was observed for the UK049 patient (Fig. 4I versus 5I; Supplemental Fig. S5 versus S6). Moreover, the metabolic effect of WGP on NC tissue slices was mostly opposite to that on the paired CA tissue slices for both patients, including PPP, the Krebs cycle, and GSH, Ala, and glycogen synthesis (Figs. 2–5). One notable exception was the release of 13C3-lactate into the medium (Supplemental Fig. S2), for which both NC and CA tissue slices of UK049 responded negatively to WGP (i.e., decreased glycolytic flux in these tissues).
β-Glucan Enhances Necrosis and Reduces Mitotic Index in UK021 CA Lung Tissue Slices but Not in UK049 CA Lung Tissue Slices
We were intrigued by the above-described distinctly perturbed metabolic networks in tumor tissue slices obtained from two patient individuals and asked the question if such metabolic differences lead to different tissue pathological outcomes. We used two molecular markers, RIP-1 and PCNA, to examine the effect of WGP on tissue necrosis and mitotic index, respectively. As shown in Figure 6A, the extent of necrosis increased, whereas the mitotic index decreased in UK021 CA tissue slices in response to WGP. In contrast, UK049 CA tissue slices did not become more necrotic while exhibiting a higher mitotic index with the WGP treatment (Fig. 6B). The effect on necrosis and mitotic index elicited by WGP in UK021 CA tissue slices is unexpected based on the enhancement of glycolysis, Krebs cycle, PPP, NADPH/GSH production, and itaconate buildup by WGP (Figs. 2 and 4). Instead, it is consistent with the polarization of TAM to the M1-like phenotype (Ghesquiere et al. 2014). The necrotic response could reflect oxidative stress resulting from abnormal Krebs cycle activity. The enhanced PPP activity as well as NADPH and GSH production in UK021 (Figs. 2 and 4) but not in UK049 CA tissue slices (Figs. 3 and 5) is consistent with this notion. Moreover, an opposite relationship—that is, decreased Krebs cycle activity, GSH synthesis (Fig. 2), and necrotic response (Fig. 6A)—was noted for the UK021 NC tissue slice. Together, these data suggest that the UK021 CA lung tissue slice responds to WGP by enhancing Krebs cycle and thus oxidative stress, which in turn induces necrotic tissue injuries and antioxidant production.
Figure 6.

β-Glucan enhances necrosis and reduces proliferation potential in UK021 CA tissue slices but not in UK049 CA tissue slices. After the WGP treatment, a small portion of the tissue slices were preserved in formalin and subjected to fluorescence-based histochemical staining separately for PCNA (a mitotic index marker) and RIP-1 (a necrosis marker) (cf. green fluorescence in A and B). All PCNA and RIP-1 stained slides were also stained with DAPI for nuclei (blue fluorescence in A and B), as described in Methods. All images were acquired at 60× magnification. The green fluorescence intensity from three to seven image frames of each slide was averaged for the bar graphs in A and B, with the error bars in B representing standard error of the average of averaged fluorescence intensity from two replicate slices. The mitotic index for the UK021 CA tissue slice was reduced, along with an increase in necrosis in response to WGP (A). The mitotic index for the UK049 CA tissue slices responded positively to WGP in contrast to that for the UK021 CA tissue slice, with little change in necrosis (B). The UK021 NC tissue slices had much lower mitotic index than the CA counterpart with a negative response of necrosis to the WGP treatment (A). For the UK049 NC tissue slices, both mitotic index and extent of necrosis were lower than those in the CA counterpart with little change in either parameter in response to WGP.
The increase in mitotic index of UK049 CA tissue slices by WGP is also unexpected based on the attenuated glycolytic activity (Supplemental Fig. S2) and lack of changes in other metabolic activities observed, except for the increased synthesis of citrate (Fig. 3E). Further investigation into the citrate-mediated metabolic network is needed to better understand its connection to mitotic index.
Differential Macrophage Activation May Contribute to Distinct Perturbations of Ex Vivo Tissue Metabolic Network and Injuries Induced by β-Glucan
We then asked if TAMs in UK021 and UK049 CA lung tissue slices could contribute to the differential metabolic responses described above. Therefore, we probed TAM distribution and phenotypes after WGP treatment in the two patients’ lung tissue slices. As shown in Supplemental Figure S7, both NC and CA lung tissues of UK021 maintained a higher number of CD68+ macrophages than the counterpart tissue slices of UK049 under both control and WGP treatments, as the case for their corresponding freshly resected bulk tissues (Fig. 1C,D). The expression of M1-phenotype marker iNOS was evident in CD68+ TAM and non-CD68+ cells (e.g., cancer cells) in both UK021 (Fig. 7A) and UK049 (Fig. 7B) CA tissue slices. Again, we used the iNOS/CD68 ratio as the measure of iNOS expression normalized to the abundance of CD68+ macrophages. We found that this ratio was enhanced in UK021 CA lung tissues in response to WGP but not in UK049 CA lung tissues with no change in the corresponding NC lung tissues (compare bar graphs in Fig. 7A,B). It is also interesting to note that the iNOS/CD68 ratio was much higher in UK021 than UK049 CA tissue slices after WGP treatment, which was opposite to the case for the corresponding bulk tissues (Fig. 1C,D). Together, these data suggested that WGP activated iNOS expression and possibly M1-like phenotype of TAM in UK021 CA tissue slices but failed to do so in UK049 CA tissue slices.
If so, WGP-induced TAM activation could underlie the activated metabolic networks described above in the UK021 CA lung tissue. iNOS expression is closely linked to NO production, which in excess can be tumoricidal (Vannini et al. 2015). We further surmise that TAM activation could contribute to the decreased mitotic index by inducing NO production via iNOS expression and production of reactive oxygen species (ROS) via the Krebs cycle, leading to necrosis (cf. Fig. 6A). Further investigations will be needed to substantiate this notion.
DISCUSSION
The two NSCLC patients UK021 and UK049 investigated had the same tumor type (adenocarcinoma) and staging (pT3N0M0, stage IIB) but differed in tumor grade, gender, and age (cf. Table 1). As β-glucan is a known activator of macrophages, the clinical diagnosis of COPD for UK021 but not for UK049 is of particular relevance. This difference in comorbidity presumably accounted for the higher number of TAMs (Fig. 1C,D; Supplemental Fig. S1), lower mitotic index, and higher level of necrosis (Fig. 1A,B) in UK021 than in UK049 CA lung tissues. The extensive chronic inflammation could also underlie the more suppressive TAM phenotype (lower iNOS/CD68 ratio; Fig. 1C,D) in the UK021 CA lung tissue.
The abundance of TAMs as well as their more suppressive state in the UK021 CA lung tissue can be related to the metabolic responses of their cultured slices to β-glucan (WGP), which were distinct from those of the cultured UK049 CA lung tissue slices with a lot fewer but possibly more active starting TAMs. WGP activated glycolysis, the Krebs cycle, PPP, and the production of NADPH, glutathione, and glycogen in UK021 (Figs. 2 and 4; Supplemental Figs. S2 and S3) but not in UK049 CA lung tissue slices (Figs. 3 and 5; Supplemental Figs. S2 and S4). These metabolic effects reflected the response of the whole tissue, which involved the complex interactions of cancer and stromal cells in the TME. Although several of these enhanced pathways and product generation (e.g., glycolysis, Krebs cycle intermediate succinate, NADPH, and GSH) are known to be associated with cancer cell metabolism, the response to the immune modulator β-glucan is also consistent with the polarization to inflammatory or M1 phenotype of macrophages in response to microbial infection or proinflammatory cytokines (Ghesquiere et al. 2014). In addition, the activation of these metabolic networks was accompanied by the buildup of itaconate in the UK021 but not UK049 CA lung tissue slices. Itaconate has been shown to stimulate murine macrophages toward M1 phenotype (Strelko et al. 2011; Michelucci et al. 2013). Furthermore, the M1-like state of TAM induced by WGP in the UK021 CA lung tissue slice is consistent with its iNOS overexpression and increased necrosis; the latter is possibly a result of excess production of NO and Krebs cycle-mediated ROS.
WGP could also perturb the cancer cell metabolism in these CA tissue slices, not just macrophages. However, four lines of evidence argued against a significant contribution of activated cancer cell metabolism to the observed metabolic response of UK021 CA tissue slices. First, lung adenocarcinoma A549 cell growth was not affected by WGP (data not shown) and therefore it is unlikely that WGP activates these metabolic processes in the cancer cell population of adenocarcinoma tissue slices. Second, the extent of necrosis increased in the UK021 CA tissue, particularly in the cancer cell–rich region (Fig. 6A), which is inconsistent with activation of cancer cell metabolism in this tissue. Third, iNOS overexpression (Fig. 7A) along with necrosis elicited by WGP in the UK021 CA lung tissue slice suggest sustained and excess NO production (Vannini et al. 2015), which can lead to tumoricidal action (Liu et al. 1998), not metabolic activation. Finally, similar metabolic processes of isolated macrophages was strongly activated by WGP (Liu et al. 2015).
Thus, in this two-case NSCLC patient study, we demonstrated that ex vivo CA lung tissue slices were metabolically more active than their NC lung counterparts, which recapitulated the metabolic reprogramming in CA lung tissues observed in vivo (Sellers et al. 2015). We also showed that the macrophage-activator β-glucan reprogrammed lung CA tissue metabolism ex vivo in one NSCLC patient with COPD and abundance of TAMs (UK021) but not in the other patient (UK049) with no COPD and much less macrophage infiltration. We surmise that the observed reprogrammed networks could reflect β-glucan activation of M1 phenotype of human macrophages. As such, this study presents a unique opportunity for investigating metabolic responses of human tumor-associated macrophages to immune modulators in their native microenvironment on an individual patient basis. Further and more extensive investigations are underway with a larger cohort, which will help validate this case study in terms of the metabolic basis of the differential tumoricidal action of β-glucan among different NSCLC patients, ultimately leading to informed decision on the choice of β-glucan for individual NSCLC therapy.
METHODS
Human Subjects
Patients UK021 and UK049 diagnosed with primary adenocarcinoma and eligible for surgical resection were recruited, and written consent was obtained for the collection of blood, urine, and lung tissues under the University of Kentucky approved IRB protocol (IRB 14-0288-F6A).
Lung Tissue Collection, Slice Preparation, Treatment, and Extraction
Fresh CA and surrounding NC (distal and proximal to CA) lung tissues were collected in the operating room within 5 min of surgical resection, a portion of which was immediately flash-frozen in liquid N2 for metabolite analysis and a second portion was preserved in 4% buffered formalin (Fisher Scientific). The remaining tissues were thinly sliced by the surgeon at ∼0.7- to 1-mm thickness with wet weights of 8–20 mg for UK021 tissue slices and 14–29 mg for UK049 tissue slices. All tissue slices were incubated in glucose and Gln-free DMEM media (Sigma-Aldrich) supplemented with 0.45% unlabeled or 13C6-glucose (Cambridge Iosotope Laboratories), 2 mM Gln, 10% dialyzed fetal bovine serum, 50 U/mL penicillin, and 50 µg/mL streptomycin (Fisher Scientific) for 24 h at 37°C/5% CO2 with gentle rocking to facilitate nutrient supplies and waste product mixing (Sellers et al. 2015; Fan et al. 2016). One half of the cultured tissue slices also contained 100 µg/mL WGP β-glucan (Biothera) in sterile phosphate-buffered saline (PBS), whereas the other half had none to serve as controls. Culture media were sampled at 0, 9 or 11, and 24 h of incubation. At the end of incubation, tissue slices were quickly rinsed in cold PBS three times to remove medium components, blotted dry, weighed on a four-place balance for wet weight, and flash-frozen in liquid N2.
The frozen tissues were then homogenized in 60% cold CH3CN in a ball mill (Precellys-24, Bertin Technologies) for denaturing proteins and optimizing extraction. Polar metabolites were extracted by the solvent partitioning method with a final CH3CN:H2O:CHCl3 (2:1.5:1, v/v) ratio, as described previously (Fan 2012). The polar extracts were lyophilized before reconstitution in 50% D2O for NMR analysis or in H2O for IC-FTMS analysis. The culture media were deproteinated in 80% acetone at −20°C for 0.5 h before centrifugation, lyophilization, and reconsititution in 50% D2O for 1H NMR analysis.
Histochemical and Image Analysis
The tissue specimens were formalin-fixed and paraffin-embedded, sliced as 4-μm thick tissue sections, deparaffinized in Xylene Substitute (A5597, Sigma-Aldrich), and rehydrated through graded alcohols. Antigen retrieval was carried out using 10 mM sodium citrate buffer (pH 6.0) in a microwave (heated to boiling, maintained at a subboiling temperature for 10 min, and then cooled at room temperature for 30 min). Methanol permeabilization was performed for PCNA staining by incubating tissue sections in ice-cold 100% methanol at −20°C for 10 min followed by PBS rinse for 5 min. Nonspecific binding was blocked with 10% appropriate host serum, 3% BSA, and 0.3% Triton X-100 in 1× PBS at room temperature for 1 h, followed by incubation with primary antibodies at 4°C for overnight. Tissue sections were then rinsed three times with PBS for 5 min each before incubation with fluorescent-conjugated secondary antibodies in the dark at room temperature for 1 h, followed by three rinses with PBS for 5 min each.
Primary antibodies used were as follows: PCNA (D3H8P) XP Rabbit mAb (1:800) and RIP-1 (D94C12) XP Rabbit mAb (1:100, Cell Signaling Technology); CD68 (KP1) (1:200, mouse monoclonal, Santa Cruz Biotechnology); iNOS (1:200, rabbit polyclonal PA3-030A, ThermoFisher Scientific). Secondary antibodies used were as follows: goat anti-rabbit IgG (H + L) Alexa Fluor 488 (1:100), donkey anti-mouse IgG Alexa Fluor 594 (undiluted); rabbit anti-rat IgG (H+L) Alexa Fluor 594 (1:200) from Thermo Fisher Scientific; goat anti-mouse IgG H&L Alexa Fluor 488 (1:200) (Abcam). The antibody dilution buffer was 1% BSA and 0.3% Triton X-100 in 1× PBS.
Stained slides were mounted in ProLong Gold Antifade Mountant with DAPI (P-36931, Thermo Fisher Scientific) and fluorescent images were acquired using a laser scanning confocal microscope Olympus FluoView FV1000 with a 60× oil immersion lens (UPLSAPO 60XO).
Gomori Trichrome Staining was performed using Richard-Allan Scientific Gomori Trichrome (Blue Collagen) (87020, Thermo Scientific) following the manufacturer's standard staining protocol. The collagen fibers were stained blue, the cytoplasm and muscle fibers were stained red, and the nuclei were stained dark blue-to-black. Slides were scanned using an Aperio ePathology scanner under a 20× objective and viewed by Aperio ImageScope software.
For image quantification, positively labeled cells were counted in at least three 217 × 212-µm fields from the selected lung slices. For all stained slides, three to eight image frames were captured from each color channel at 12-bit digital resolution using the appropriate fluorescence filter set, under identical exposure and magnification settings. The image frames were then analyzed using the ImageJ program (National Institutes of Health). Region of interest (ROI) of each image frame was quantified by thresholding individual frames and using the function “Analyze particles” to calculate the percentage of the image area exceeding the set threshold. This value was averaged over multiple frames before the ratio of the “green” (PCNA, RIP-1, or iNOS) or “red” (CD68) fluorescence to the “blue” DAPI fluorescence was calculated.
NMR Analysis
The polar extracts were analyzed in 1.7-mm tubes on an automated 14.1 T NMR spectrometer equipped with a 3-mm HCN cold probe (Agilent Technologies). 1H NMR data were acquired using a simple one-pulse method with a 90° observe pulse, water presaturation, 6-sec interpulse delay, and 512 transients. 13C-edited 1H heteronuclear single quantum coherence (HSQC) spectra were acquired with 90° observe pulse, 2-sec interpulse delay, and an adiabatic 1H decoupling scheme. The 1H resonance intensities in 1D 1H and HSQC spectra were quantified by using the peak deconvolution routine of the MesReNova software and calibrated against the peak intensity of the methyl resonance of d6-2,2-dimethyl-2-silapentane-5-sulfonate (DSS) (an internal standard added at 27.5 nmol) for 1D 1H or that of the methyl resonance of lactate (quantified from 1D 1H spectra) for 1D HSQC.
Ion Chromatography–Mass Spectrometric Analysis
Polar extracts were reconstituted in ultrapure deionized water (EMD Millipore) in a volume based on tissue wet weight. All analyses were performed on a Dionex ICS-5000+ ion chromatography interfaced to a Thermo Fusion Orbitrap Tribrid mass spectrometer (IC-FTMS) (Thermo Fisher Scientific). Ion chromatography (Wang et al. 2014) was performed using an IonPac AS11-HC-4 μm RFIC&HPIC (2 × 250 mm) column and an IonPac AG11-HC-4 μm guard column (2 × 50 mm). The column flow rate was kept at 0.38 mL/min with column temperature at 35°C and 0.06 mL/min methanol added postcolumn as a makeup solvent to aid vaporization in the heated electrospray ionization (HESI) unit. The HESI vaporizer temperature was 400°C with sheath gas set at 35 Arb and auxiliary nitrogen flow at 4 Arb. The column was initially equilibrated for 8 min with 1 mM KOH, and then followed by 1 mM KOH for 2 min after 5 μL of sample was injected. The KOH gradient program used to elute samples included ramping up to 40 mM KOH from 2 to 25 min, and to 100 mM from 25 to 39.1 min, at 100 mM to 50 min, and ramping down to 1 mM KOH at 50.1 min and at 1 mM KOH to 52.5 min. KOH suppression was achieved with a Dionex AERS 500 2 mm suppressor with an external AXP pump supplying regenerant at a flow rate of 0.75 mL/min and injected into the Orbitrap mass spectrometer via HESI. Mass spectra were recorded at a resolution of 450,000 (achieving a resolution of ∼360,000 at 400 m/z) from 50 to 750 m/z mass scan range, with detection in the negative ion mode voltage using the following settings: HESI = 2800 V; ion transfer tube temperature = 300°C; automatic gain control (AGC) = 2 × 105; maximal injection time = 100 msec. Peak areas were integrated and exported to Excel via the Thermo TraceFinder (version 3.3) software package. Peak areas were corrected for natural abundance as previously described (Moseley 2010).
ADDITIONAL INFORMATION
Data Deposition and Access
The raw analytical data and relevant metadata have been deposited at the Metabolomics Consortium Data Repository and Coordinating Center (DRCC; http://www.metabolomicsworkbench.org/data/index.php) under project number PR000292.
Ethics Statement
Written consent was obtained for the collection of human tissue, blood, and urine samples under an IRB approved protocol (IRB 14-0288-F6A) at the University of Kentucky.
Acknowledgments
We thank Teresa Cassel, Yan Zhang, Penghui Lin, Ramon Sun, Timothy Scott, Anh-Thu Le, and Maria Scavo for assistance on the collection, experimentation, processing, and extraction of human tissue slices and media; we also thank Penghui Lin for assistance in the medium NMR analysis.
Author Contributions
T.W.-M.F. designed the experiments, collected samples, analyzed/interpreted data, and wrote the manuscript; A.N.L. designed experiments, collected samples, and contributed to the manuscript writing; M.O.W., Q.S., and R.M.H. performed IC-FTMS analysis and contributed to the manuscript writing; H.S. and J.T.-C. performed histochemical analysis and contributed to the manuscript writing; and A.M. and J.T.M. contributed to experimental design, performed the lung surgeries, and procured thin tissue slices for ex vivo studies.
Funding
This work was supported in part by the National Institutes of Health (NIH) P01 CA163223-01A1, NIH 1R21ES025669-01, NIH 1U24DK097215-01A1, the Edith D. Gardner Endowed Chair (to T.W.-M.F.), and the Carmen L. Buck endowed chair (to A.N.L.). NMR and mass spectrometry data were recorded at the Center for Environmental and Systems Biochemistry supported by the University of Kentucky and by National Cancer Institute (NCI) Cancer Center Support Grant (P30 CA177558).
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Belenky P, Bogan KL, Brenner C. 2007. NAD+ metabolism in health and disease. Trends Biochem Sci 32: 12–19. [DOI] [PubMed] [Google Scholar]
- Benoit M, Desnues B, Mege JL. 2008. Macrophage polarization in bacterial infections. J Immunol 181: 3733–3739. [DOI] [PubMed] [Google Scholar]
- Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, Witkiewicz AK, Heiden MG, Migneco G, Chiavarina B, et al. 2010. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 9: 1960–1971. [DOI] [PubMed] [Google Scholar]
- Caneba CA, Yang L, Baddour J, Curtis R, Win J, Hartig S, Marini J, Nagrath D. 2014. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis 5: e1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. 2011. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci 108: 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW-M. 2012. Sample preparation for metabolomics investigation. In The handbook of metabolomics: pathway and flux analysis, methods in pharmacology and toxicology (ed. Fan TW-M, Lane AN, Higashi RM), pp. 7–27. Springer Science, New York. [Google Scholar]
- Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM. 2009. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol Cancer 8: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, Lane AN, Higashi RM. 2016. Stable isotope resolved metabolomics studies in ex vivo tissue slices. Bio-protocol 6: e1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. 2013. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17: 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghesquiere B, Wong BW, Kuchnio A, Carmeliet P. 2014. Metabolism of stromal and immune cells in health and disease. Nature 511: 167–176. [DOI] [PubMed] [Google Scholar]
- Graham DB, Becker CE, Doan A, Goel G, Villablanca EJ, Knights D, Mok A, Ng AC, Doench JG, Root DE, et al. 2015. Functional genomics identifies negative regulatory nodes controlling phagocyte oxidative burst. Nat Commun 6: 7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudewicz PW. 1975. Mechanisms and regulation of macrophage glycogen metabolism. In Physiology, p. 114 Loyola University Chicago, Chicago, IL. [Google Scholar]
- Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, et al. 2013. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci 110: 3919–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Ho P-C, Bihuniak Jessica D, Macintyre Andrew N, Staron M, Liu X, Amezquita R, Tsui Y-C, Cui G, Micevic G, Perales Jose C, et al. 2015. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162: 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakutani R, Adachi Y, Takata H, Kuriki T, Ohno N. 2012. Essential role of Toll-like receptor 2 in macrophage activation by glycogen. Glycobiology 22: 146–159. [DOI] [PubMed] [Google Scholar]
- Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR, et al. 2015. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. 2012. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithner K, Wohlkoenig C, Stacher E, Lindenmann J, Hofmann NA, Galle B, Guelly C, Quehenberger F, Stiegler P, Smolle-Juttner FM, et al. 2014. Hypoxia increases membrane metallo-endopeptidase expression in a novel lung cancer ex vivo model—role of tumor stroma cells. BMC Cancer 14: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Wang CH, Chen TC, Lin HC, Yu CT, Kuo HP. 1998. Increased level of exhaled nitric oxide and up-regulation of inducible nitric oxide synthase in patients with primary lung cancer. Br J Cancer 78: 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Luo F, Ding C, Albeituni S, Hu X, Ma Y, Cai Y, McNally L, Sanders MA, Jain D, et al. 2015. Dectin-1 activation by a natural product β-glucan converts immunosuppressive macrophages into an M1-like phenotype. J Immunol 195: 5055–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C, Madden C, et al. 2012. Metabolism of [U-13C]glucose in human brain tumors in vivo. NMR Biomed 25: 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, et al. 2013. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci 110: 7820–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley H. 2010. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinformatics 11: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. 2012. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AJ, Prendergast GC. 2007. Indoleamine 2,3-dioxygenase in immune suppression and cancer. Curr Cancer Drug Targets 7: 31–40. [DOI] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. 2010. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. 2009. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J 33: 118–126. [DOI] [PubMed] [Google Scholar]
- Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. 2011. The tissue microlocalisation and cellular expression of CD163, VEGF, HLA-DR, iNOS, and MRP 8/14 is correlated to clinical outcome in NSCLC. PLoS One 6: e21874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, Boscá L. 2010. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185: 605–614. [DOI] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M II, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. 2015. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 125: 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. 2013. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. 2008. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118: 3930–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern R, Shuster S, Neudecker BA, Formby B. 2002. Lactate stimulates fibroblast expression of hyaluronan and CD44: the Warburg effect revisited. Exp Cell Res 276: 24–31. [DOI] [PubMed] [Google Scholar]
- Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, Roberts MF. 2011. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 133: 16386–16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. 2013. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496: 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayanithy V, Babatunde V, Dickson EL, Wong P, Oh S, Ke X, Barlas A, Fujisawa S, Romin Y, Moreira AL, et al. 2014. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp Cell Res 323: 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini F, Kashfi K, Nath N. 2015. The dual role of iNOS in cancer. Redox Biol 6: 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenta S, Schroeder T, Mueller-Klieser W. 2004. Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem 11: 2195–2204. [DOI] [PubMed] [Google Scholar]
- Wang J, Christison TT, Misuno K, Lopez L, Huhmer AF, Huang Y, Hu S. 2014. Metabolomic profiling of anionic metabolites in head and neck cancer cells by capillary ion chromatography with orbitrap mass spectrometry. Anal Chem 86: 5116–5124. [DOI] [PubMed] [Google Scholar]
- Warburg O. 1924. Is the respiration of cancer cells normal? Biochem Zeitschr 51. [Google Scholar]
- Warburg O. 1956. On the origin of cancer cells. Science 123: 309–314. [DOI] [PubMed] [Google Scholar]
- Wysoczynski M, Ratajczak MZ. 2009. Lung cancer secreted microvesicles: underappreciated modulators of microenvironment in expanding tumors. Int J Cancer 125: 1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Hanai JI, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. 2014. Targeting lactate dehydrogenase-A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab 19: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. 2007. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178: 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanakou M, Germenis AE, Karanikas V. 2007. Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunol Lett 111: 69–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.