

Abstract
Whole-exome sequencing is increasingly used for diagnosis and identification of appropriate therapies in patients. Here, we present the case of a 3-yr-old male with a lifelong and severe transfusion-dependent anemia of unclear etiology, despite an extensive clinical workup. Given the difficulty of making the diagnosis and the potential side effects from performing interventions in patients with a congenital anemia of unknown etiology, we opted to perform whole-exome sequencing on the patient and his parents. This resulted in the identification of homozygous loss-of-function mutations in the EPB41 gene, encoding erythrocyte protein band 4.1, which therefore causes a rare and severe form of hereditary elliptocytosis in the patient. Based on prior clinical experience in similar patients, a surgical splenectomy was performed that resulted in subsequent transfusion independence in the patient. This case illustrates how whole-exome sequencing can lead to accurate diagnoses (and exclusion of diagnoses where interventions, such as splenectomy, would be contraindicated), thereby resulting in appropriate and successful therapeutic intervention—a major goal of precision medicine.
Keywords: chronic hemolytic anemia, congenital hemolytic anemia
INTRODUCTION
There is an extremely sophisticated understanding of the molecular genetics underlying congenital forms of anemia. This group includes disorders affecting red blood cell (RBC) production, RBC function, or some combination of these pathologies (Sankaran and Weiss 2015). For most cases of these anemias, the range of molecular lesions has been extensively characterized, and only a minority of cases have no known molecular basis (Sankaran and Gallagher 2013). Despite this sophisticated understanding, however, the diagnosis of congenital forms of anemia primarily relies on clinical features and laboratory testing. Genetic testing is not routinely performed. However, in some cases, even after performing a range of clinical and laboratory tests, the exact diagnosis remains unclear. Molecular genetic testing can be extremely valuable in such cases.
The diagnostic uncertainty in such cases represents a significant limitation for treatment, because specific interventions may cause unintended harm. For example, in certain forms of anemia, particularly those involving defects in RBC production (erythropoiesis), there can be significantly increased iron absorption that can result in iron overload when coupled with regular transfusion regimens and inadequate chelation (Sankaran and Weiss 2015). Some anemias can significantly benefit from removal of the spleen (splenectomy), but this can potentially result in side effects—most notably pulmonary hypertension, severe thromboembolic disease, and cardiovascular disease—in patients with specific forms of anemia, including some hemoglobin disorders, as well as hereditary hydrocytosis and xerocytosis (Stewart et al. 1996; Karimi et al. 2011).
Here, we present the case of a child with a transfusion-dependent congenital anemia in which whole-exome sequencing (WES) ultimately identified the diagnosis of a rare and severe form of hereditary elliptocytosis (HE) due to homozygous loss-of-function mutations in EPB41. HE results from defects in the horizontal interactions of the RBC membrane skeleton that cause increased RBC breakdown. Defects in α- and β-spectrins (SPTA1 and SPTB) and erythrocyte protein band 4.1 (EPB41) are the most common causes of HE, which generally displays an autosomal-dominant inheritance (Birkenmeier and Barker 2004; Delaunay 2007; Da Costa et al. 2013; Gallagher 2013). Most individuals with HE are asymptomatic or have mild forms of anemia, but severe transfusion-dependent forms of anemia have been rarely reported (Tchernia et al. 1981; Dalla Venezia et al. 1992; Conboy et al. 1993; Baklouti et al. 2011). In this case, the correct diagnosis that resulted from genetic testing led to definitive treatment with a surgical splenectomy and allowed the patient to become transfusion-independent.
RESULTS
Clinical Presentation and Family History
A 3-yr-old male from Kuwait presented to our hospital for evaluation of a lifelong, transfusion-dependent anemia of unclear etiology. At birth, he initially presented with jaundice requiring exchange transfusion. He was found 2 months later to be pale and lethargic and have a hemoglobin level in the 2–3 g/dL range. He subsequently required transfusions every 1–2 months to maintain his hemoglobin level at >7–8 g/dL. Although his father had been jaundiced intermittently throughout adulthood and his mother had an asymptomatic anemia, neither had a formal diagnosis nor had ever required transfusions. The boy's diagnosis remained unknown despite an extensive workup; thus, the optimal treatment approach remained unclear.
Although the broad differential diagnosis for anemia is relatively straightforward (red cell destruction, blood loss, insufficient red cell production, or a combination of these factors), definitive diagnosis in this patient was complicated by his dependence on monthly transfusions, which confounded much of the testing obtained, even in periods distal from the blood transfusions. The patient had massive splenomegaly (>5 cm below the costal margin and crossing the midline) and frontal bossing, suggestive of either erythroid hyperplasia or extramedullary erythropoiesis. The lactate dehydrogenase (450–550 unit/L) and unconjugated bilirubin (total bilirubin 2–7 mg/dL with conjugated bilirubin 0.3 mg/dL) were elevated, suggestive of hemolysis and/or ineffective erythropoiesis. Multiple negative Coombs tests and the lifelong nature of this anemia made an autoimmune etiology unlikely. The remaining possible etiologies included a hemolytic anemia secondary to an intrinsic membrane defect, a hemoglobinopathy, an enzymatic defect (such as G6PD or pyruvate kinase deficiency), or a form of ineffective erythropoiesis (such as the congenital dyserythropoietic anemias).
In this case, definitive diagnosis was felt to be important as splenectomy could potentially improve or eliminate the patient's transfusion dependence but could also result in clinical complications with certain diagnoses, such as hereditary hydrocytosis or xerocytosis (Delaunay et al. 1999). The extensive workup that spanned a number of years and institutions included a normal hemoglobin electrophoresis, normal incubated osmotic fragility, normal eosin-5-malemide binding test by flow cytometry making hereditary spherocytosis less likely, and normal RBC enzyme evaluation that made an enzymopathy less likely (including normal enzymatic activity of pyruvate kinase, glucose phosphate isomerase, hexokinase B, adenosine deaminase, adenylate kinase, phosphofructokinase, phosphoglycerate kinase, triosephosphate isomerase, and pyrimidine 5′ nucleotidase). Gel electrophoresis of red cell membrane proteins suggested a partial erythrocyte protein band 4.1 (herein band 4.1R) deficiency consistent with HE; however, individuals with partial band 4.1R deficiency have a mild hemolytic anemia that alone would not explain this patient's transfusion dependence. In addition, this defect may have been secondary to another etiology, as can occur with some forms of congenital dyserythropoietic anemia (Wickramasinghe and Wood 2005).
Given the child's frequent RBC transfusions, the extent to which the band 4.1R deficiency was contributing to the anemia was unclear. Evaluation of the peripheral blood smear demonstrated elliptocytes with bizarre morphology (Fig. 1). A bone marrow aspirate revealed erythroid elements that had ragged cellular borders and extensive blebbing—findings suggestive of a membrane defect—but these findings could not narrow down the differential diagnosis further. Importantly, no signs of dyserythropoiesis were observed. Having reached the limit of traditional diagnostic testing, the patient and his parents underwent WES to attempt to identify a definitive diagnosis (Table 1).
Figure 1.

(A,B) Peripheral blood smears obtained 2 mo prior to splenectomy demonstrate a dimorphic population with predominance of normochromic, normocytic cells with interspersed population demonstrating anisopoikilocytosis with frequent elliptocytes and other unique morphologies. This was taken prior to a monthly transfusion but likely contains a significant fraction of transfused blood. (C,D) Peripheral blood smears obtained 2 mo postsplenectomy with marked red cell anisopoikilocytosis and polychromasia. Numerous elliptocytes can be seen here with some residual transfused blood present. The smears were imaged with a 100× objective and stained using Wright–Giemsa.
Table 1.
Whole-exome sequencing coverage
| Sample | Total aligned reads | Average read coveragea | Coding genesa ≥10-fold coverage (%) | Red cell disorder genes ≥10-fold coverage (%) |
|---|---|---|---|---|
| Proband | 41,999,140 | 42.8 | 92.8 | 92.7 |
| Father | 33,564,334 | 31.3 | 87.5 | 86.0 |
| Mother | 34,716,160 | 33.5 | 88.2 | 85.6 |
aBased on the Consensus Coding DNA Sequence (CCDS) database.
Genomic Analysis
We examined the WES data for rare variants in known genes implicated in the major congenital RBC disorders that could potentially fit the phenotype observed in the proband with either an autosomal-recessive or de novo inheritance pattern. After filtering for variants in the Exome Aggregation Consortium (ExAC), which consists of exome-sequencing data from 60,706 unrelated individuals, we tested for variants in the genes implicated in congenital red cell disorders that result in anemia (ANK1, SPTB, SPTA1, SLC4A1, EPB42, EPB41, PIEZO1, KCNN4, GLUT1, G6PD, PKLR, NT5C3A, HK1, GPI, PGK1, ALDOA, TPI1, PFKM, ALAS2, FECH, UROS, CDAN1, SEC23B, KIF23, KLF1, GATA1, HBB, HBA1, and HBA2). Assuming homozygous recessive inheritance, there were no rare (defined as no homozygotes present in ExAC) damaging (missense or predicted loss-of-function [LOF]) mutations in any of the red cell disorder genes except for EPB41. Similarly, testing for rare damaging compound heterozygous mutations in these genes yielded no variants (allele frequency [AF] < 0.1% in ExAC). Assuming de novo inheritance, there were no rare (AF < 0.01% in ExAC) damaging mutations in any of the erythroid disorder genes.
Interpretation of the family's WES data revealed that the male proband inherited two copies of a rare 7-bp deletion of the gene encoding erythrocyte protein band 4.1 (EPB41) that is predicted to cause complete deficiency of the protein as a result of a frameshift mutation (Table 2). Complete deficiency of band 4.1R has previously been associated with severe phenotypes of HE in rare patients (Tchernia et al. 1981; Dalla Venezia et al. 1992; Conboy et al. 1993; Baklouti et al. 2011). Consistent with this genetic finding, we were able to confirm a near complete absence of glycophorin C on the membrane of the abnormal elliptocytes, which is a finding that would occur specifically in the setting of complete band 4.1R deficiency (Fig. 2; Reid et al. 1990). Flow cytometry analysis confirmed a marked specific deficiency of surface expression of glycophorin C in patient red cells and normal levels of surface expression on transfused normal red cells.
Table 2.
Rare and damaging mutations in red cell disorder genes
| Gene | Mutation | Proband | Father | Mother | Predicted effect | ExAC AF (%) |
|---|---|---|---|---|---|---|
| EPB41 | Chr1:29344900 CGAATCAG>C c.444_450delGAATCAG p.Asn149Profs |
Two copies | One copy | One copy | Frameshift | 0 |
| ANK1 | Chr8:41554011 C>T c.2830G>A p.Ala944Thr |
One copy | One copy | Zero copies | Missensea; A944T | 0.08 |
ExAC, Exome Aggregation Consortium; AF, allele frequency.
aPredicted probably/possibly deleterious by PolyPhen HDIV (P), LRT (D), MutationTaster (D), and fathmm-MKL (D) and predicted benign/neutral by SIFT (T), PolyPhen HVAR (B), Mutation Assessor (N), fathmm (T), PROVEAN (N), CADD (3.6), DANN (0.49), MetaSVN (T), and MetaLR (T).
Figure 2.
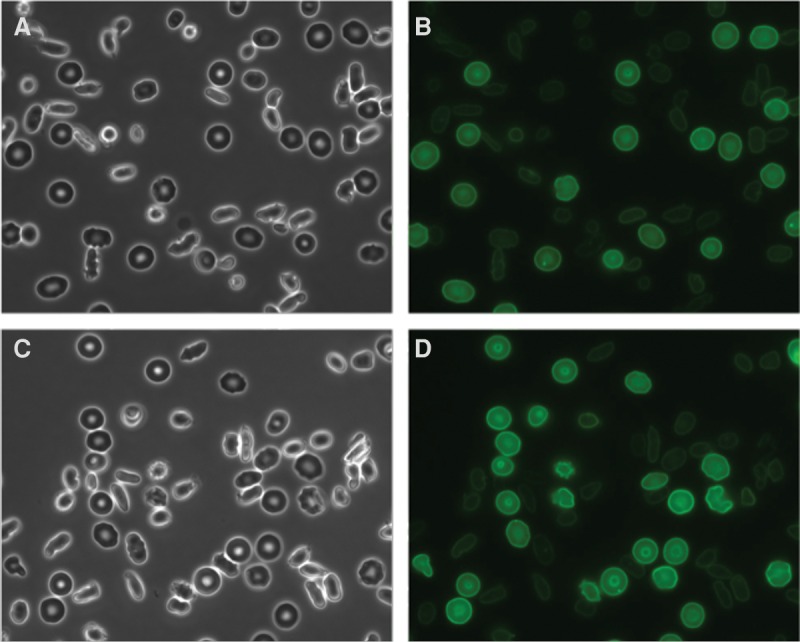
Microscopic images of representative fields of red blood cells (RBCs) from the EPB41-deficient patient. (A,C) Phase-contrast and (B,D) corresponding fluorescent images of Alexa 488 anti-GPC labeled RBCs. The images were taken using a fluorescent microscope (Nikon Ti) with a 60× oil immersion objective.
To look for potential disease-modifying mutations, we investigated semirare (AF < 0.1% or AC [allele count] < 121 in ExAC) damaging variants in the red cell disorder genes where one allele was present in the proband and one allele was present in either the mother or father. The only mutation fitting these criteria was a paternal ANK1 missense mutation that was predicted to be deleterious by some algorithms and benign by others (Table 2). This paternally inherited variant in erythrocyte ankyrin, encoded by the gene ANK1, which plays a key role in membrane integrity and stability, results in a threonine substitution for an alanine residue at position 944 in the ankyrin ZU5N domain (Ala944Thr). Ala944 has been shown to be involved in van der Waals interactions with the BC loop of spectrin, and there is evidence to suggest that amino acids larger than alanine at the 944 position cause steric clashes with residues in the BC loop (Ipsaro and Mondragon 2010). The clinical significance of this heterozygous variant in a protein known to be a key component of the RBC membrane skeleton is unknown.
Immediate Treatment Outcomes
With the WES diagnosis of complete EPB41 deficiency causing HE (and possible modification or worsening of the phenotype by the rare variant in the ANK1 gene), splenectomy was undertaken and was successful without complications. Following the procedure, the patient became transfusion-independent. Subsequent hemoglobin values ranged from 11.5 to 12.5 g/dL with reticulocyte counts of 2%–3%. There was also a notable alteration in the morphology on the blood smear with a variety of bizarre morphologies noted following the splenectomy and a larger extent of variation in RBC size and shape (Fig. 1). Further treatment or inventions were not necessary, and the patient was able to return home without any concerns. For several years following the splenectomy, the patient continues with stable hemoglobin levels and has not required subsequent transfusions.
DISCUSSION
Although most individuals with HE have no symptoms or only mild anemia and generally present with RBC morphology clearly indicating the presence of elliptocytes, this patient's particularly severe phenotype, variable morphology, and reliance on regular transfusions made the diagnosis difficult. As a result of WES and a definitive genetic diagnosis of EPB41 loss-of-function homozygous mutations, splenectomy was undertaken and the patient no longer required transfusions. Importantly, definitive diagnosis with WES excluded diagnoses that could be worsened with splenectomy, such as hereditary xerocytosis or specific hemoglobin disorders, before the procedure.
Analysis of the patient's exome found an additional ANK1 variant that may have contributed to deranged RBC membrane skeletal interactions and severe fragility. The RBC membrane is supported by a complex web of interacting proteins; spectrins (the most commonly mutated protein in HE) exist predominantly as tetramers and contribute to the structural hexagonal network of the red cell membrane skeleton, whereas band 4.1R (which our patient completely lacked) mediates interactions between these spectrin units and actin, another RBC membrane skeletal protein. Ankyrins further mediate connections between spectrin, actin, and other components of the membrane (Giorgi et al. 2001), but variants in ANK1 have not previously been implicated in any reported case of HE. Although the functional significance of the ANK1 variant identified here is currently unknown, the importance of modifier mutations that affect the severity and presentation of disease is increasingly appreciated in other red cell disorders such as hereditary spherocytosis and erythropoietic porphyria (To-Figueras et al. 2011; van Zwieten et al. 2015).
Case series of next-generation sequencing in diagnostic dilemmas have shown that this technology permits diagnosis in at least 25% of cases (Yang et al. 2013; Lee et al. 2014), including anemias with a presumed genetic component (Lyon et al. 2011). In this case, following a definitive molecular diagnosis, our patient was able to receive an appropriate therapy with significant clinical improvement. Beyond our patient's individual experience, sequencing his exome facilitated description of a novel variant in EPB41 leading to the phenotype of severe HE. This case illustrates the value of clinical WES for diagnosis and therapy (Yang et al. 2014). We suggest that in many complex congenital anemias, WES may be a cost-effective approach that would allow definitive diagnosis to be reached in challenging patients requiring transfusions or lacking a diagnosis through standard clinical testing.
MATERIALS AND METHODS
Sequencing and Analysis
DNA was extracted from maternal, paternal, and proband blood samples for exome sequencing. Samples were prepared as a sequencing library (Illumina) and enriched for exonic sequences using the NimbleGen Exome Enrichment protocol (SeqCap EZ VCRome 2.0). The captured libraries were sequenced using an Illumina HiSeq 2000 Sequencer and paired-end 100-bp reads were obtained. The reads were mapped to the human genome assembly University of California at Santa Cruz hg19 using Burrows–Wheeler alignment (BWA version 0.5.8) (Li and Durbin 2009). Coverage across the CCDS plus an additional 20 bp on the side of each exon (downloaded from the UCSC Genome Browser on December 13, 2015) was calculated using BEDTools (version 2.17) (Quinlan and Hall 2010). Single-nucleotide polymorphisms (SNPs) and small insertions/deletions were called with SAMtools (version 0.1.7) (Li et al. 2009). The variant call file (VCF) containing these variants was annotated with Variant Effect Predictor v83 and the dbNSFP (Database for Nonsynonymous SNPs’ Functional Predictions) v3.1 (McLaren et al. 2010; Liu et al. 2015). The Genome Analysis Toolkit was used to filter variants meeting the specified annotations (missense and LOF), inheritance (recessive, de novo, or parentally inherited modifier), and population allele frequency (based on the ExAC v0.3 release of 60,706 individuals) (McKenna et al. 2010; Exome Aggregation Consortium et al. 2015). The predicted deleteriousness of each missense variant was obtained from dbNSFP, and a summary of these algorithms and their output is available at https://sites.google.com/site/jpopgen/dbNSFP. Sanger sequencing verified the pathogenic EPB41 mutation in the family.
Glycophorin C Expression Analysis
Whole blood from the EPB41-deficient patient (∼50 µL) was washed three times in PBS and 1 µL of packed red cells was resuspended in 200 µL PBS. The cell suspension was incubated at room temperature for 30 min with 1 µL of Alexa 488 conjugated glycophorin C monoclonal antibody (GPC; R&D Systems). The red cells were subsequently washed and resuspended in 1 mL PBS + 0.5% BSA, 200 µL of which is placed into an observation chamber. Phase contrast and fluorescent images were then taken of each representative field using an inverted Nikon Ti fluorescence microscope with 60× 1.4 NA oil immersion objective. Cell surface expression of glycophorin C was assessed by flow cytometric analysis with a FACS Canto flow cytometer (Becton Dickinson), and flow data overlay plots were produced using the CellQuest Pro software (Becton Dickinson).
ADDITIONAL INFORMATION
Data Deposition and Access
Our patient consent does not permit patient sequence data to be uploaded to a data repository. The EPB41 loss-of-function variant reported has been deposited in the ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) database under accession number SCV000262628.1.
Ethics Statement
The Institutional Review Board at Boston Children's Hospital approved this research protocol (IRB number is 10-02-0053, “Manton Center for Orphan Disease Research Gene Discovery Core”). Written informed consent was received from both parents.
Acknowledgments
We thank the family for their interest in this work and willingness to participate in the research study.
Author Contributions
J.N.L., R.F.G, M.C.T., P.B.A., and V.G.S contributed to patient recruitment and phenotyping. J.N.L., J.C.U., N.M., S.E.L., P.B.A., and V.G.S. contributed to sequence data analysis and interpretation, J.N.L., J.C.U., J.H., N.M., S.E.L., and V.G.S. contributed to functional evaluation of the variant. J.N.L., J.C.U., and V.G.S contributed to writing the initial draft of the manuscript. All authors contributed to revising the manuscript and reviewing the final draft.
Funding
This work was supported by the National Institutes of Health grants R01 DK103794 (National Institute of Diabetes and Digestive and Kidney Diseases) and R21 HL120791 (National Heart, Lung, and Blood Institute) (to V.G.S.) and R01 DK 26263 (to N.M.).
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Baklouti F, Morinière M, Haj-Khélil A, Fénéant-Thibault M, Gruffat H, Couté Y, Ninot A, Guitton C, Delaunay J. 2011. Homozygous deletion of EPB41 genuine AUG-containing exons results in mRNA splicing defects, NMD activation and protein 4.1R complete deficiency in hereditary elliptocytosis. Blood Cells Mol Dis 47: 158–165. [DOI] [PubMed] [Google Scholar]
- Birkenmeier CS, Barker JE. 2004. Hereditary haemolytic anaemias: unexpected sequelae of mutations in the genes for erythroid membrane skeletal proteins. J Pathol 204: 450–459. [DOI] [PubMed] [Google Scholar]
- Conboy JG, Chasis JA, Winardi R, Tchernia G, Kan YW, Mohandas N. 1993. An isoform-specific mutation in the protein 4.1 gene results in hereditary elliptocytosis and complete deficiency of protein 4.1 in erythrocytes but not in nonerythroid cells. J Clin Invest 91: 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Costa L, Galimand J, Fenneteau O, Mohandas N. 2013. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev 27: 167–178. [DOI] [PubMed] [Google Scholar]
- Dalla Venezia N, Gilsanz F, Alloisio N, Ducluzeau M-T Jr, Benz EJ, Delaunay J. 1992. Homozygous 4.1 (–) hereditary elliptocytosis associated with a point mutation in the downstream initiation codon of protein 4.1 gene. J Clin Invest 90: 1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay J. 2007. The molecular basis of hereditary red cell membrane disorders. Blood Rev 21: 1–20. [DOI] [PubMed] [Google Scholar]
- Delaunay J, Stewart G, Iolascon A. 1999. Hereditary dehydrated and overhydrated stomatocytosis: recent advances. Curr Opin Hematol 6: 110–114. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O’Donnell-Luria A, Ware J, Hill A, et al. 2015. Analysis of protein-coding genetic variation in 60,706 humans. BioRxiv 10.1101/030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PG. 2013. Abnormalities of the erythrocyte membrane. Pediatr Clin North Am 60: 1349–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi M, Cianci CD, Gallagher PG, Morrow JS. 2001. Spectrin oligomerization is cooperatively coupled to membrane assembly: a linkage targeted by many hereditary hemolytic anemias. Exp Mol Pathol 70: 215–230. [DOI] [PubMed] [Google Scholar]
- Ipsaro JJ, Mondragon A. 2010. Structural basis for spectrin recognition by ankyrin. Blood 115: 4093–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M, Musallam KM, Cappellini MD, Daar S, El-Beshlawy A, Belhoul K, Saned MS, Temraz S, Koussa S, Taher AT. 2011. Risk factors for pulmonary hypertension in patients with β thalassemia intermedia. Eur J Intern Med 22: 607–610. [DOI] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, et al. 2014. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312: 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate shirt read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, Boerwinkle E. 2015. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human non-synonymous and splice site SNVs. Hum Mutat 37: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon GJ, Jiang T, Van Wijk R, Wang W, Bodily PM, Xing J, Tian L, Robison RJ, Clement M, Lin Y, et al. 2011. Exome sequencing and unrelated findings in the context of complex disease research: ethical and clinical implications. Discov Med 12: 41–55. [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. 2010. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26: 2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid ME, Takakuwa Y, Conboy J, Tchernia G, Mohandas N. 1990. Glycophorin C content of human erythrocyte membrane is regulated by protein 4.1. Blood 75: 2229–2234. [PubMed] [Google Scholar]
- Sankaran VG, Gallagher PG. 2013. Applications of high-throughput DNA sequencing to benign hematology. Blood 122: 3575–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Weiss MJ. 2015. Anemia: progress in molecular mechanisms and therapies. Nat Med 21: 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GW, Amess JA, Eber SW, Kingswood C, Lane PA, Smith BD, Mentzer WC. 1996. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol 93: 303–310. [DOI] [PubMed] [Google Scholar]
- Tchernia G, Mohandas N, Shohet SB. 1981. Deficiency of skeletal membrane protein band 4.1 in homozygous hereditary elliptocytosis: implications for erythrocyte membrane stability. J Clin Invest 68: 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To-Figueras J, Ducamp S, Clayton J, Badenas C, Delaby C, Ged C, Lyoumi S, Gouya L, de Verneuil H, Beaumont C, et al. 2011. ALAS2 acts as a modifier gene in patients with congenital erythropoietic porphyria. Blood 118: 1443–1451. [DOI] [PubMed] [Google Scholar]
- van Zwieten R, van Oirschot BA, Veldthuis M, Dobbe JG, Streekstra GJ, van Solinge WW, Schutgens REG, van Wijk R. 2015. Partial pyruvate kinase deficiency aggravates the phenotypic expression of band 3 deficiency in a family with hereditary spherocytosis. Am J Hematol 90: E35–E39. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe SN, Wood WG. 2005. Advances in the understanding of the congenital dyserythropoietic anaemias. Br J Haematol 131: 431–446. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. 2013. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. 2014. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312: 1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]