

Abstract
Shugoshin 1 (SGO1) is required for accurate chromosome segregation during mitosis and meiosis; however, its other functions, especially at interphase, are not clearly understood. Here, we found that downregulation of SGO1 caused a synergistic phenotype in cells overexpressing MYCN. Downregulation of SGO1 impaired proliferation and induced DNA damage followed by a senescence-like phenotype only in MYCN-overexpressing neuroblastoma cells. In these cells, SGO1 knockdown induced DNA damage, even during interphase, and this effect was independent of cohesin. Furthermore, MYCN-promoted SGO1 transcription and SGO1 expression tended to be higher in MYCN- or MYC-overexpressing cancers. Together, these findings indicate that SGO1 plays a role in the DNA damage response in interphase. Therefore, we propose that SGO1 represents a potential molecular target for treatment of MYCN-amplified neuroblastoma.
Shugoshin 1 (SGO1) is required to ensure the accuracy of chromosome segregation during mitosis and meiosis1. During Meiosis I in fission or budding yeast, Sgo1 protects pericentromeric cohesin for cleavage from separase, a role assigned to Sgo2 in mammalian cells. During mitosis in vertebrate cells, Sgo1 protects pericentromeric cohesin from removal from the prophase pathway, thereby protecting the cell from untimely sister chromatid separation2. The human genome encodes two Shugoshin paralogs, SGO1 and SGO2, which function during chromosome biorientation by promoting recruitment of proteins involved in the correction of erroneous kinetochore–microtubule attachments, such as the chromosomal passenger complex (CPC) and mitotic centromere-associated kinesin (MCAK)3. SGO1 expression is regulated by YAP/TAZ/TEAD (i.e., the Hippo pathway) and AP-1 (activator protein-1, a dimer of JUN and FOS proteins)4. To date, no other regulators of SGO1 transcription have been reported. SGO1 also plays important roles in various cancers5,6,7,8,9; in particular, defects in SGO1 induce premature chromosome segregation, followed by chromosomal instability (CIN). The molecular mechanism underlying CIN involves dysfunction of the inner centromere–Shugoshin (ICS) network, which coordinates sister chromatid cohesion and kinetochore–microtubule attachment10. However, the role of SGO1 during interphase in cancer cells in general, and in neuroblastoma in particular, remains unclear.
The cohesin complex, which contains Structural maintenance of chromosome 1A (SMC1A), SMC3, RAD21, and Stromal antigen 2 (STAG2), forms a ring-like structure that holds sister chromatids together11. Cohesin is involved in DNA replication via interaction with minichromosome maintenance (MCM) proteins that stabilize chromatin loops and regulate the frequency of origin firing12. In human cells, cohesin is also involved in DNA repair: it is recruited by RAD50–MRE11 to DNA double strand break (DSB) sites after irradiation and facilitates homologous recombination (HR) by holding sister chromatids together13. Cohesin also plays other important roles. For example, in ES cells, cohesin, Mediator, and Nipbl regulate transcription by forming DNA loops that bring enhancers and promoters closer together14. Furthermore, cohesin mutations have been detected in various cancers, including colorectal cancer, glioblastoma, Ewing’s sarcoma, melanoma, and acute myeloid leukemia (AML). These mutations promote tumorigenesis by inducing genome instability due to defects in DNA replication and DNA damage repair, as well as chromosome mis-segregation11.
MYCN is a MYC family protein and neural tissue-specific transcription factor that contains a β-helix-loop-helix domain15. The MYC-binding DNA sequence motif, known as the E-box (CANNTG)16, is present in the promoters of many target genes, including some that encode DNA damage response (DDR) proteins17,18,19,20,21.
Although MYCN cannot transform cells on its own22,23, it is associated with the malignant phenotype of several human malignancies. MYCN is amplified in ~25% of cases of neuroblastoma, the most common extracranial solid tumor seen during childhood, and MYCN amplification correlates with poor prognosis. Because MYC or MYCN is required for fundamental cellular processes, MYC or MYCN inhibitors may cause undesirable side effects. Identifying the gene(s) which shows synthetic (dosage) lethal interactions24 with MYCN or MYC amplification may help the development of promising strategies for the treatment of MYCN- or MYC-driven cancers because inhibiting genes that show synthetic lethality with MYC or MYCN amplification would selectively kill cancer cells25,26,27,28,29,30,31,32,33,34,35,36.
We previously reported that the condensin subunit SMC2 is a target of MYCN, and that SMC2 downregulation causes a synergistic phenotype in conjunction with MYCN amplification or overexpression35. In that study, we showed that SMC2 regulates transcription of DDR genes in cooperation with MYCN.
Here, we demonstrate that MYCN overexpression/amplification and SGO1 knockdown synergistically inhibit cell proliferation. The growth defect in SGO1-knockdown/MYCN-overexpressing/amplified cells is the result of persistent DNA damage, which leads to a senescence-like phenotype. In MYCN-overexpressing neuroblastoma cells, SGO1 knockdown induced DNA damage even in interphase, and this phenotype was independent of cohesin. In addition, we found that SGO1 is a transcriptional target of MYCN, and that SGO1 expression correlates with MYCN or MYC expression in various cancers. These results suggest that SGO1 represents a potential molecular target for therapeutics against MYCN- or MYC-overexpressing cancers.
Results
SGO1 expression is elevated in MYCN- or MYC-overexpressing cancers and cell lines
In a previous study, we used microarray data (GEO accession: GSE43419) to identify genes induced during progression of neuroblastoma in MYCN-Tg mice35. Among these genes, we focused on Sgo1 (Fig. S1a). To confirm the microarray results, we performed quantitative RT-PCR on RNA from ganglia of wild-type (wt), hemizygous, and homozygous MYCN-Tg mice (Fig. 1a). As expected, Sgo1 mRNA levels in precancerous and tumor samples were high. Next, we measured SGO1 expression in neuroblastoma samples from patients (GSE19274) using the R2 bioinformatics platform (http://r2.amc.nl). Consistent with the expression pattern in MYCN-Tg mice, SGO1 expression was elevated in human MYCN-amplified tumors (Fig. S1b). Likewise, SGO1 mRNA and protein levels were higher in MYCN-amplified neuroblastoma cell lines (IMR32, SK-N-BE, and NB39) than in a neuroblastoma cell line harboring a single copy of MYCN (SK-N-AS) (Fig. 1b). Based on these findings, we investigated the relationship between MYCN or MYC and SGO1/SGO2 expression levels using The Cancer Genome Atlas (TCGA) pan-cancer gene expression datasets (Fig. S1c). Expression of MYCN correlated with that of SGO1/SGO2, except in lower-grade gliomas, and SGO1/SGO2 expression tended to be higher in some MYC-overexpressing cancers. Together, these results showed that SGO1 is elevated in MYCN- or MYC-overexpressing cancers, including neuroblastoma cell lines showing MYCN amplification.
Figure 1. Sgo1 expression increases with neuroblastoma progression, and SGO1 expression is elevated in MYCN-amplified neuroblastoma cell lines.

(a) Quantitative RT-PCR analyses of Sgo1 mRNA levels in precancerous lesions from four hemizygous MYCN-Tg mice (1.6 or 2 wks old), tumor lesions from three hemizygous MYCN-Tg mice (9 or 10 wks old) and three homozygous MYCN-Tg mice (6 or 6.1 wks old), and ganglia from three wild-type mice (1.6 or 2 wks old). (b) SGO1 mRNA (left upper : semi-quantitative, left bottom : quantitative qPCR analysis) and protein (right) levels in neuroblastoma cell lines.
SGO1 is a potential novel transcriptional target of MYCN
To determine whether MYCN regulates SGO1 mRNA levels, we measured changes in SGO1 mRNA levels using SH-EP cells harboring a single copy of MYCN. MYCN overexpression induced SGO1 upregulation at both the mRNA and protein levels (Fig. 2a). In addition, SGO1 protein levels fell when MYCN was downregulated in IMR32 cells (Fig. 2b). Since MYC family transcriptional factors bind E-boxes, we searched for the latter within the SGO1 genome sequence and found four (E-box1–4) in the 4 kb region upstream of the SGO1 start codon and one in a SGO1 intron (Fig. 2c). To determine whether MYCN binds to the E-box sequences upstream of SGO1, we next performed chromatin immunoprecipitation (ChIP) assays. ChIP analysis revealed that MYCN associated most strongly with E-box1 (P = 0.035), and tended to associate with E-box2 (P = 0.056). It also associated with E-box3, but much less strongly (Fig. 2d). The association between MYCN and E-box4 was minimal, and was essentially no stronger than its association with the genomic region located >20 kb upstream of the SGO1 start codon used as a negative control. These results indicated that SGO1 is a potential novel transcriptional target of MYCN through binding to E-box1 and E-box2.
Figure 2. SGO1 is a potential transcriptional target of MYCN.

(a) MYCN overexpression induced SGO1 mRNA (left) and protein (right) expression in SH-EP cells (MYCN single-copy neuroblastoma cell line). (b) SGO1 protein levels were reduced in MYCN-downregulated IMR32 cells, but the SMC3 protein level was unchanged in these cells. Samples were harvested 3 days after non-target or MYCN-targeted shRNA lentivirus infection. Quantification of proteins on Western blots using ImageJ software. (c) Positions of E-box sequences associated with SGO1. Black circles, E-boxes; white boxes, 5′- or 3′-UTRs; gray boxes, exons. (d) ChIP analysis revealed enrichment of MYCN in E-box1 and E-box2 upstream of SGO1. A sequence 20 kb upstream of the SGO1 gene was used as a negative control. Data show the percentage of target DNA precipitated with control IgG or MYCN antibody, and are expressed as the means ± SE of at least three independent experiments.
SGO1 downregulation impairs cell proliferation and causes MYCN-overexpressing neuroblastoma cells to accumulate at G2/M
Next, to determine whether SGO1 affects proliferation of neuroblastoma cells, we knocked down SGO1 in MYCN-overexpressing SH-EP cells using short hairpin RNA (shRNA). SGO1 knockdown severely inhibited the proliferation of MYCN-overexpressing SH-EP cells, but had a much smaller effect in control SH-EP cells (Fig. 3a, upper panel). We confirmed the knockdown efficiency of SGO1 in each cell line (Fig. 3a, bottom panel). We also examined the impact of SGO1 knockdown on several neuroblastoma cell lines and found that SGO1 knockdown severely inhibited the proliferation of MYC-amplified cell line (IMR32). The effect of SGO1 knockdown was much smaller in a cell line harboring a single copy of MYCN (SK-N-AS) (Fig. 3b). Next, we examined cell viability in the presence of caffeine to see whether it inhibited the DDR. Although viability was lower than that in the absence of caffeine, SGO1 knockdown still severely inhibited the proliferation of MYCN-overexpressing SH-EP cells; the effect on control SH-EP cells was much smaller (Fig. S2). These results indicated that overexpression/amplification of MYCN and downregulation of SGO1 exert a synergistic effect on cell proliferation.
Figure 3. SGO1 knockdown and MYCN overexpression induce G2/M accumulation.

(a) SGO1 knockdown inhibited cell proliferation only in MYCN-overexpressing SH-EP cells. The number of the cells was counted 6 days after non-target or SGO1-targeted shRNA lentivirus infection. Upper panel, relative viability; lower panel, SGO1 knockdown efficiency. Data are expressed as the mean ± SE of at least three independent experiments. (b) SGO1 knockdown inhibited cell proliferation only in MYCN-amplified neuroblastoma cell lines. Number of cells counted at 4, 5, 6, 7, or 8 days after non-target or SGO1-targeted shRNA lentivirus infection. Upper panel, relative viability; bottom panel, SGO1 knockdown efficiency. (c) Flow cytometry profiles of control and SGO1-knockdown cells at 48 hrs after non-target or SGO1-targeted shRNA lentivirus infection. Lower panel shows SGO1 knockdown efficiency.
We next performed flow cytometry analysis to further investigate the mechanisms underlying inhibition of cell proliferation in SGO1-knockdown cells. A larger fraction of the cell population accumulated in G2/M phase in SGO1-knockdown cells than in control cells transfected with a non-targeting shRNA (Fig. 3c, left panels and Table. S1). The effect of SGO1 knockdown was larger in MYCN-overexpressing cells than in CMV-Venus control cells (Fig. 3c, right panels). Together, these observations indicate that SGO1 is required for efficient progression through G2/M phase in MYCN-overexpressing neuroblastoma cells.
Downregulation of cohesin induces a growth defect in MYCN-overexpressing cells, but does not induce G2/M arrest
Next, we measured the expression levels of cohesin subunits in MYCN-Tg mice (GSE43419). With the exception of Stag2, expression of all cohesin subunits increased during neuroblastoma progression (Fig. S3a), as in the case of Sgo1 (Fig. S1a). To determine whether this expression pattern is also present in humans, we assessed the expression levels of cohesin subunits in neuroblastoma patients using R2. With the exception of SMC1A, expression of cohesin subunits was upregulated in MYCN-amplified tumors from patients (Fig. S3b). Thus, expression of cohesin subunits is elevated when MYCN levels are high.
To determine whether cohesin plays a role in the growth defect of SGO1-knockdown cells, we knocked down cohesin subunits and monitored the effect on proliferation. Like the SGO1 knockdown, cohesin subunit knockdowns impaired cell proliferation in MYCN-overexpressing cells (Fig. S4a,b), but had no inhibitory effect in control cells (Fig. S4c,d). In contrast to SGO1 knockdown, however, cohesin subunit knockdown did not significantly alter the cell-cycle profile, even in MYCN-overexpressing neuroblastoma cells (Fig. S5a,b). In addition, SGO1 knockdown had no effect on the protein levels of SMC3 in either MYCN-amplified/non-amplified or MYCN-overexpressing/control cells (Fig. S5c). Thus, knockdown of cohesin subunits and MYCN overexpression synergistically inhibit neuroblastoma cell growth, but this effect is not mediated by gross changes in the cell-cycle distribution of the population. Therefore, knockdown of cohesin subunits and SGO1 inhibits growth via different mechanisms; however, further studies are needed to examine this in detail. Based on these findings, we concluded that the phenotype of SGO1-knockdown MYCN-overexpressing cells was distinct from that of cohesin subunit knockdown on the background of MYCN overexpression.
Poly ADP-ribose polymerase (PARP) inhibitors show synergistic responses to cohesin knockdown37. To determine whether PARP inhibitors also have inhibitory effects, we examined the viability of SGO1-knockdown cells in the presence or absence of MYCN overexpression. Indeed, SGO1 knockdown and the PARP inhibitor synergistically inhibited proliferation (P = 0.007) (Fig. S6). We detected no significant effect of PARP inhibition on the growth of SGO1-knockdown MYCN-overexpressing cells (P = 0.36), possibly because the viability of these cells was already quite low.
SGO1 downregulation induces DNA damage in MYCN-overexpressing neuroblastoma cells
To determine whether DNA damage was responsible for the observed impairment in proliferation, we assessed the levels of DNA damage in SGO1-knockdown MYCN-amplified cells. SGO1 knockdown increased the frequency of γ-H2AX foci, which mark sites of DNA damage, in the MYCN-amplified cell lines IMR32 and NB39 (Fig. 4a,b). We identified shRNA-infected NB39 cells using a relevant fluorescence (mRFP) probe (Fig. S7). Approximately 30–50% of non-target shRNA-infected NB39 or IMR32 cells displayed γ-H2AX foci (Fig. 4b). The mechanism underlying induction of DNA damage in non-target lentivirus-infected MYCN-amplified cells is unknown. Despite this, the results suggest that knockdown of SGO1 induces additional DNA damage in MYCN-amplified cells (P = 0.018 and P = 0.002, respectively, in Fig. 4b). We next used U2OS cells expressing ECFP-Geminin and EYFP-53BP138 to determine the cell-cycle phase in which the DNA damage occurred. Geminin is a cell-cycle indicator: its levels are low in G1 phase, gradually increase as the cell cycle progresses, reach a maximum level at mitosis, and rapidly decrease during cytokinesis. Meanwhile, 53BP1 is an indicator of DNA damage that is excluded from irradiation-induced foci during mitosis39. As with the frequency of γ-H2AX foci, more 53BP1-positive cells were present in SGO1-knockdown MYCN-overexpressing cells than in SGO1-knockdown control cells (Fig. 4c). Nocodazole treatment led to a significant increase in the number of 53BP1-positive cells among the Geminin-positive SGO1-knockdown MYCN-overexpressing cell population (Fig. 4c,d). As noted above, Geminin and 53BP1 double-positive cells must be in interphase. These results indicated that DNA damage occurred even in interphase in some SGO1-knockdown MYCN-overexpressing cells.
Figure 4. SGO1 knockdown and MYCN overexpression induce DNA damage that is repaired primarily by NHEJ.

(a) Immunofluorescence of γ-H2AX and DAPI staining of IMR32 (MYCN-amplified) and SK-N-AS (MYCN-single copy) cells infected with non-targeting or SGO1-specific shRNA. Images were acquired 3 days after infection. (b) Percentage of γ-H2AX-positive cells in the cell lines shown in (a), as well as in NB39 (MYCN-amplified) and SH-EP (MYCN-single copy) neuroblastoma cells. Data represent means ± SE of three independent repeats. (c) DSB formation in SGO1-knockdown MYCN-overexpressing U2OS cells expressing EYFP-53BP1 and ECFP-Geminin 48 hrs after virus infection. Nocodazole (0.9 μg/ml) was added for 16 hrs to arrest cells in G2/M phase, and cells were counterstained with DAPI. (d) Percentage of 53BP1-positive cells relative to the total number of Geminin-positive cells in (c). Data are expressed as the mean ± SE of four independent experiments. (e) NHEJ efficiency in SGO1-knockdown MYCN-overexpressing H1299dA3-1 #1 cells and HR efficiency in SGO1-knockdown MYCN-overexpressing DR-U2OS cells. NU7026 (a DNA-PK inhibitor) was used as a negative control for the NHEJ assay. shBRCA1 and shBRCA2 were used as a negative control for the HR assay.
Why did DNA damage occur only in MYCN-overexpressing cells when SGO1 was knocked down? One possibility is that more DNA damage occurred in these cells; alternatively, DNA repair could have been more efficient in control cells. To answer this question, we performed non-homologous end joining (NHEJ)40 and HR assays41 in cell lines that express GFP when the corresponding type of repair takes place. In the NHEJ assay, SGO1 knockdown tended to increase the proportion of GFP-positive cells in both MYCN-overexpressing and control cell populations (Fig. 4e, NHEJ assay). In the HR assay, the proportion of GFP-positive cells was much lower than in the NHEJ assay (Fig. 4e, HR assay), suggesting that DNA damage induced by SGO1 knockdown is mainly repaired by NHEJ. In addition, the number of GFP-positive cells in the HR assay was higher in control cells than in MYCN-overexpressing cells. Because HR is active primarily in G2 phase, the GFP-positive cells in the HR assay might reflect cells that were damaged in interphase.
Taken together, these data demonstrate that SGO1 knockdown induced DNA damage even in interphase, and that most of the damage was repaired primarily by NHEJ. In MYCN-overexpressing cells, however, persistent SGO1 knockdown-induced damage caused growth arrest. Although the underlying mechanism remains unknown, one possibility is that SGO1 knockdown induces greater DNA damage, which exceeds the capacity for repair.
Downregulation of SGO1 induces a senescence-like phenotype in MYCN-overexpressing neuroblastoma cells
We next investigated how SGO1-knockdown MYCN-overexpressing cells halt their proliferation. For this purpose, we first performed a TdT-mediated dUTP nick-end labeling (TUNEL) assay to detect apoptosis in MYCN-amplified IMR32 cells. However, TUNEL-positive cells were barely detectable (Fig. 5a). Therefore, we assayed for senescence-associated β-galactosidase (SA-β-GAL), a widely used biomarker of senescence in mammalian cells. Similar to the pattern of γ-H2AX foci (Fig. 4a), the proportion of SA-β-GAL positive cells was greater in IMR32 cells than in the control cell line SK-N-AS (Fig. 5b,c). In addition, expression of p16 and p21 was upregulated in SGO1-knockdown IMR32 cells, but less so in SGO1-knockdown SK-N-AS cells (Fig. 5d). These results demonstrate that SGO1-knockdown MYCN-overexpressing cells stop proliferation by entering senescence-like phenotype rather than undergoing apoptosis.
Figure 5. SGO1 knockdown together with MYCN overexpression induces senescence-like phenotype, but not apoptosis.

(a) TUNEL staining of control or SGO1-knockdown IMR32 cells 6 days after lentivirus infection. (b) MYCN-overexpressing or control SH-EP cells infected with lentiviral vectors expressing shRNA against SGO1 were assayed for SA-β-gal activity 6 days after infection. (c) The percentage of SA-β-gal-positive cells shown in (b). Values represent the mean ± SE of three fields. (d) Relative expression of p16 and p21 in the cells shown in (b). Values are expressed as the mean ± SE.
Discussion
Here, we showed that simultaneous downregulation of SGO1 and upregulation of MYCN induces higher levels of DNA damage, which in turn halts cell proliferation and causes cells to express a senescence-like phenotype (Fig. 6). MYCN induced DNA damage in MYCN-amplified cells, most likely via the production of ROS and replicative stress. On the other hand, since SGO1 is also overexpressed in these cells, the DDR (probably HR) may also be highly activated. As a consequence, cells are viable. In this situation, knockdown of SGO1 impairs the DDR response, resulting in a senescence-like phenotype (Fig. 6). It is still unclear whether this synergistic effect is due to an increased rate of damage or a reduced rate of repair.
Figure 6. Schematic model of the synergistic phenotype of SGO1 knockdown and MYCN overexpression.
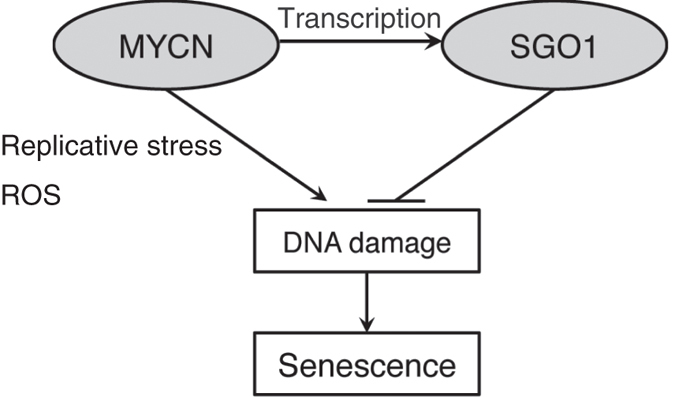
The phenotype of SGO1 knockdown varied depending on cell type, potentially due to differences in the expression level of MYC or MYCN. In this regard, SGO1 knockdown alone induces cellular senescence in human fibroblasts42, possibly because growing fibroblasts express a high level of MYC43. Subsequently, SGO1 knockdown halts the proliferation of HCT116, NB-4, EOL-1, and U937 cell lines5,9, all of which express MYC. Together, these observations support the notion of a strong genetic interaction between MYC and SGO1.
SGO1 knockdown induced DNA damage in MYCN-overexpressing neuroblastoma cells (Fig. 4a,b), resulting in G2/M accumulation (Fig. 3b). This damage was repaired by NHEJ to a greater extent than by HR (Fig. 4d). The higher level of DNA damage in SGO1-knockdown cells suggests that SGO1 positively regulates the HR pathway. If HR were fully functional in SGO1-knockdown cells, the number of GFP-positive cells detected in the HR assay would be comparable to that observed in the NHEJ assay. Together, these observations suggest that DNA damage induced by SGO1 downregulation is not fully repaired in G2/M phase by HR, resulting in accumulation at G2/M.
In SGO1-knockdown MYCN-overexpressing cells, some DNA damage occurred in interphase (Fig. 4c). Several mechanisms might be responsible for induction of DSBs in interphase. First, as demonstrated previously in HeLa cells, SGO1 protects cohesin on chromosomal arms during interphase via the HP1-SGO1 pathway44. When SGO1 is knocked down, cohesin is released from both the chromosomal arm and pericentromeric regions even in interphase, potentially resulting in DNA damage. Second, SGO1 is involved in the DNA repair system, and as noted above may positively regulate HR. Moreover, SGO1 interacts with Ku7045, a subunit of DNA-PK holoenzyme, which is essential for recognition of DSBs in NHEJ. The Ku complex (Ku70–Ku80), the DNA damage sensor for NHEJ, also functions as a DNA damage sensor for HR in the absence of the Mre11-Rad50-Nbs1 (MRN) complex, the usual damage sensor for HR46, suggesting that the Ku complex plays a role in DNA repair beyond NHEJ. Third, SGO1 may regulate the expression of DDR proteins; if so, SGO1 knockdown would decrease the abundance of DDR factors and compromise the efficiency of repair, thereby increasing the frequency of DSBs. Consistent with this hypothesis, SGO1 interacts with condensin in budding yeast47, and SMC2, a subunit of condensin, regulates transcription of DDR genes in cooperation with MYCN35.
Finally, we showed that, in addition to being a target of YAP/TAZ/TEAD (the Hippo pathway) and AP-1, SGO1 is also a target of MYCN (Fig. 2). Like many other genes involved in replication and cell-cycle control, MYC itself is also a target of YAP/TAZ4, suggesting that both MYCN or MYC and SGO1 can amplify signals from the Hippo pathway. Therefore, we propose that SGO1 is a potential molecular target for the treatment of MYCN-amplified neuroblastoma and tumors bearing YAP/TAZ hyperactivation.
Methods
Mice
MYCN Tg mice48 and wild type mice were maintained in the animal facility at Nagoya University Graduate School of Medicine, where they were housed in a controlled environment and provided with standard nourishment and water. Normal ganglia and precancerous and tumor tissues from wild type, hemizygous, and homozygous MYCN Tg mice were dissected and minced before total RNA was extracted. All methods were performed in accordance with the relevant guidelines and regulationswas approved by the Animal Care and Use Committee of Nagoya University Graduate School of Medicine, Nagoya, Japan.
Gene expression array data
Previously published data35 were used for the expression analyses. These data were deposited in the Gene Expression Omnibus database of NCBI (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE43419.
Cell lines and lentiviruses
Cell lines used in this study are listed in Supplementary Table S2. Cells were cultured as previously described35,40,38,41. For the knockdown experiments, replication-defective and self-inactivating lentiviruses were prepared as previously described35. We used a 2nd generation packaging system based on psPAX2 and pMD2.G vectors.
Plasmids and primers
Commercially available non-targeting (Sigma) and SGO1- or cohesin subunit-specific shRNA vectors (Thermo Scientific Open Biosystems) were used. The shRNA sequences are listed in Table S3. The primers used in this study are listed in Supplementary Table S4.
Isolation of RNA, RT-PCR, and qPCR
Total RNA was isolated from cells using the RNeasy Plus Mini kit (Qiagen) or the ISOGEN II reagent (Nippongene). cDNA was prepared using ReverTra Ace qPCR RT kit with gDNA Remover (TOYOBO). Quantitative PCR analyses were performed using the KAPA SYBR Fast qPCR kit (KAPA Biosystems) on an Applied Biosystems 7500 Real-Time PCR System. Relative gene expression levels were determined using the ΔΔCT method.
Protein preparation and immunoblotting
Whole-cell lysates were prepared using Cell Lytic M solution (C2978, Sigma) containing a protease inhibitor cocktail (11697498001, Roche) and subjected to SDS-PAGE. Immunoblotting was performed as previously described35. Western blots were analyzed using ImageJ software.
Antibodies
The following antibodies were used: anti-SGO1 mouse monoclonal (ab58023, Abcam); anti-SGO1 rabbit polyclonal (GTX117103, GeneTex, and ab21633, Abcam); anti-MYCN mouse monoclonals OP13 (Calbiochem), NB200-109 (Novus Biologicals), and sc-53993 (Santa Cruz Biotechnology); anti-MYCN rabbit polyclonal (#9405, Cell Signaling Technology); anti-phospho-histone H2A.X (Ser139) mouse monoclonal (JBW301, Millipore); anti-SMC3 rabbit polyclonal (A300-060A, Bethyl laboratories); anti-β-ACTIN (8H10D10) mouse monoclonal (#3700, Cell Signaling Technology); normal mouse IgG (#12–371, Millipore); goat anti-mouse IgG (H+L) secondary antibody, Alexa Fluor® 488 conjugate (A-11001, Thermo Fisher Scientific); anti-mouse IgG, HRP-linked (#7076S, Cell Signaling Technology); and anti-rabbit IgG, HRP-linked (#7074S, Cell Signaling Technology) antibodies.
ChIP
Cultured sub-confluent SH-EP cells expressing Venus or MYCN were treated with 1% (v/v) formaldehyde for 10 min at room temperature. Cross-linking was stopped by addition of glycine to a final concentration of 125 mM. The cells were washed with cold PBS and then harvested. The cells were pelleted, frozen at −70 °C, and then lysed by mechanical disruption. The Halo-ChIP system (Promega) was then used according to the manufacturer’s instructions, with a slight modification: an anti-MYCN antibody and Dynabeads-protein G (Dynal) were used. DNA was purified twice using phenol-CIAA and then PCR-amplified using primers spanning the MYCN-binding site upstream of the SGO1 gene. Cells immunoprecipitated with control IgGs were used as a negative control.
Cell viability
MYCN-overexpressing or control Venus-expressing SH-EP cells were infected with non-target or shSGO1 lentivirus. 4, 5, 6, 7, or 8 days later, the cells were counted using Cell Counting Kit-8, according to the manufacturer’s protocol (Dojindo Laboratories).
FACS
DNA content was determined on a FACSCalibur instrument (BD Bioscience), and data were analyzed using Cell Quest software.
Immunofluorescence analysis
Cells were grown on glass coverslips in 4 well plates and then fixed with 4% paraformaldehyde for 1 hr at room temperature. Fixed cells were permeabilized with PBS containing 1% NP-40 for 10 min at room temperature and incubated with an anti-γ-H2AX antibody (1:1000 dilution) for 1 hr at room temperature, followed by an Alexa Fluor 488-conjugated anti-mouse IgG (Molecular Probes) (1:1000 dilution) for 30 min at room temperature. After incubation with 4′6-diamidino-2-phenylindole (0.1 μg/ml) for 5 min, cells were mounted in FluorSave Reagent (Millipore). All images were obtained using BZ-9000 (Keyence) or LSM710 (Carl-Zeiss) and subsequently processed using MetaMorph or Zen software and Adobe Photoshop.
NHEJ assay
The NHEJ assay was performed as described40. Briefly, H1299dA3-1#1 cells40 were cultured in 24 well plates, infected with shRNAs, and transfected with the I-SceI expression plasmid (pCBASce). After 72 hrs, cells were harvested and single cell suspensions were analyzed in a FACS Aria III cytometer (Becton Dickinson). NU7026 (35 μM; a DNA-PK inhibitor) treatment was used as a negative control.
HR assay
The HR assay was performed as previously described41. Briefly, DR-U2OS cells41 were cultured in 24 well plates and infected with shRNAs and transfected with I-SceI expression plasmid (pCBASce). After 72 hrs, cells were harvested and single cell suspensions were analyzed using a FACS Aria III cytometer (Becton Dickinson).
Image analysis
Cells were infected with lentivirus to introduce shSGO1 and induce MYCN overexpression. After 24 hrs, nocodazole (0.9 μg/ml) was added to arrest cells at G2/M. After another 24 hrs, cells were counterstained with DAPI. Cells were then examined under an LSM710 microscope (Carl-Zeiss), and images were analyzed using Image Zen software (Zeiss).
TUNEL assay
The TUNEL assay was performed using the APO-DIRECT kit (BD Pharmingen), and the cells were analyzed on a FACSCanto flow cytometer (BD Pharmingen) using the Diva software.
β-galactosidase assay
SA-β-GAL assays were performed using the Senescence Detection kit (K320-250, Biovision).
Statistical analysis
Results are expressed as the mean ± SE. Homoscedasticities were checked using the f test. Statistical significance was evaluated with a 2-tailed, unpaired t test and significance set at P < 0.05.
Additional Information
How to cite this article: Murakami-Tonami, Y. et al. SGO1 is involved in the DNA damage response in MYCN-amplified neuroblastoma cells. Sci. Rep. 6, 31615; doi: 10.1038/srep31615 (2016).
Supplementary Material
Acknowledgments
We thank Y. Watanabe (The University of Tokyo) for critical reading of the manuscript. We also thank H. Suzuki (The Jikei University School of Medicine) for anti-SGO1 antibody; D. Trono (Swiss Federal Institute of Technology, Switzerland) for providing the lentiviral packaging and production plasmids psPAX2 and pMD2.G; H. Miyoshi (Keio University) and A. Miyawaki (RIKEN Brain Science Institute, Japan) for providing the CSII-CMV-RfA-IRES2-Venus, CSII-CMV-Venus and CSII-CMV-mRFP vectors; the Cell Bank of RIKEN BioResource Center (Japan), the Health Science Research Resource Bank (Japan), JCRB (Japan), ATCC, M. Schwab (German Cancer Research Center [DKFZ]), and H. Chiba (Fukushima Medical University) for neuroblastoma cells; T. Kohno (National Cancer Center) for H1299dA3-1 cells and I-SceI plasmid ; M. Jasin (Memorial Sloan Kettering Cancer Center) for DR-U2OS cells and plasmids; G. Lahav (Harvard Medical School) for U2OS cells expressing EYFP-53BP1 and ECFP-Geminin; F. Ohka and A. Hatanaka (Nagoya City University) for technical support with the TCGA data analysis; and T. Nawashiro (Nagoya University) and Y. Suyama (Kinjo Gakuin University) for technical assistance. We are grateful to M. Foiani (IFOM) for communicating unpublished results, and to all members of our laboratory for support and helpful discussion. This work was supported by JSPS KAKENHI Grant Numbers 15K08325 and 25290053 (to Y.M-T.), the research fund of Aichi Cancer Research Foundation (to Y.M-T.), a Grant-in-Aid from the National Cancer Center Research and Development Fund (22–4) (to K.K.), Health and Labour Sciences Research Expenses for Commission, Applied Research for Innovative Treatment of Cancer (H26-applied-general-006) from the Ministry of Health, Labour and Welfare (to K.K.), and the Tailor-made Medical Treatment Program (no. 15km0305013h0101) from the Japan Agency for Medical Research and development (to K.K.).
Footnotes
Author Contributions Y.M.-T., H.M. and K.K. conceived the project. Y.M.-T. performed most of the experimental procedures and analyzed the data. H.I. assisted with flow cytometry analysis. R.Y. assisted with Western blotting. M.I. assisted with cell-cycle profile analysis. S.I. assisted with PARP inhibitor experiments. J.K. helped the R2 data analysis. I.T. performed the statistical analysis. S.K. and K.K. provided the MYCN-Tg mice. Y.K. assisted with the TCGA data analysis. Y.M.-T., H.M., T.M., Y.S. and K.K. analyzed the data. Y.M.-T. and H.M. wrote the manuscript. K.K. supervised the project.
References
- Watanabe Y. & Kitajima T. S. Shugoshin protects cohesin complexes at centromeres. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 360, 515–521, discussion 521, doi: 10.1098/rstb.2004.1607 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima T. S. et al. Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 441, 46–52, doi: 10.1038/nature04663 (2006). [DOI] [PubMed] [Google Scholar]
- Tanno Y. et al. Phosphorylation of mammalian Sgo2 by Aurora B recruits PP2A and MCAK to centromeres. Genes & development 24, 2169–2179, doi: 10.1101/gad.1945310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nature cell biology 17, 1218–1227, doi: 10.1038/ncb3216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaizumi M. et al. Human Sgo1 downregulation leads to chromosomal instability in colorectal cancer. Gut 58, 249–260, doi: 10.1136/gut.2008.149468 (2009). [DOI] [PubMed] [Google Scholar]
- Liu L. et al. Lentivirus-mediated siRNA interference targeting SGO-1 inhibits human NSCLC cell growth. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine 33, 515–521, doi: 10.1007/s13277-011-0284-0 (2012). [DOI] [PubMed] [Google Scholar]
- Yamada H. Y. et al. Haploinsufficiency of SGO1 results in deregulated centrosome dynamics, enhanced chromosomal instability and colon tumorigenesis. Cell cycle 11, 479–488, doi: 10.4161/cc.11.3.18994 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S. et al. SGOL1 variant B induces abnormal mitosis and resistance to taxane in non-small cell lung cancers. Scientific reports 3, 3012, doi: 10.1038/srep03012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Ikezoe T., Nishioka C. & Yokoyama A. A novel treatment strategy targeting shugoshin 1 in hematological malignancies. Leukemia research 37, 76–82, doi: 10.1016/j.leukres.2012.10.002 (2013). [DOI] [PubMed] [Google Scholar]
- Tanno Y. et al. The inner centromere-shugoshin network prevents chromosomal instability. Science 349, 1237–1240, doi: 10.1126/science.aaa2655 (2015). [DOI] [PubMed] [Google Scholar]
- Losada A. Cohesin in cancer: chromosome segregation and beyond. Nature reviews. Cancer 14, 389–393, doi: 10.1038/nrc3743 (2014). [DOI] [PubMed] [Google Scholar]
- Guillou E. et al. Cohesin organizes chromatin loops at DNA replication factories. Genes & development 24, 2812–2822, doi: 10.1101/gad.608210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. S., Krasieva T. B., LaMorte V., Taylor A. M. & Yokomori K. Specific recruitment of human cohesin to laser-induced DNA damage. The Journal of biological chemistry 277, 45149–45153, doi: 10.1074/jbc.M209123200 (2002). [DOI] [PubMed] [Google Scholar]
- Kagey M. H. et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435, doi: 10.1038/nature09380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress T. R., Sabo A. & Amati B. MYC: connecting selective transcriptional control to global RNA production. Nature reviews. Cancer 15, 593–607, doi: 10.1038/nrc3984 (2015). [DOI] [PubMed] [Google Scholar]
- Meyer N. & Penn L. Z. Reflecting on 25 years with MYC. Nature reviews. Cancer 8, 976–990, doi: 10.1038/nrc2231 (2008). [DOI] [PubMed] [Google Scholar]
- Menssen A. & Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proceedings of the National Academy of Sciences of the United States of America 99, 6274–6279, doi: 10.1073/pnas.082005599 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P. C. et al. Genomic targets of the human c-Myc protein. Genes & development 17, 1115–1129, doi: 10.1101/gad.1067003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. et al. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proceedings of the National Academy of Sciences of the United States of America 100, 8164–8169, doi: 10.1073/pnas.1332764100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang Y. C., Teng S. C., Su Y. N., Hsieh F. J. & Wu K. J. c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. The Journal of biological chemistry 278, 19286–19291, doi: 10.1074/jbc.M212043200 (2003). [DOI] [PubMed] [Google Scholar]
- Perini G., Diolaiti D., Porro A. & Della Valle G. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proceedings of the National Academy of Sciences of the United States of America 102, 12117–12122, doi: 10.1073/pnas.0409097102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K., Era T., Takebe A., Jakt L. M. & Nishikawa S. ARID3B induces malignant transformation of mouse embryonic fibroblasts and is strongly associated with malignant neuroblastoma. Cancer research 66, 8331–8336, doi: 10.1158/0008-5472.CAN-06-0756 (2006). [DOI] [PubMed] [Google Scholar]
- Huang M. & Weiss W. A. Neuroblastoma and MYCN. Cold Spring Harbor perspectives in medicine 3, a014415, doi: 10.1101/cshperspect.a014415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLornan D. P., List A. & Mufti G. J. Applying synthetic lethality for the selective targeting of cancer. The New England journal of medicine 371, 1725–1735, doi: 10.1056/NEJMra1407390 (2014). [DOI] [PubMed] [Google Scholar]
- Cole K. A. et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America 108, 3336–3341, doi: 10.1073/pnas.1012351108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund A., Stromvall K., Li Y., Forshell L. P. & Nilsson J. A. Chk2 deficiency in Myc overexpressing lymphoma cells elicits a synergistic lethal response in combination with PARP inhibition. Cell cycle 10, 3598–3607, doi: 10.4161/cc.10.20.17887 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima M. et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proceedings of the National Academy of Sciences of the United States of America 109, 9545–9550, doi: 10.1073/pnas.1121119109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M. et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nature structural & molecular biology 18, 1331–1335, doi: 10.1038/nsmb.2189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar J. J. et al. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proceedings of the National Academy of Sciences of the United States of America 106, 12968–12973, doi: 10.1073/pnas.0901418106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T. et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer cell 15, 67–78, doi: 10.1016/j.ccr.2008.12.005 (2009). [DOI] [PubMed] [Google Scholar]
- Beierle E. A. et al. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. The Journal of biological chemistry 282, 12503–12516, doi: 10.1074/jbc.M701450200 (2007). [DOI] [PubMed] [Google Scholar]
- Rottmann S., Wang Y., Nasoff M., Deveraux Q. L. & Quon K. C. A TRAIL receptor-dependent synthetic lethal relationship between MYC activation and GSK3beta/FBW7 loss of function. Proceedings of the National Academy of Sciences of the United States of America 102, 15195–15200, doi: 10.1073/pnas.0505114102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler J. D. et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353, doi: 10.1126/science.1212728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer cell 5, 501–512 (2004). [DOI] [PubMed] [Google Scholar]
- Murakami-Tonami Y. et al. Inactivation of SMC2 shows a synergistic lethal response in MYCN-amplified neuroblastoma cells. Cell cycle 13, 1115–1131, doi: 10.4161/cc.27983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu T. Y. et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388, doi: 10.1038/nature14985 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil N. J., van Pel D. M. & Hieter P. Synthetic lethality and cancer: cohesin and PARP at the replication fork. Trends in genetics : TIG 29, 290–297, doi: 10.1016/j.tig.2012.12.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanam K., Kafri R., Loewer A. & Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Molecular cell 47, 320–329, doi: 10.1016/j.molcel.2012.05.052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S., Belotserkovskaya R. & Jackson S. P. DNA damage signaling in response to double-strand breaks during mitosis. The Journal of cell biology 190, 197–207, doi: 10.1083/jcb.200911156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara H. et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 30, 2135–2146, doi: 10.1038/onc.2010.592; (2011). [DOI] [PubMed] [Google Scholar]
- Xia B. et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular cell 22, 719–729, doi: 10.1016/j.molcel.2006.05.022 (2006). [DOI] [PubMed] [Google Scholar]
- Chetaille P. et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nature genetics 46, 1245–1249, doi: 10.1038/ng.3113 (2014). [DOI] [PubMed] [Google Scholar]
- Waters C. M., Littlewood T. D., Hancock D. C., Moore J. P. & Evan G. I. c-myc protein expression in untransformed fibroblasts. Oncogene 6, 797–805 (1991). [PubMed] [Google Scholar]
- Chu L. et al. The spatiotemporal dynamics of chromatin protein HP1alpha is essential for accurate chromosome segregation during cell division. The Journal of biological chemistry 289, 26249–26262, doi: 10.1074/jbc.M114.581504 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C. G. et al. Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature 441, 53–61, doi: 10.1038/nature04664 (2006). [DOI] [PubMed] [Google Scholar]
- Hartlerode A. J., Morgan M. J., Wu Y., Buis J. & Ferguson D. O. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nature structural & molecular biology 22, 736–743, doi: 10.1038/nsmb.3072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijlbergen K. F. et al. Shugoshin biases chromosomes for biorientation through condensin recruitment to the pericentromere. eLife 3, e01374, doi: 10.7554/eLife.01374 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss W. A., Aldape K., Mohapatra G., Feuerstein B. G. & Bishop J. M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. The EMBO journal 16, 2985–2995, doi: 10.1093/emboj/16.11.2985 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.