

ABSTRACT
Searching for new regulators of autophagy involved in selective dopaminergic (DA) neuron loss is a hallmark in the pathogenesis of Parkinson disease (PD). We here report that an endoplasmic reticulum (ER)-associated transmembrane protein SLC35D3 is selectively expressed in subsets of midbrain DA neurons in about 10% TH (tyrosine hydroxylase)-positive neurons in the substantia nigra pars compacta (SNc) and in about 22% TH-positive neurons in the ventral tegmental area (VTA). Loss of SLC35D3 in ros (roswell mutant) mice showed a reduction of 11.9% DA neurons in the SNc and 15.5% DA neuron loss in the VTA with impaired autophagy. We determined that SLC35D3 enhanced the formation of the BECN1-ATG14-PIK3C3 complex to induce autophagy. These results suggest that SLC35D3 is a new regulator of tissue-specific autophagy and plays an important role in the increased autophagic activity required for the survival of subsets of DA neurons.
KEYWORDS: autophagy, BECN1-ATG14-PIK3C3 complex, dopaminergic neuron, neurodegeneration, Parkinson disease, SLC35D3
Introduction
Macroautophagy (or autophagy) is a crucial process for cellular homeostasis. Many studies have demonstrated that basal autophagy is essential for neuronal survival.1-3 Neurodegenerative diseases, such as Alzheimer disease, Parkinson disease (PD) and schizophrenia, are associated with impaired autophagy.3,4 Several PD genes are known to function in regulating autophagy or mitophagy.5 Dopaminergic (DA) neurons are often overloaded by protein aggregates, damaged mitochondria, or reactive oxygen species, which requires higher autophagic activities for their clearance to maintain cellular homeostasis or cell survival. Treatment with rapamycin is able to prevent the loss of TH (tyrosine hydroxylase)-positive neurons in the 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP) mouse model of PD likely via activation of the autophagic pathway.6 It is likely that genes specifically expressed in DA neurons to regulate autophagy could be involved in protecting DA neuron loss.
A variety of molecules involved in regulating the autophagic process have been identified.7 Currently, in mammalian cells, studies are focusing on the elucidation of the roles of newly-defined autophagic regulators in several crucial steps of autophagic flux such as autophagy initiation, as well as autophagosome formation, and fusion with lysosomes, in basal or induced autophagy.8 Autophagy is triggered by many intrinsic and extrinsic cellular factors. During autophagy initiation, inhibition of MTOR (mechanistic target of rapamycin [serine/threonine kinase]) is required for starvation-induced autophagy.9 Both AMP-activated protein kinase (AMPK) and MTOR regulate autophagy through opposing phosphorylation of ULK1 (unc51-like autophagy activating kinase 1),10,11 which is a core component of the ULK complex containing ULK1, ULK2, ATG13 (autophagy related 13), RB1CC1 (RB1 inducible coiled-coil 1) and ATG101.12 Another critical initiation complex is the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which contains core components, namely BECN1 (Beclin 1, autophagy related), ATG14, PIK3C3/VPS34 and PIK3R4/VPS15.7,13-16 The PtdIns3K complex is required for the generation of PtdIns3P for the formation of the phagophore. The scaffold protein AMBRA1 (autophagy/Beclin 1 regulator 1) links ULK1 and BECN1.17 ULK1 induces autophagy through phosphorylating BECN1 and activating PIK3C3 activity.18 The PtdIns3P effectors, ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1) and WIPI1 (WD repeat domain, phosphoinositide interacting 1)-WIPI2, are recruited to the phagophore to promote membrane nucleation and thereafter expansion during autophagosome formation.19
Previous studies have characterized a recessive null mutation of the Slc35d3 (solute carrier 35 member D3) gene in the ros (roswell mutant) mouse, which carries a spontaneous intracisternal A particle insertion at the first exon of the Slc35d3 gene to disrupt its function.20 SLC35D3 is predicted to be an orphan nucleotide sugar transporter or a fringe connection-like protein with 10 transmembrane domains. It has been reported that SLC35D3 is involved in the biogenesis of platelet dense granules.20,21 Moreover, Slc35d3 is specifically expressed in the substantia nigra (SN), striatum, and olfactory bulb in mouse brain.22 It functions as an ER chaperone for DRD1 (dopamine receptor D1) trafficking to the plasma membrane of the medium spiny neurons in the striatum. Loss of SLC35D3 reduces the number of DRD1 receptors on the plasma membrane and causes imbalance in the activity controlling loop in the basal ganglia, which leads to the development of abnormal locomotor activity, obesity and metabolic syndrome in mice and humans.22 In this study, we explored the function of SLC35D3 in the midbrain. We found that SLC35D3 is differentially expressed in subsets of DA neurons in the SNc and VTA, which could confer protection from neurodegeneration by regulating autophagy.
Results
SLC35D3 is selectively expressed in subsets of DA neurons in the SNc and VTA and the ros mutant exhibits DA neuron loss
We have previously determined that SLC35D3 is selectively expressed in the SN and VTA, striatum, and olfactory bulb by immunoblotting.22 To investigate its function in the SN and VTA, we explored its expression pattern in this region. Using human diphtheria toxin receptor-green fluorescent protein (DTR-GFP) transgenic mice (3 mo old), driven by the SLC35D3 promoter,23 we found that SLC35D3 was highly expressed in projections of DRD1-expressing medium spiny neurons enriched in the substantia nigra pars reticulata (SNr) (Fig. 1A), which is in agreement with a previous report.24 In addition, we detected GFP signals in cell bodies of neurons colabeled with TH in the SNc and VTA (Fig. 1B). We then quantified the percentage of SLC35D3+ cells in the SNc and VTA. In the SNc, an average of 9.46% TH+ cells were SLC35D3+, and an average of 83.12% SLC35D3+ cells were TH+. By contrast, in the VTA, an average of 22.06% TH+ cells were SLC35D3+, and an average of 74.06% SLC35D3+ cells were TH+ (Fig. 1C). In the SNc, we observed that SLC35D3+ cells were almost exclusively located in the median SNc compared to the lateral part (Fig. 1B and D). There was little difference in the antero-posterior repartition of the SLC35D3+ cells. These results suggest that SLC35D3 is expressed in a subset of DA neurons in the VTA or in the SNc.
Figure 1.
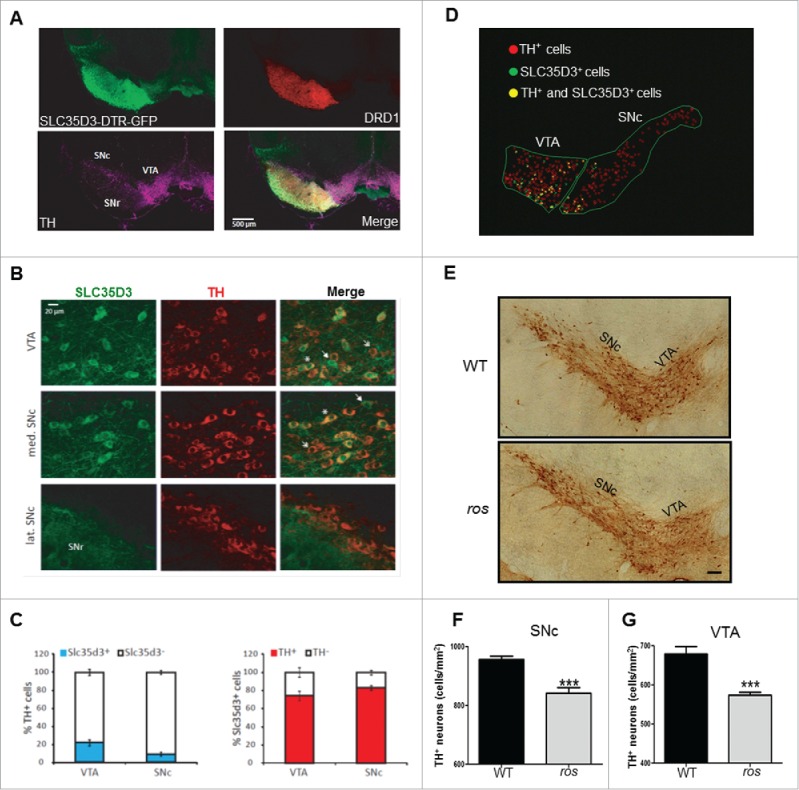
SLC35D3 is differentially expressed in subsets of DA neurons in the midbrain and the ros mice show reduced TH+ neurons. (A) In our DTR-GFP transgenic mice (3 month old, 2 males), the Slc35d3-promoter driven GFP signals were present in the projections of striatal DRD1-positive neurons in the substantia nigra pars reticutala (SNr), and in the cell bodies of the TH-positive neurons located in the substantia nigra pars compacta (SNc) and in the ventral tegmental area (VTA). (B) Colabeling of SLC35D3-GFP (green) and TH (red) in the cell bodies of the neurons located in the VTA, medial SNc and lateral SNc. Note that only a portion of TH+ neurons express SLC35D3 (asterisk), some neurons express SLC35D3 but not TH (arrow), some neurons express TH but not SLC35D3. (C) In the SNc, 9.46 ± 2.08 % TH+ cells was SLC35D3+, and 83.12 ± 2.21 % SLC35D3+ cells was TH+. By contrast, in the VTA, 22.06 ± 3.35 % TH+ cells was SLC35D3+, and 74.06 ± 5.10 % SLC35D3+ cells was TH+. The quantifications were performed on 2 mice and the number of cells was counted on 40-µm thick section (n=8 sections per mouse, both hemispheres). (D) Schematic distribution pattern of different population neurons in the midbrain (right hemisphere) from the images in (B). Note that in the SNc, SLC35D3+ cells were almost exclusively located in the median SNc compared to the lateral part. (E) Immunohistochemical detection of TH on the coronal brain sections of the midbrain at 6 mo of age in both wild-type (WT) and ros mice. Scale bar: 100 µm. (F) Ros mice showed a 11.9 % reduction in cell density of TH+ neurons in the SNc. WT: 956 ± 11 cells/mm2; ros: 842 ± 19 cells/mm2, n = 5; ***, P < 0.001. (G) Ros mice showed a 15.5% reduction in cell density of TH+ neurons in the VTA. WT: 679 ± 19 cells/mm2, ros, 574 ± 8 cells/mm2, n = 5; ***, P < 0.001.
We then investigated whether the number of TH+ cells changes in the ros mutant mice with loss of SLC35D3 protein.22 We observed an average of 11.9 % reduction of TH+ cells in the SNc, and an average of 15.5% reduction of TH+ cells in the VTA (Fig. 1E to G) in 6-mo-old ros mice. This fraction of cell loss matches well with the percentage of SLC35D3+ cells in the SNc or VTA. In addition, we observed an average of 11.1 % reduction of TH+ cells in the SNc in 3-mo-old ros mice (WT: 738 ± 16 cells/mm2; ros: 664 ± 18 cells/mm2. P = 0.004), suggesting no drastic increase of neuron loss in older mice.
Impaired autophagy in ros midbrain
Autophagy dysfunction is well known to cause neuronal cell loss. SLC35D3 is localized to the ER membrane,22 which likely functions in autophagic origin. We therefore tested whether autophagy is altered in these dopaminergic neurons. By immunoblotting, we found a significant decrease of the lipidated form of the autophagic marker MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β), LC3B-II, in the SN and VTA in ros mice compared with wild-type mice (Fig. 2A and B). In contrast, LC3B-II levels in 2 other SLC35D3-expressing brain subregions, the striatum and olfactory bulb, and in the non-SLC35D3-expressing cerebellum were not significantly changed (Fig. S1), suggesting the alteration of LC3B-II is restricted to the SN and VTA region, implicating a tissue-specific impairment of autophagy. The lower autophagic activity resulted in the increase of SQSTM1/p62 (sequestosome 1), a receptor and substrate of autophagy, in the SN and VTA in ros mice (Fig. 2C and D). Furthermore, we observed reduced MAP1LC3 or LC3A/B/C (LC3 in short) puncta in the cell bodies of TH+ neurons in both the SNc and VTA of the ros mice (Fig. 2E to G).
Figure 2.
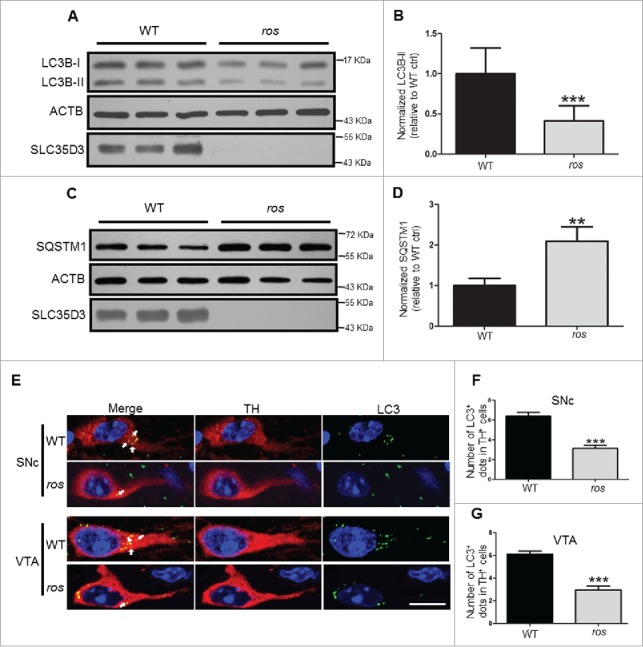
Reduced LC3B-II levels in ros midbrain. (A, B) Steady-state level of LC3B-II in the midbrain containing the SN and VTA was lower in ros mice compared with wild type (WT). ACTB/β-actin was used as a loading control. Bars represent mean ± SEM, WT: n = 12, ros: n=11; ***, P < 0.001. (C, D) Steady-state level of insoluble SQSTM1 in the SN and VTA was increased in ros mice compared with WT mice. ACTB was used as a loading control. Bars represent mean ± SEM, n = 3; **, P < 0.01. (E) Representative images of LC3 puncta (green) stained with anti-LC3 antibody in TH-labeled (red) neurons. Arrows show the LC3 -positive dots. Scale bar: 10 μm. (F) The number of autophagosomes in TH+ neurons from the SNc was reduced in ros mice. 225 neurons were counted from 5 WT mice, 238 neurons were counted from 5 ros mice. WT, 6.40 ± 0.39 dots/TH+ neuron; ros, 3.15 ± 0.29 dots/TH+ neuron; ***, P < 0.001. (G) The number of autophagosomes in TH+ neurons from the VTA was reduced in ros mice. 239 neurons were counted from 5 WT mice, 261 neurons were counted from 5 ros mice. WT, 6.10 ± 0.27 dots/TH+ neuron; ros, 2.95 ± 0.34 dots/TH+ neuron; ***, P < 0.001.
To further explore whether dopaminergic neuron loss is specific, we did immunostaining assays of GAD1/GAD67 (glutamate decarboxylase 1), a marker of γ-aminobutyric acid (GABA) neurons, and GFAP (glial fibrillary acidic protein), a marker of glial cells, respectively. We showed less GAD1+ cells and less GFAP+ cells in the SNc and VTA compared to the SNr region (Fig. S2A and S3A). We then quantified these cell numbers in the SNc or VTA in both WT and ros mutant. There was no significant change in the GAD1+ cells (Fig. S2B and C) or GFAP+ cells between these two groups (Fig. S3B and C). Furthermore, we colabeled the GAD1+ cells with LC3 and quantified the cytosolic LC3 dots (Fig. S4A). There was no significant change in the number of LC3 dots in the GAD1+ cells in the SNc and VTA from the ros mice compared to the WT mice (Fig. S4B). As GFAP is an intermediate filament protein of astrocytes (Fig. S4C), it is difficult to verify the cytosolic LC3 dots in the GFAP labeled glial cells, precluding us to quantify the LC3 dots directly in the slices. However, we confirmed that there was no expression of SLC35D3 in the cultured mouse primary astrocytes both from WT and ros mice, as well as a human glioma cell line, U-87 MG (Fig. S4D). In addition, there was no significant difference of expressed GAD1 or GFAP in the SNc and VTA between ros and WT mice (Fig. S4E and F), suggesting that it is unlikely to affect the GABA+ cells or glial cells in the midbrain due to the loss of SLC35D3. Taken together, our results suggest that the altered autophagy is likely restricted to the DA neurons in the SNc and VTA region, and that SLC35D3 plays a role in tissue- and cell-type specific (i.e. DA neurons in the midbrain) autophagy.
To further confirm that SLC35D3 is involved in autophagy, we transfected human embryonic kidney (HEK) 293T cells with Flag-tagged vectors. We have previously reported that SLC35D3 is not expressed in HEK293T cells.22 Expression of Flag-SLC35D3 in these cells increased the cytosolic GFP-LC3 puncta (Fig. 3A and B) and the protein level of LC3B-II (Fig. 3E and F), but decreased the level of SQSTM1 (Fig. 3G and H). Similarly, transfection of Flag-SLC35D3 in the SH-SY5Y (a dopaminergic-like cell line) neuroblastoma cells increased the cytosolic LC3 puncta (Fig. 3C and D). These results suggest that autophagic activity is reduced in the midbrain DA neurons and SLC35D3 plays an important role in regulating autophagy.
Figure 3.
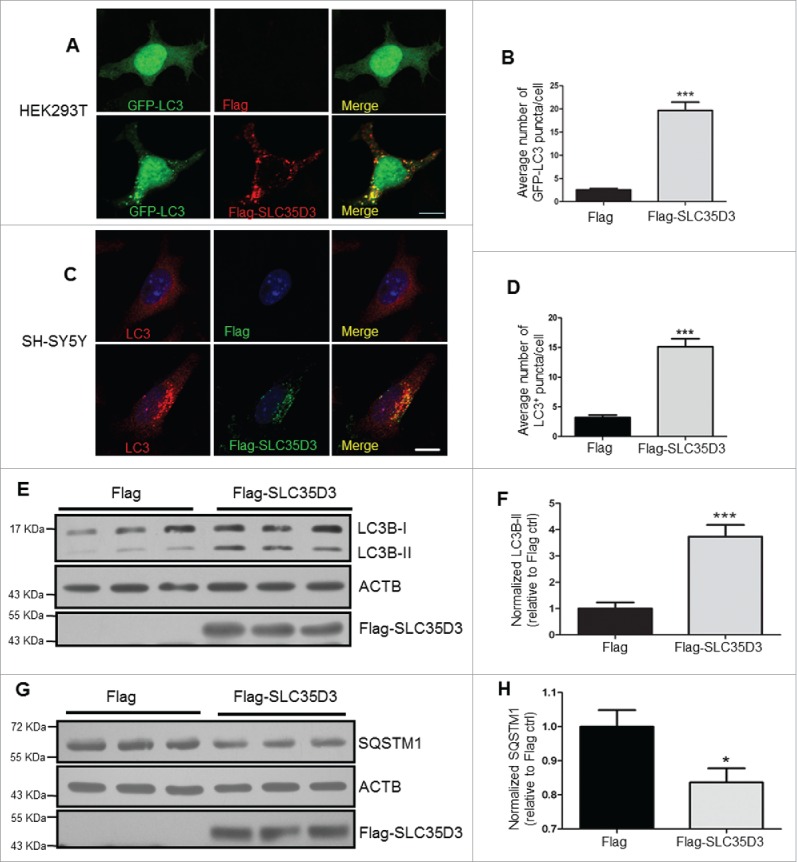
Expression of SLC35D3 increases autophagic activity. (A, B) In HEK293T cells, cotransfection of GFP-LC3 and Flag-SLC35D3 increased LC3 puncta. Scale bar: 10 µm; for statistics, at least 50 cells were counted; ***; P < 0.001. (C, D) In SH-SY5Y cells, transfection of Flag-SLC35D3 increased LC3 puncta. Scale bar: 10 µm; for statistics, at least 50 cells were counted; ***, P < 0.001. (E, F) Steady-state level of LC3B-II in the SLC35D3-transfected cells was increased compared to the control cells. ACTB was used as a loading control. Bars represent mean ± SEM, n = 11; ***, P < 0.001. (G, H) Steady-state level of insoluble SQSTM1 in the SLC35D3-transfected cells was decreased compared to the control cells. ACTB was used as loading control. Bars represent mean ± SEM, n = 5; *, P < 0.05.
SLC35D3 functions in autophagy induction
To determine how SLC35D3 acts on the autophagic flux, we treated the cells with bafilomycin A1 (BAF1) and rapamycin. We did not find an increase of LC3B-II when treated with rapamycin, an inducer of autophagic flux (Fig. 4A and B). However, the LC3B-II level was enhanced when treated with BAF1 (Fig. 4C and D), an inhibitor of late-stage autophagic flux, suggesting that SLC35D3 functions in the early stage of autophagic flux. ZFYVE1 is involved in the initiation of autophagy and serves as a marker for omegasomes.25 We found that the number of ZFYVE1-positive puncta was increased when cotransfected with SLC35D3 (Fig. 4E and F). Furthermore, we observed that SLC35D3 was partially colocalized with the autophgic origin marker ZFYVE1, and the fraction of colocalized GFP-LC3 and SLC35D3 was higher than the fraction of colocalized ZFYVE1 and SLC35D3 (Fig. 5A), suggesting that the increase of LC3 puncta is not dependent on the transfection of ZFYVE1. In addition, ATG14 is a key component of the autophagy initiation complex (AIC). We found the interaction between SLC35D3 and ATG14 by coimmunoprecipitation (coIP) assays (Fig. 5B). Our gradient fractionation assays further confirmed that SLC35D3 coexisted with the ER marker SEC61B (Sec61 translocon β subunit), but not with the lysosomal marker LAMP1 (lysosomal associated membrane protein 1). SLC35D3 coexisted partially with ATG14, which had a broader distribution spanning the lysosome marker LAMP1 (Fig. 5C), supporting its involvement in the autolysosome formation.26 These results suggest that SLC35D3 functions in autophagy induction.
Figure 4.

SLC35D3 functions in the early stage of autophagic flux. (A, B) Immunoblotting of LC3B-II in the HEK293T cells transfected with Flag or Flag-SLC35D3 plasmid and treated with DMSO or rapamycin (100 nM) for 4 h. ACTB was used as a loading control. There were no significant changes (N.S., P > 0.05) of LC3B-II after rapamycin treatment. Bars represent mean ± SEM, n = 6. (C, D) Immunoblotting of LC3B-II in the HEK293T cells transfected with Flag or Flag-SLC35D3 plasmid and treated with DMSO or BAF1 (400 nM) for 4 h. ACTB was used as a loading control. The increase of LC3B-II by SLC35D3 expression was further enhanced after BAF1 treatment. Bars represent mean ± SEM, n = 6. **, P < 0.01. (E, F) Expression of SLC35D3 leads to an increase of omegasomes in the HeLa cell. Representative images of GFP-ZFYVE1 puncta in HeLa cells cotransfected with GFP-ZFYVE1 and Flag or GFP-ZFYVE1 and Flag-SLC35D3 plasmids. Scale bar, 10 μm. Quantitative analysis of GFP-ZFYVE1 puncta in the transfected cells showed an increase of GFP-ZFYVE1 dots when cotransfected with Flag-SLC35D3 (55 cells were counted; ***, P < 0.001).
Figure 5.
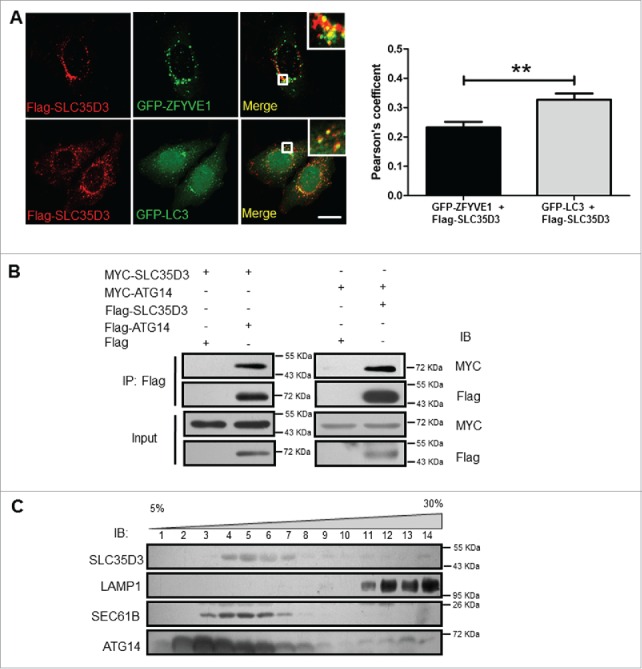
SLC35D3 is localized to the autophagy initiation site. (A) Partial colocalization of SLC35D3 with ZFYVE1 or GFP-LC3. HeLa cells were cotransfected with GFP-ZFYVE1/Flag-SLC35D3 or GFP-LC3/Flag-SLC35D3. Cells were fixed and permeabilized after 18 h. SLC35D3 (red) partially colocalizes with the ZFYVE1 or LC3 (green). Insets showed the enlarged white rectangle area. Scale bar: 10 µm. Quantification of colocalization represented by the Pearson correlation coefficient showed that Flag-SLC35D3 colocalized more with GFP-LC3 than with GFP-ZFYVE1. **, P < 0.01, n = 50 cells. (B) Interactions between SLC35D3 and ATG14. CoIP assays showed that Flag-ATG14 coprecipitated with MYC-SLC35D3 (left panel), and Flag-SLC35D3 coprecipitated with MYC-ATG14 (right panel). The Flag-empty vector was used as a negative control. (C) OptiPrep (5–30% [w/v]) gradient assay showed that SLC35D3 coexisted with the ER marker SEC61B, but segregated with the lysosome marker LAMP1. ATG14 had a broader distribution overlapping with SLC35D3 and the lysosomal fractions. Fractions 1 and 14 correspond to the top and bottom ends of the gradient, respectively. IB, immunoblotting; IP, immunoprecipitation.
SLC35D3 interacts with the BECN1-ATG14-PIK3C3 complex to promote autophagy
To further explore how SLC35D3 functions in autophagy induction, we tested whether SLC35D3 interacts with other components of the AIC complex. We found that SLC35D3 interacted with BECN1 and PIK3C3 by coIP assays (Fig. 6A and B). We confirmed that SLC35D3 interacted with ATG14, BECN1 and PIK3C3 by the Flag affinity isolation or endogenous affinity isolation assays (Fig. 6C and D). In contrast, due to the loss of SLC35D3 in ros brain extracts, the endogenous affinity isolation assays by using the anti-SLC35D3 antibody did not pull down the AIC components compared with the WT mice (Fig. 6E). We further verified the interaction between SLC35D3 and the AIC complex by using the strep affinity isolation assay (Fig. S5A and B). Our sedimentation assay showed that SLC35D3 did not coreside in the same fractions with PIK3C3 or BECN1 (Fig. S5C), suggesting that SLC35D3 is not a core component, but a regulatory component, of the AIC complex.
Figure 6.
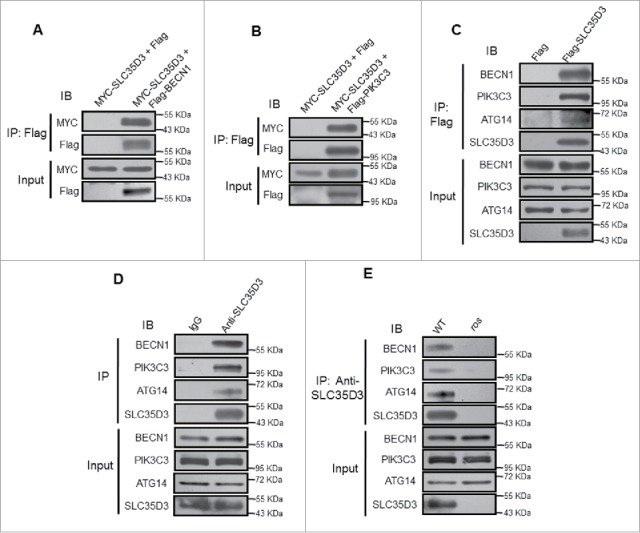
SLC35D3 interacts with the BECN1-ATG14-PIK3C3 complex. (A) Coimmunoprecipitation (coIP) assays showed that Flag-BECN1 coprecipitated with MYC-SLC35D3. The Flag-empty vector was used as a negative control. (B) coIP assays showed that Flag-PIK3C3 coprecipitated with MYC-SLC35D3. The Flag-empty vector was used as a negative control. (C) Affinity isolation assays showed that Flag-SLC35D3 pulled down BECN1, PIK3C3 and ATG14. The Flag-empty vector was used as a negative control. (D) Endogenous IP assays of brain lysates showed that SLC35D3 precipitated BECN1, PIK3C3 and ATG14. IgG was used as a negative control. (E) Endogenous IP assays of brain lysates showed that SLC35D3 precipitated BECN1, PIK3C3 and ATG14 in wild-type brain but not in ros brain. All the above IP assays were repeated independently at least 3 times. IB, immunoblotting; IP, immunoprecipitation.
To understand why expression of SLC35D3 increased autophagy in the transfected cells, we tested whether SLC35D3 enhances the AIC formation to increase autophagic activity. Indeed, the expression of SLC35D3 in cultured cells enhanced BECN1-PIK3C3 and BECN1-ATG14 interactions (Fig. 7A). Likewise, ATG14-BECN1 and ATG14-PIK3C3 interactions were increased when SLC35D3 was expressed (Fig. 7B). Inversely, the BECN1-ATG14 and BECN1-PIK3C3 interactions were reduced in the SN and VTA of ros mice compared to the wild-type mice (Fig. 7C). This suggests that loss of SLC35D3 in the midbrain ros mice reduces autophagic activity and that the SN and VTA neurons require higher autophagic activity for their survival under physiological conditions.
Figure 7.
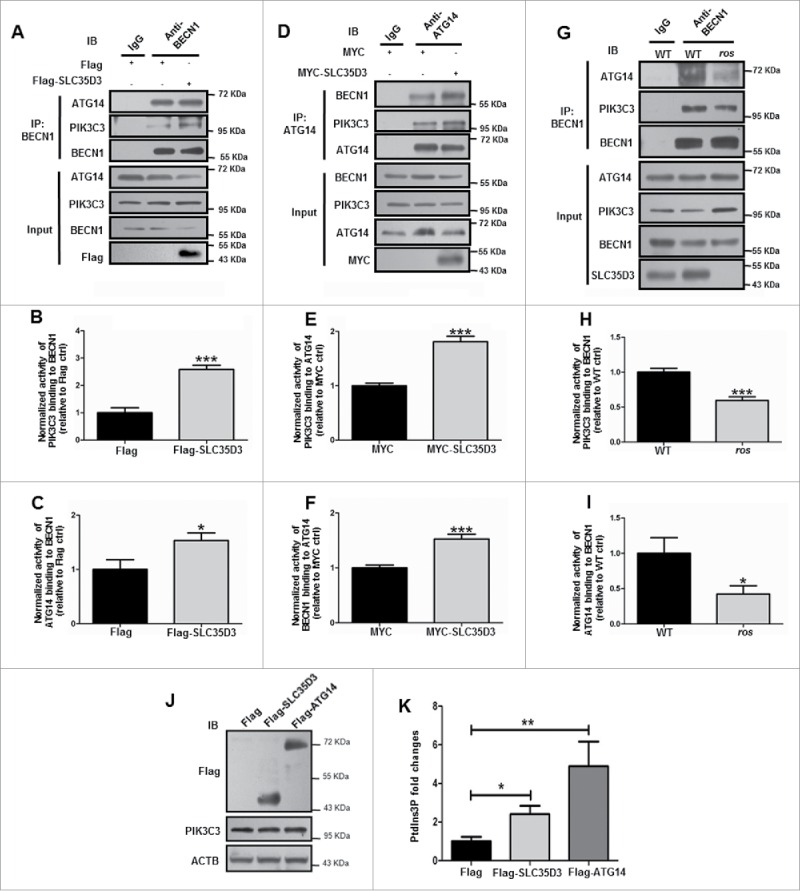
Expression of SLC35D3 enhances the BECN1-ATG14-PIK3C3 complex formation. (A) Expression of Flag-SLC35D3 enhanced BECN1-PIK3C3 and BECN1-ATG14 interactions in HEK293T cells. (B, C) The quantification data of 3 independent experiments in (A) are shown. Bars represent mean ± SEM, n = 3. *, P < 0.05; ***, P < 0.001. (D) Expression of MYC-SLC35D3 increased ATG14-BECN1 and ATG14-PIK3C3 interactions in HKE293T cells. (E, F) The quantification data of 3 independent experiments in (D) are shown. Bars represent mean ± SEM, n = 3. ***, P < 0.001. (G) Reduced BECN1-PIK3C3 and BECN1-ATG14 interactions in the SN and VTA of 6-mo-old ros mice. (H, I) The quantification data of 3 independent experiments in (G) are shown. Bars represent mean ± SEM, n = 3. **, P < 0.01; ***, P < 0.001. (J) PIK3C3 activity was detected by analyzing PtdIns3P production using ELISA assay as described in Materials and Methods. The expression of indicated proteins was verified by immunoblotting. (K) The PtdIns3P fold changes were measured based on the concentration of PtdIns3P and normalized to the Flag control group. Both the expression of Flag-SLC35D3 and Flag-ATG14 enhanced the PIK3C3 activities. Bars represent mean ± SEM, n = 9; *, P < 0.05; **, P < 0.01. IP: immunoprecipitation.
Overexpression of ATG14 led to the increased PIK3C3 activities.27 Likewise, we found that the expression of SLC35D3 resulted in increased PIK3C3 activities (Fig. 7D). Furthermore, we did not find apparent changes of the phospho (p)-RPS6KB1 (ribosomal protein S6 kinase B1), p-EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) and p-AMPK in the SLC35D3-expressing cultured cells (Fig. S6), suggesting that the enhanced PIK3C3 activity is not dependent on the MTOR or AMPK activity. Taken together, our results suggest that SLC35D3 promotes autophagy by enhancing BECN1-ATG14-PIK3C3 complex formation in the DA neurons during autophagy initiation.
Discussion
Autophagy is prone to be activated to protect cells from dying, and especially is crucial for the survival of post-mitotic neurons. The selective neuron loss in PD is a long-standing mystery. In this study, we have provided evidence to support the notion that genes selectively expressed in DA neurons may confer the resistance to neuronal loss. SLC35D3 is expressed in subsets of DA neurons and plays an important role in autophagy induction by enhancing the interaction with the autophagy initiation complex, BECN1-ATG14-PIK3C3. Under physiological conditions, SLC35D3 increases autophagic activity, which is required for the protection of neurons from cell death. Thus, the SLC35D3-expressing DA neurons which have higher autophagic activities are likely viable under physiological conditions or when subject to PD-risk factors or other stressors. When SLC35D3 is null, the SLC35D3-expressing DA neurons are susceptible to cell death.
We have shown that SLC35D3 is involved in autophagy induction through the interaction with the BECN1-ATG14-PIK3C3 complex, which is independent of the MTOR and AMPK pathway. Enhancing the interactions of the BECN1-ATG14-PIK3C3 components will increase the PtdIns3K activity during phagophore nucleation.18,27 Unlike other inducers, we found that this SLC35D3 mediated regulatory mechanism functions specifically in the DA neurons as we did not find changes of LC3B-II levels in the striatum and olfactory bulb, the other 2 subregions with SLC35D3 expression (Fig. S1). In addition, there is no apparent change in the number of SLC35D3-expressing D1 neurons,22 suggesting that the striatal D1 neurons are not affected by this autophagic pathway. Likewise, we did not find changes in the GAD1+ GABA interneurons or in the GFAP+ glial cells in the SNc and VTA regions, suggesting that the impairment of autophagy is tissue-specific and cell-autonomous. Similarly, our recent work in other mouse mutants indicates the tissue-specific impairment of autophagy in hilar mossy cells or Purkinje cells.3,28
We have shown that SLC35D3 is not a core component of the BECN1-ATG14-PIK3C3 complex, suggesting that it is not an essential component for autophagy induction. This is evident from the fact that autophagy induction can be triggered in SLC35D3 nonexpressing cells. Therefore, the expression of SLC35D3 increases autophagic activity, which is required for the survival of specific cells such as DA neurons. Our results suggest that increasing autophagic activity in DA neurons could be a therapeutic design in the treatment of PD patients to prevent progressive neuronal loss.
Our previous results have shown that SLC35D3 is localized to the ER and acts as a chaperone for DRD1 in the striatal neurons. Loss of SLC35D3 in the ros mice results in lowered locomotor activities and less energy expenditure.22 The lesions of DA neurons in the SNc and VTA may likewise contribute to this motor dysfunction. Thus, it is not exclusive that the DA neuron loss contributes to the development of the features in obesity and metabolic syndrome of the ros mice and human subjects.22 Interestingly, an overweight phenotype is more prevalent in PD patients, and body mass index is a risk factor for PD.29,30 Our results suggest that obese patients with SLC35D3 mutations could experience motor dysfunction due to the partial loss of midbrain DA neurons.
Materials and methods
Mice
The ros (roswell) mutant (slc35d3−/-)20 and control C3H/HeSnJ mice (wild-type, WT) were originally obtained from Dr. Richard T. Swank's laboratory and bred in the animal facility of the Institute of Genetics and Developmental Biology (IGDB), Chinese Academy of Sciences. All procedures were approved by the Institutional Animal Care and Use Committee of IGDB. To confirm the genotypes of slc35d3−/- and wild-type mice, we developed a PCR method of genotyping based on the nature of the insertional mutation in the Slc35d3 gene.20 The human diphtheria toxin receptor-green fluorescent protein (DTR-GFP) transgenic mice were generated as previously described.23 All the animal procedures were approved by Institut de Biologie du Développement de Marseille (France).
SN/VTA tissue preparation and TH-positive neuron counting
The fresh whole mouse brain was isolated and put into an adult mouse brain coronal matrix. Midbrain region was separated from the whole brain immediately. Then the SN and VTA sub-brain regions were dissected under a mouse brain matrix (Ted Pella, USA) according to the adult mouse brain map. The SN and VTA brain tissues were used for blotting immediately.
The number of TH+ neurons, GFAP+ cells, and GAD1+ neurons were counted in both sides of SNc or VTA from every sixth 20-µm-thick transverse sections throughout the entire SNc and VTA regions for each mouse. Each section was viewed under lower magnification (4×) and outlined. The number of TH+, GFAP+, and GAD1+ cells were counted at higher magnification (10×). The areas of SNc or VTA sampled for counting were measured using the Image-Pro+ 6.0 (Media Cybernetics, MD, USA). Total TH+, GFAP+, and GAD1+ cells within the area of SNc or VTA were counted. Then the number of TH+, GFAP+, and GAD1+ cells neurons per mm2 were calculated.
Antibodies, constructs and reagents
The polyclonal rabbit anti-mouse SLC35D3 antiserum for immunoblotting (IB) was prepared using the purified C-terminal (322 to 422 aa) peptide as an antigen as described previously.22 Monoclonal mouse anti-Flag (Sigma-Aldrich, F3165) antibody and polyclonal (pAb) rabbit anti-MYC/c-MYC (Sigma-Aldrich, C3956) antibody were used for IB (1:5000) and immunocytochemical (ICC) analysis (1:1000), respectively. Monoclonal mouse anti-TH for immunohistochemistry (IHC) (1:1000) was purchased from Sigma-Aldrich (T1299). Rabbit anti-TH monoclonal antibody was used for immunofluorescence (IF) (1:1000; AB152, Chemicon). Rabbit anti-LC3B pAb (Sigma-Aldrich, L7543) was used for IB (1:5000). Anti-LC3 pAb which recognizes LC3A/B/C (MBL, PM036) was used for ICC (1:1000). Anti-BECN1 pAb (MBL, PD017) was used for IB (1:6000) and immunoprecipitation (IP) (1:100). BECN1 antibody (Novus Biologicals, NBP100085) was used for IP (1:100). Anti-ATG14 antibody (MBL, M184-3) was used for IB (1:2000) and IP (1:100). ATG14 antibody (Cell Signaling Technology [CST], 5504) was used for IB (1:1000). Anti-SQSTM1/p62 antibody (MBL, PM045) was used for IB (1:2000). Rabbit anti-PIK3C3 antibody (Invitrogen, 38–2100) was used for IB (1:1000). SEC61B (Abcam, ab78276) and LAMP1 (BD Biosciences, CD107) antibodys were used for IB (1:5000 and 1:1000, respectively). p-RPS6KB1 (CST, Thr389, 9205), RPS6KB1 (CST, 2708), p-EIF4EBP1 (CST, Thr37/46, 2855) and EIF4EBP1 (CST, 9452) antibodies were used for IB (1:2000). p-AMPK (CST, Ser485, 2537), AMPK (CST, 2532) antibodies were used for IB (1:1000). Mouse monoclonal anti-ACTB/β-actin (Sigma-Aldrich, A5441) was used for IB (1:20000). Mouse anti-GFAP monoclonal antibody was used for IF and IB (1:1000, IF03L, Millipore). Mouse anti-GAD1 monoclonal antibody was used for IF (1:100, MAB5406, Millipore) and IB (1:1000). Alexa Fluor 594-conjugated donkey anti-mouse (R37115) and donkey anti-rabbit IgG (H+L) (A-21209), and Alexa Fluor 488-conjugated donkey anti-mouse (R37118) and goat anti-rabbit IgG (H+L) (A-11006) were purchased from Molecular Probes (Invitrogen).
The entire coding region of mouse Slc35d3 (NCBI RefSeq NM_029529) was subcloned into the pCMV-tag2B vector (Stratagene) with a Flag-tag and the pEGFP-C2 vector with an EGFP-tag and the pCMV-tag3B vector with a MYC-tag. The Flag-BECN1 and Flag-PIK3C3 vectors were gifts from Dr. Yeguang Chen (Tsinghua University). ATG14 was subcloned into the pCMV-tag3B vector with a MYC-tag. BAF1 was from Fermentek Ltd (CSA 88899–55–2). Rapamycin was obtained from Sigma-Aldrich (R0395).
Cell culture and transfection
HEK293T, HeLa, U-87 MG or SH-SY5Y cells were grown in DMEM medium from HyClone (SH30022.01B) supplemented with 10% fetal bovine serum (GIBCO, 10099–141) and 1% penicillin and streptomycin at 37°C under 5% CO2. Transfected HEK293T, HeLa, U-87 MG or SH-SY5Y cells grew to confluency on 24-well plates. Cells were harvested 48 h after transfection and lysed in a RIPA buffer (Pierce, 89900) and protease inhibitors (Sigma-Aldrich, P8340). Cell lysates were centrifuged at 12,000 × g for 15 min, and the supernatant fraction was collected for immunoblotting.
Primary astrocytes were isolated from neonatal mouse brain by the following procedures. After removing the meninges from the brain, the whole brain except cerebellum was transferred into a Petri dish. After cutting the brain tissue into pieces, 2 mL 0.25% trypsin was added for incubating 7 min at 37°C. The reaction was stopped by adding 5 mL DMEM/F12 medium. The cell suspension was then centrifuged at 150 × g for 5 min (Cence, L500, Changsha). The mixed glial cell cultures were plated with 10 mL of culture media and flasks were incubated in a 5% CO2 incubator at 37°C for 10 to 12 d. After the mixed glial cultures were confluent, flasks were placed in a shaker (THZ-032, Shengneng Bocai, Shanghai) at 180 rpm for 5 h at 37°C. The upper layer of media (containing high purity of microglia cells) was removed without disrupting the lower astrocyte layer adherent on the flask surface. Cultured astrocytes were fixed or collected for immunostaining or immunoblotting.
Immunoblotting
Cell lysates, immunoprecipitates or tissue lysates were combined with loading buffer and subjected to 8–15% SDS polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblotting procedures were described previously.22
Coimmunoprecipitation (coIP) and affinity isolation assays
Transfected HEK293T cells were harvested and lysed in cell lysis buffer with protease inhibitor cocktail (Sigma-Aldrich, P8340) for 1 h at 4°C. All the procedures for coIP and affinity isolation assays are described previously.31
Immunohistochemical and immunofluorescence staining
Mice were perfused through the heart with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) under deep pentobarbital anesthesia. The brains were removed, and 20-µm frozen sections in the coronal plane were prepared for immunohistochemical staining (IHC). The procedures are described previously.22 For immunofluorescence staining of brain sections, all procedures are described previously.3,32
Immunofluorescence confocal imaging
Cells transfected with the indicated plasmids were grown on glass coverslips in 24-well plates until 30 to 50% confluence; 18 to 20 h after transfection, they were fixed and examined with the procedures described previously.22
OptiPrep gradient analyses and sucrose sedimentation assay
OptiPrep gradient analyses and sucrose sedimentation assays were performed by following the procedures described previously.22,32 For OptiPrep gradient analyses, briefly, mouse SN and VTA were immediately homogenized with 1 mL HB lysis buffer (250 mM sucrose [Sigma-Aldrich, S9378], 20 mM Tris-HCl, pH 7.4, 1 mM EDTA) followed by centrifugation at 1000 × g for 15 min. Cytosolic extracts were placed onto the top of an 11 ml continuous 5% to 30% OptiPrep (Sigma-Aldrich, D1556) gradient in HB buffer. The gradient was centrifuged at 100,000 × g for 14 h in a MLA-55 rotor (Beckman Coulter). Fourteen fractions were collected and analyzed by immunoblotting.
For sucrose sedimentation assay, briefly, mouse SN and VTA were homogenized in a sample buffer containing 0.3 M Tris-HCl, pH 7.5, 1 mM EGTA, 1 mM dithiothreitol, 0.5 mM MgCl2 and protease inhibitor cocktail (Sigma-Aldrich, P8340) followed by centrifugation at 5000 × g for 5 min and at 12,000 × g for 90 min at 4°C to obtain a cytosolic extract. The supernatant (0.6 mg of total protein) was fractioned by ultracentrifugation on a Beckman MLA-55 rotor at 100,000 × g for 14 h on a 5–20% (w/v) sucrose gradient (12 mL). Nineteen fractions were collected and analyzed by immunoblotting.
PtdIns3K complex purification
HEK293 cells were cotransfected with the PtdIns3K complex coexpression plasmid (pMlink-ATG14-PIK3C3-PIK3R4-BECN1) and the plasmid expressing SLC35D3 with or without a strep-tag (pCAG-strep-SLC35D3 or pCAG-SLC35D3). Cells were harvested 72 h after transfection and incubated with the lysis buffer (100 mM Tris, pH 8.0, 150 mM NaCl, proteinase inhibitor cocktail, 2 mM DTT and 1% Triton X-100 [Sigma-Aldrich, T8787]). The lysates were then subjected to centrifugation at 10000 g for 15 min at 4°C. The supernatant were incubated with Strep-Tactin Sepharose (IBA GmbH, 2–1201–010) for 2 h at 4°C. The resin was washed 5 times with the wash buffer (100 mM Tris, pH 8.0, 300 mM NaCl, 2 mM DTT and 0.1% Triton X-100). Then the complexes were eluted with 2× SDS-PAGE sample buffer and detected by Coomassie brilliant blue staining and western blotting.
PIK3C3 kinase assay
PIK3C3 kinase assay was performed as described previously.13,27 HEK293T cells transfected with or without Flag-SLC35D3 or Flag-ATG14 plasmids were immunoprecipitated by ATG14 antibody to pull down the PtdIns3K complex and the immunoprecipitated fractions were subjected to PIK3C3 kinase activity assay. PIK3C3 activity was measured with the PtdIns3K ELISA Kit (Echelon, K-3000). Briefly, 20 μl kinase reaction buffer (10 mM Tris, pH 8, 100 mM NaCl, 1 mM EDTA, 10 mM MnCl2), 4 μl of 500 μM phosphatidylinositol substrate and 1 μl of 1.25 mM ATP were added into the beads immunoprecipitated by ATG14 antibody and incubated at 37°C for 1 h. Then the reaction mixture was quenched with 5 μl of 100 mM EDTA, diluted with 90 μl H2O and 80 μl PtdIns3P detection buffer. Finally the quenched reaction mixture and the PtdIns3P detector protein were added to the PtdIns3P-coated microplate for competitive binding to the PtdIns3P detector protein. The amount of PtdIns3P detector protein bound to the plate was determined through colorimetric detection of absorbance at 450 nm by the ELISA Reader machine (High Creation Technology CO. LTD, HR801, Shenzhen). The concentration of PtdIns3P in the reaction mixture was calculated as reversed to the amount of PtdIns3P detector protein bound to the plate.
Data analysis
All data were obtained from at least 3 independent experiments. Data were expressed as mean ± SEM and statistical significance was tested by the Student t test.
Supplementary Material
Abbreviations
- ACTB
actin, β
- AMPK
AMP-activated protein kinase
- AIC
autophagy initiation complex
- ATG
autophagy related
- BAF1
bafilomycin A1
- BECN1
Beclin 1, autophagy related
- coIP
coimmunoprecipitation
- DA neuron
dopaminergic neuron
- ZFYVE1
zinc finger FYVE-type containing 1
- ER
endoplasmic reticulum
- GAD1
glutamate decarboxylase 1
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescent protein
- IB
immunoblotting
- MAP1LC3/LC3
microtubule associated protein 1 light chain 3
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- pAb
polyclonal antibody
- PD
Parkinson disease
- PIK3C3
phosphatidylinositol 3-kinase catalytic subunit type 3
- PtdIns3K
phosphatidylinositol 3-kinase
- ros
roswell mutant mouse
- SLC35D3
solute carrier family 35 member D3
- SNc
substantia nigra pars compacta
- SNr
substantia nigra pars reticulata
- SQSTM1
sequestosome 1
- TH
tyrosine hydroxylase
- ULK
unc51-like autophagy activating kinase
- VTA
ventral tegmental area
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are very thankful to Dr. Richard T. Swank who provided the ros mutant for this study and proofread this manuscript. We thank Ms. Siqi Zhao for the assistance in primary culturing of astrocytes.
Funding
This work was partially supported by grants from National Natural Science Foundation of China (91539204; 31230046), National Basic Research Program of China (2013CB530605), and from Chinese Academy of Sciences (KJZD-EW-L08).
References
- [1].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- [2].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/ 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- [3].Yuan Y, Wang H, Wei Z, Li W. Impaired autophagy in hilar mossy cells of the dentate gyrus and its implication in schizophrenia. J Genet Genomics 2015; 42:1-8; PMID:25619597; http://dx.doi.org/ 10.1016/j.jgg.2014.12.001 [DOI] [PubMed] [Google Scholar]
- [4].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/ 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- [5].Funke C, Schneider SA, Berg D, Kell DB. Genetics and iron in the systems biology of Parkinson's disease and some related disorders. Neurochem Int 2013; 62:637-52; PMID:23220386; http://dx.doi.org/ 10.1016/j.neuint.2012.11.015 [DOI] [PubMed] [Google Scholar]
- [6].Liu K, Shi N, Sun Y, Zhang T, Sun X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res 2013; 38:201-7; PMID:23117422; http://dx.doi.org/ 10.1007/s11064-012-0909-8 [DOI] [PubMed] [Google Scholar]
- [7].Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- [9].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/ 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011; 7:643-4; PMID:21460621; http://dx.doi.org/ 10.4161/auto.7.6.15123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468-76; PMID:19270693; http://dx.doi.org/ 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005; 1:46-52; PMID:16874027; http://dx.doi.org/ 10.4161/auto.1.1.1542 [DOI] [PubMed] [Google Scholar]
- [15].Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360-72; PMID:18843052; http://dx.doi.org/ 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al.. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/ 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- [17].Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al.. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191:155-68; PMID:20921139; http://dx.doi.org/ 10.1083/jcb.201002100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/ 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yamamoto A, Yue Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci 2014; 37:55-78; PMID:24821313; http://dx.doi.org/ 10.1146/annurev-neuro-071013-014149 [DOI] [PubMed] [Google Scholar]
- [20].Chintala S, Tan J, Gautam R, Rusiniak ME, Guo X, Li W, Gahl WA, Huizing M, Spritz RA, Hutton S, et al.. The Slc35d3 gene, encoding an orphan nucleotide sugar transporter, regulates platelet-dense granules. Blood 2007; 109:1533-40; PMID:17062724; http://dx.doi.org/ 10.1182/blood-2006-08-040196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Meng R, Wang Y, Yao Y, Zhang Z, Harper DC, Heijnen HF, Sitaram A, Li W, Raposo G, Weiss MJ, et al.. SLC35D3 delivery from megakaryocyte early endosomes is required for platelet dense granule biogenesis and is differentially defective in Hermansky-Pudlak syndrome models. Blood 2012; 120:404-14; PMID:22611153; http://dx.doi.org/ 10.1182/blood-2011-11-389551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Z, Hao CJ, Li CG, Zang DJ, Zhao J, Li XN, Wei AH, Wei ZB, Yang L, He X, et al.. Mutation of SLC35D3 causes metabolic syndrome by impairing dopamine signaling in striatal D1 neurons. PLoS Genet 2014; 10:e1004124; PMID:24550737; http://dx.doi.org/ 10.1371/journal.pgen.1004124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Revy D, Jaouen F, Salin P, Melon C, Chabbert D, Tafi E, Concetta L, Langa F, Amalric M, Kerkerian-Le Goff L, et al.. Cellular and behavioral outcomes of dorsal striatonigral neuron ablation: new insights into striatal functions. Neuropsychopharmacology 2014; 39:2662-72; PMID:24903652; http://dx.doi.org/ 10.1038/npp.2014.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci 2006; 9:443-52; PMID:16491081; http://dx.doi.org/ 10.1038/nn1654 [DOI] [PubMed] [Google Scholar]
- [25].Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/ 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al.. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015; 520:563-6; PMID:25686604; http://dx.doi.org/ 10.1038/nature14147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma B, Cao W, Li W, Gao C, Qi Z, Zhao Y, Du J, Xue H, Peng J, Wen J, et al.. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res 2014; 24:912-24; PMID:24980960; http://dx.doi.org/ 10.1038/cr.2014.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhen Y, Li W. Impairment of autophagosome-lysosome fusion in the buff mutant mice with the VPS33A (D251E) mutation. Autophagy 2015; 11:1608-22; PMID:26259518; http://dx.doi.org/ 10.1080/15548627.2015.1072669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Morales-Briceno H, Cervantes-Arriaga A, Rodriguez-Violante M, Calleja-Castillo J, Corona T. Overweight is more prevalent in patients with Parkinson's disease. Arq Neuropsiquiatr 2012; 70:843-6; PMID:23175195; http://dx.doi.org/ 10.1590/S0004-282X2012001100004 [DOI] [PubMed] [Google Scholar]
- [30].Hu G, Jousilahti P, Nissinen A, Antikainen R, Kivipelto M, Tuomilehto J. Body mass index and the risk of Parkinson disease. Neurology 2006; 67:1955-9; PMID:17159100; http://dx.doi.org/ 10.1212/01.wnl.0000247052.18422.e5 [DOI] [PubMed] [Google Scholar]
- [31].Zhang A, He X, Zhang L, Yang L, Woodman P, Li W. Biogenesis of lysosome-related organelles complex-1 subunit 1 (BLOS1) interacts with sorting nexin 2 and the endosomal sorting complex required for transport-I (ESCRT-I) component TSG101 to mediate the sorting of epidermal growth factor receptor into endosomal compartments. J Biol Chem 2014; 289:29180-94; PMID:25183008; http://dx.doi.org/ 10.1074/jbc.M114.576561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang H, Yuan Y, Zhang Z, Yan H, Feng Y, Li W. Dysbindin-1C is required for the survival of hilar mossy cells and the maturation of adult newborn neurons in dentate gyrus. J Biol Chem 2014; 289:29060-72; PMID:25157109; http://dx.doi.org/ 10.1074/jbc.M114.590927 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.