

ABSTRACT
Inhibition of prosurvival BCL2 family members can induce autophagy, but the mechanism is controversial. We have provided genetic evidence that BCL2 family members block autophagy by inhibiting BAX and BAK1, but others have proposed they instead inhibit BECN1. Here we confirm that small molecule BH3 mimetics can induce BAX- and BAK1-independent MAP1LC3B/LC3B lipidation, but this only occurred at concentrations far greater than required to induce apoptosis and dissociate canonical BH3 domain-containing proteins that bind more tightly than BECN1. Because high concentrations of a less-active enantiomer of ABT-263 also induced BAX- and BAK1-independent LC3B lipidation, induction of this marker of autophagy appears to be an off-target effect. Indeed, robust autophagic flux was not induced by BH3 mimetic compounds in the absence of BAX and BAK1. Therefore at concentrations that are on target and achievable in vivo, BH3 mimetics only induce autophagy in a BAX- and BAK1-dependent manner.
KEYWORDS: ABT-199, ABT-263, ABT-737, autophagy, BCL2, BECN1
Introduction
Prosurvival BCL2 family members (BCL2, BCL2L1/Bcl-XL, MCL1, BCL2A1/Bfl1/A1 and BCL2L2/Bcl-w) inhibit mitochondrial-mediated (intrinsic) apoptosis by binding to pro-apoptotic BH3-only proteins, such as BCL2L11/Bim, and the multidomain apoptosis effector proteins BAX and BAK1.1 BH3 mimetic compounds such as ABT-737, Navitoclax (ABT-263) and Venetoclax (ABT-199), bind to prosurvival BCL2 family members to displace pro-apoptotic BCL2 family members or “activator” BH3-only proteins, initiating activation of BAX and BAK1 (Fig. 1A). When activated, BAX and BAK1 induce mitochondrial outer membrane permeabilization, releasing CYCS (cytochrome c, somatic) from the mitochondria. CYCS then enables formation of the apoptosome, which subsequently cleaves and activates caspases, causing rapid cell death.
Figure 1.
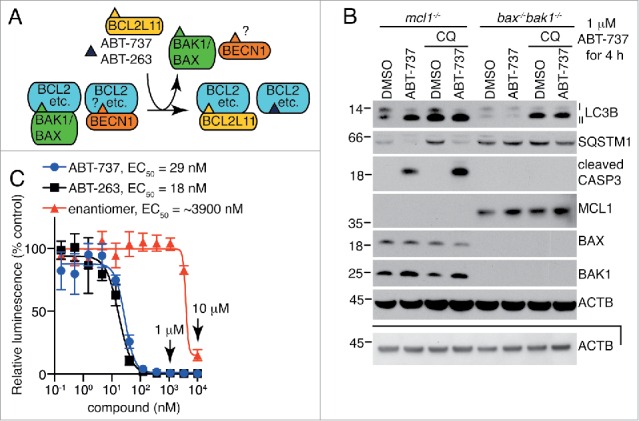
Autophagy induced by the BH3 mimetic ABT-737 is dependent on BAX and BAK1 at 1 μM, a concentration well above that required to inhibit the BCL2 family of proteins. (A) The BH3-only protein BCL2L11 and the BH3 mimetics ABT-737 and ABT-263 compete off BH3 domain-containing proteins such as BAX and BAK1, and possibly BECN1, from the pro-survival BCL2 family members. (B) Autophagy induced by 1 μM ABT-737 is dependent on BAX and BAK1. Representative western blot of an LC3B turnover assay in MEFs treated with 1 μM ABT-737 and/or 10 μM chloroquine (CQ) for 4 h prior to lysis. Another independent experiment is available in Fig. S1B. The lower ACTB panel is of the same samples, diluted, and run on a separate gel. Molecular weight markers are indicated on the left. (C) The number of metabolically active viable mcl1−/− MEFs as measured by the CellTiter-Glo® Luminescent Cell Viability Assay after a 24-h treatment with ABT-737, ABT-263, or the enantiomer of ABT-263. The mean luminescence of compound-treated cells compared to vehicle-treated cells ± SEM are shown (n = 3 independent experiments, using 2 independent cell lines).
In addition to controlling apoptosis, BCL2 family members can also regulate autophagy, but how they do so is controversial.2,3 On the one hand, it has been proposed that prosurvival BCL2 family members such as BCL2 and BCL2L1 can directly bind to a BH3-like domain of BECN1/Beclin 1 to inhibit autophagy.4-6 In this model, the BH3-only proteins, BH3 mimetics, BAX, BAK1, and BECN1 all compete to bind to the same groove on the prosurvival BCL2 family proteins (Fig. 1A). On the other hand, we provided evidence that inhibition of prosurvival BCL2 family members only induce autophagy by an indirect mechanism that requires the presence of BAX or BAK1. Activation of BAX or BAK1 triggers both autophagy and apoptosis,7,8 although it is yet to be determined whether BAX and BAK1 promote autophagy by a direct or indirect mechanism. Furthermore, inhibiting BCL2, BCL2L1, and BCL2L2 by overexpression of the BH3-only protein BCL2L11 is unable to induce LC3B lipidation in cells lacking BAX and BAK1.7 Consistent with this, overexpression of BAX alone can stimulate autophagy.9
Recently, it was reported that high concentrations of the BH3 mimetic ABT-737 could trigger markers of autophagy in cells lacking BAX and BAK1, presumably by releasing BECN1 from BCL2.10 Because this release takes up to 48 h,10 we attempted to determine if this was an on-target effect of this small molecule. When BAX and BAK1 were absent, we only saw MAP1LC3B/LC3B lipidation by BH3-mimetic compounds at very high concentrations (10–50 µM), which are much greater than those required to inhibit the prosurvival BCL2 proteins and induce apoptosis in sensitive cells. Furthermore, while these high levels of compounds induced this marker of autophagy, they did not induce strong autophagic flux. Our results suggest that the ability of BH3-mimetic compounds to induce LC3B lipidation in the absence of BAX and BAK1 is an off-target effect and is unrelated to displacement of BECN1 from BCL2.
Results
To investigate the association of BH3-mimetic-induced apoptosis with autophagy, we used mcl1−/− MEFs. These cells are highly dependent on BCL2L1 for their survival and hence die in response to ABT-737, which targets both BCL2L1 and BCL2 with high affinity. As we have previously reported, LC3B lipidation (generating LC3B-II) induced by 1 μM of the BH3 mimetic ABT-737 occurred in the presence of the effectors of mitochondrial-mediated apoptosis, BAX and BAK1, but not in cells that lacked them (Fig. S1A).7,8 Addition of chloroquine to inhibit the degradation of LC3B-II via autophagy did not further increase LC3B-II protein levels in bax−/−bak1−/− cells, confirming autophagy was not being induced in these cells, as it was in the cells that bore BAX and BAK1, but lacked MCL1 (Fig. 1B, S1B). The concentration of ABT-737 used in these experiments (1 μM) was well above that required to eliminate viable, metabolically-active mcl1−/− MEFs after 24 h, as assessed by loss of ATP (Fig. 1C). As expected, robust cleavage of CASP3 (caspase 3) was observed in these cells after 4 h (Fig. 1B, S1B).
It was recently reported that even higher concentrations (10 μM) of ABT-737 could induce autophagy in cells in a BAX- and BAK1-independent manner.10 We decided to test this by looking at markers of autophagy after exposing cells to high concentrations of ABT-737, as well as the clinical analog ABT-263.11 As expected,12 both ABT-737 and ABT-263 induced apoptosis of mcl1−/− MEFs within 4 h, as indicated by propidium iodide uptake and cleavage of CASP3, and increased conversion of LC3B-I to LC3B-II was also observed (Fig. 2A, S1C). CASP3 was not cleaved in wild-type (wt) MEFs during this time, presumably due to the presence of MCL1, but the compounds were also able to cause some LC3B conversion, albeit to a lesser extent (Fig. 2A).12 In addition, 10 μM ABT-263 induced LC3B lipidation by 4 h in cells lacking BAX and BAK1 (Fig. 2A). While we were unable to definitively reproduce the previously reported results with 10 μM ABT-737,10 we did see robust LC3B-II formation at even higher concentrations (50 μM) (Fig. 2B, S1D); this discrepancy is most likely due to slight differences in activities of compound batches and/or different sensitivities of the specific MEF lines used in each study. SQSTM1/p62 protein levels were not reduced at these concentrations of ABT-737. Nonetheless, at high concentrations both ABT-737 and ABT-263 were able to induce LC3B lipidation regardless of MCL1, BAX and BAK1 expression at high concentrations, which is consistent with previously reported data.10
Figure 2.

BAX- and BAK1-independent LC3B lipidation by BH3 mimetics is unrelated to BH3 mimetic activity and has only minimal effects on autophagic flux. (A) BAX- and BAK1-independent LC3B lipidation induced by high dose (10 µM) of ABT-737, ABT-263 and enantiomer does not correlate with their affinities for the prosurvival BCL2 family members. Western blot of MEFs following a 4 h treatment with 10 μM BH3 mimetics. (B) Robust LC3B-II formation with very high concentrations (50 µM) of ABT-737 and A-1331852 (A-1852) in bax−/−bak1/− MEFs. Western blot of cells lysed following a 4-h treatment. The lower ACTB panel is of the same samples, diluted, and run on a separate gel. Another independent experiment is available in Fig. S1D. (C) Western blot of MEFs after a 4-h treatment with a panel of BH3 mimetics at 10 μM. Another independent experiment is available in Fig. S1F. (D) LC3B is not efficiently degraded by autophagy after treatment with ABT-263 or its enantiomer in bax−/−bak1−/− MEFs. Western blot of MEFs treated with 10 μM ABT-263, enantiomer, or chloroquine for 4 h prior to lysis. Additional examples in Figs. S1G-H using 2 bax−/−bak1−/− lines generated from independent mice. (E-F) Autolysosome formation is very weak after treatment with 10 μM BH3 mimetics or 0.1 μM bafilomycin A1 (BafA1) in bax−/−bak1−/− MEFs. Cells expressing mCherry-EGFP-LC3B were treated with compound or starved in HBSS for 24 h prior to analysis via flow cytometry. Above: Quantification of the percentage of mCherry+ cells that were GFPhigh (not in an acidic lysosomal environment). The mean of 4–6 independent experiments ± SEM is shown using 2 independent cell lines. Below: Representative dot plots. A full representation of (F) is available in Fig. S2B. Gating strategy is shown in Fig. S2A.
Because 10 μM is almost a 100-fold greater concentration of either ABT-737 or ABT-263 than is required to completely kill cells as indicated by Celltitre-Glo assays (Fig. 1C),7,12,13 we hypothesized that LC3B lipidation at this very high concentration is an off-target effect of the compounds, unrelated to their BH3 mimetic activity. Indeed, prolonged treatment with 10 μM ABT-263 induced death of MEFs lacking BAX and BAK1 (Fig. S1E), which is contrary to its expected mode of action.12 To further test this theory we compared induction of LC3B lipidation by ABT-263 to that caused by its enantiomer A-900526, which has more than a 100-fold greater EC50 and greatly reduced ability to bind the BCL2 protein targets (Fig. 1C).11 Consistent with its greatly reduced ability to bind to BCL2, BCL2L1, or BCL2L2, the enantiomer did not induce cleavage and activation of CASP3 in mcl1−/− MEFs (Fig. 2A) or cause cell death within 4 h (Fig. S1C). In contrast, 10 μM enantiomer caused LC3B lipidation regardless of cell genotype (Fig. 2A), suggesting that at these very high concentrations, this effect of the compounds are not related to their capacity to bind BCL2 prosurvival family members. It is worthwhile to note that another common marker of autophagy, SQSTM1/p62 protein reduction, did not correlate with LC3B-II formation, but instead correlated better with CASP3 cleavage (Fig. 2A), suggesting this is a poor marker for autophagy when apoptosis can also be activated. We also investigated if other related BH3 mimetic compounds acted in a similar manner. Indeed, this form of BAX- and BAK1-independent LC3B lipidation was not only induced by ABT-263 and ABT-737, but also occurred with ABT-199, a BCL2 specific inhibitor, and the BCL2L1 specific inhibitor A-1331852 (A-1852) (Fig. 2B-C, S1F).14,15
To determine if the LC3B lipidation observed following treatment with 10 μM compound in the absence of BAX and BAK1 was indicative of an increase in autophagic flux, we performed LC3B-II turnover assays. As expected, chloroquine increased the amount of LC3B-II, indicating a block in autolysosomal function; however, addition of ABT-263 or its enantiomer together with chloroquine did not consistently cause a further increase in LC3B-II formation, in 2 independent cell lines (Figs. 2D, S1G, S1H), suggesting that downstream degradation was not taking place. Consistent with this result, autolysosome formation (as measured by decreased GFP fluorescence, which is pH sensitive) in cells lacking BAX and BAK1 was negligible compared to starvation by culturing in Hank's balanced salt solution (HBSS) using a mCherry-EGFP-LC3B marker (Figs. 2E, S2A). Similar to ABT-263, ABT-199 and A-1331852 (A-1852) did not induce convincing autolysosomal formation (Fig. 2F, S2B).
In contrast to experiments at 10 μM, treatments with 1 μM compound showed the expected BAX- and BAK1-dependent pro-apoptotic activity. For example, ABT-737 and ABT-263, but not the enantiomer, caused apoptosis of mcl1−/− MEFs that expressed BAX and BAK1 (Fig. S2C), while cells lacking BAX and BAK1 were completely resistant to killing by all compounds, even after 48-h treatment. Wild-type cells were protected against 1 μM ABT-737 and ABT-263 because MCL1 inhibits the activation of BAX and BAK1, as previously published.12 Under these conditions, neither ABT-263 nor its enantiomer could induce LC3B lipidation or SQSTM1 protein reduction in either WT or BAX- and BAK1-null MEFs (Fig. S2D). Combined, these data suggest that at concentrations where ABT-737 and ABT-263 act on target (i.e., 1 μM or less), they can only induce autophagy when BAX and BAK1 can be activated.
BAX- and BAK1-independent autophagy has also been reported to occur in the human colon cancer cell line HCT116.10 After treatment with 3 μM or 10 μM ABT-737 for 12 h, an increase in LC3B-I and LC3B-II protein levels was observed regardless of whether HCT116 cells expressed BAX and BAK1.10 We repeated these experiments, using BH3 mimetics ABT-737, ABT-263, and the enantiomer. As expected, there were negligible effects when ABT-737 or ABT-263 were added at lower concentrations (1 μM) to either WT or BAX−/−BAK1−/− HCT116 cells (Figs. 3A-B, S3A). In contrast, treatment of BAX−/−BAK1−/− HCT116 cells with 10 μM ABT-263 induced more robust LC3B lipidation, but a similar effect was also observed when the cells were treated with the enantiomer (Figs. 3A, S3A). An increase in the ratio of LC3B-II to LC3B-I also occurred when WT HCT116 cells were treated with the enantiomer (Figs. 3A, S3A), but high concentrations of ABT-737 and ABT-263 also caused significant cell death (Fig. 3B), making the assessment of western blots difficult. Similar to the MEFs, SQSTM1 protein levels did not decrease significantly after treatment of BAX−/−BAK1−/− HCT116 cells with 10 μM compound (Figs. 3A, S3A), and correlated with cell death in WT cells.
Figure 3.
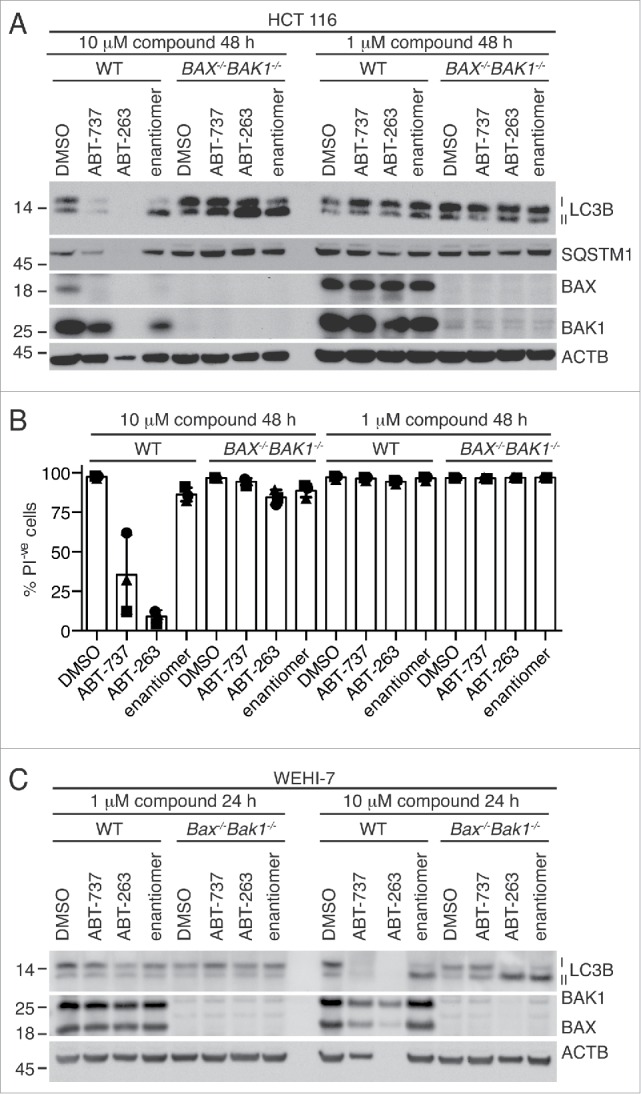
LC3B lipidation induced by ABT-263 and related compounds does not correlate with BH3 mimetic activity in BAX- and BAK1-null HCT116 cells and WEHI-7 cells. (A) Representative western blot of HCT116 cells following treatment with 10 µM or 1 µM BH3 mimetics for 48 h. Another example is shown in Fig. S3A. (B) Cell death measured by propidium iodide (PI) uptake of same samples as in (A). Data show the mean ± SEM of 3 independent experiments. “PI−ve” denotes cells that are resistant to propidium iodide staining. (C) Western blot of WEHI-7 cells with or without Bax and Bak1 deleted by CRISPR-Cas9 technology following 24 h treatment with 1 µM or 10 µM BH3 mimetics. An independent experiment is available in Fig. S3B.
We also investigated if a similar result would occur with a cell type in which Bax and Bak1 were more acutely deleted. We deleted Bax and Bak1 from the mouse thymoma line WEHI-7 using CRISPR-Cas9 technology and treated them with ABT-263 and its less active enantiomer. As with HCT116 cells, all compounds, including the enantiomer, induced LC3B lipidation at 10 μM but not 1 μM in Bax- and Bak1-deleted cells (Figs. 3C, S3B). Treatment with 10 uM of compound caused cell death of wild-type WEHI-7 cells and ABT-263 induced nonapoptotic cell death in WEHI-7 cells lacking BAX and BAK1, similar to MEFs (Fig. S3C). Therefore, these results do not appear to be due to a genetic developmental defect arising from long-term loss of BAX and BAK1.
It has been proposed that ABT-737 induces autophagy by displacing BCL2 from BECN1.16 In cells lacking BAX and BAK1, LC3B lipidation is observed after 12 h10 and in our hands after 4 h. However, reduced binding of BECN1 to BCL2 is not reported to occur until 48 h of treatment of MEFs with ABT-737.10 This suggests release of BECN1 might not be the primary cause of autophagy induction in MEFs under these circumstances. Even accounting for the difference in kinetics between experiments, we found it surprising that release of BECN1 from BCL2 would be so slow, given that ABT-737 and ABT-263 can induce apoptosis within a few hours by the same mechanism, namely by binding to the same prosurvival BCL2 family of proteins (Fig. 1A).7,12
A very slow release of BECN1 would only be expected if the BCL2-BECN1 and BCL2L1-BECN1 interactions were of much higher affinity than the comparable interactions that must be antagonized to induce apoptosis (i.e., BCL2-BAX, BCL2-BAK1, BCL2L1-BAX, BCL2L1-BAK1 and BCL2-BCL2L11, BCL2L1-BCL2L11). However, when binding affinities were compared, we found that ABT-737 bound nearly 1000 times more tightly than BECN1 to BCL2L1 (Table 1). In contrast, the BH3 domains of BCL2L11 and 2 BH3-only proteins, BCL2L11 and BAD (the latter which ABT-737 and ABT-263 mimic), bound BCL2L1 with similar affinities (Table 1). We chose to study the binding to BCL2L1 because this is the prosurvival protein for which the binding affinities are best characterized. Our data correlate very well with those previously published by separate laboratories (Table 1).17-21
Table 1.
Binding affinities of ABT-737 or the BH3 domains of the indicated proteins to BCL2L1.
To confirm these binding data and extend our studies, we performed co-immunoprecipitations using antibodies against both BCL2L1 and BCL2. While detergents such as Triton X-100, Tween 20 and NP-40 induce conformational changes in some BCL2 family proteins and can force artificial interactions with BCL2 and BCL2L1, this is not the case with CHAPS.22 Because BECN1 has been proposed to interact with BCL2 and BCL2L1 via its BH3 domain, we wanted to ensure we did not induce artificial interactions due to effects of the detergent. When we overexpressed BCL2L1 to induce maximum binding of potential binding partners, BCL2L11 and BAK1 were co-immunoprecipitated specifically with the BCL2L1 antibody, but not with the isogenic control antibody (Fig. 4A). Both BCL2L11 and BAK1 were completely out-competed by a 4-h treatment of either 1 μM or 10 μM ABT-737 prior to cell lysis (Fig. 4A). Similarly, BCL2L11, BAK1, and BAX were all co-immunoprecipitated when BCL2 was overexpressed and immunoprecipitated, and binding was equally inhibited at both concentrations of ABT-737 (Fig. 4B). In contrast, whereas a weak band of the same size as BECN1 was detected in both the BCL2 and BCL2L1 immunoprecipitations, it was also present at the same level in the negative antibody isotype controls. Moreover, the intensity of this band was unaffected by ABT-737 treatment (Fig. 4A-B). Therefore, under conditions where BH3 domain-containing proteins are expected to be in their physiological conformations, we were unable to detect specific interactions of BECN1 with BCL2 or BCL2L1 that could be displaced by ABT-737. Indeed, even in a Triton X-100-based buffer, we were unable to see specific interaction between BECN1 and overexpressed BCL2 or BCL2L1 (Fig. 4C-D). While other BH3 domain-containing proteins were pulled down and their binding was inhibited by BH3 mimetic, it is interesting to note that their association with the prosurvival proteins differed from when cells were lysed in CHAPS (Fig. 4A-D). A similar trend was observed in HCT116 cells—we could not see any specific interaction of the prosurvival protein with BECN1 (Fig. 4E). The lack of specific interaction under diverse conditions between BECN1 and BCL2 or BCL2L1 is consistent with the weak binding affinities presented in Table 1 and observed by independent groups.
Figure 4.
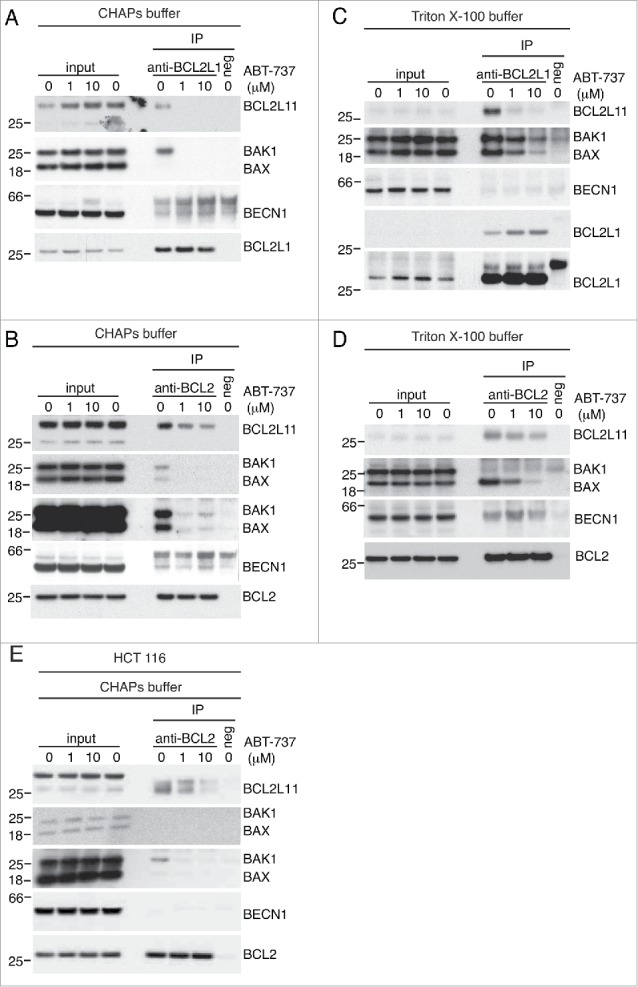
BCL2 family members, but not BECN1, are dissociated from BCL2L1 and BCL2 by ABT-737. (A, C) Wild-type MEFs overexpressing BCL2L1 were treated with the indicated concentrations of ABT-737 for 4 h prior to lysis in a (A) CHAPS-based or (C) Triton X-100-based buffer and immunoprecipitated with anti-BCL2L1 antibody or an isogenic (negative) control. (B, D) Wild-type MEFs overexpressing BCL2 were treated with the indicated concentrations of ABT-737 for 4 h prior to lysis in a (B) CHAPS-based or (D) Triton X-100-based buffer and immunoprecipitated with anti-BCL2 antibody or an isogenic (negative) control. (E) Wild-type HCT116 cells overexpressing BCL2 were treated with the indicated concentrations of ABT-737 for 4 h prior to lysis in CHAPS-based buffer and immunoprecipitated with anti-BCL2 antibody or an isogenic (negative) control.
If BAX- and BAK1-independent LC3B lipidation induced by BH3 mimetics is the result of off-target effects of the compounds, then overexpression of a BH3-only protein should produce strictly BAX- and BAK1-dependent autophagy. Consistent with this hypothesis, overexpression of BCL2L11s from a lentiviral vector with doxycycline induced LC3B lipidation, which was accompanied by induced CASP3 cleavage, within 4 h in WT MEFs, but had no effect in bax−/−bak1−/− MEFs, even after 24 h (Fig. 5A).7 Indeed, LC3B was only turned over when BAX and BAK1 were present (Fig. 5B). SQSTM1 levels decreased only in wild-type cells, although this was more pronounced in the experiment shown in Fig. 5A. Taken together, these data indicate that LC3B lipidation induced in bax−/−bak1−/− MEFs by very high levels of ABT-737 and its related compounds is independent of their function as BH3 mimetics.
Figure 5.
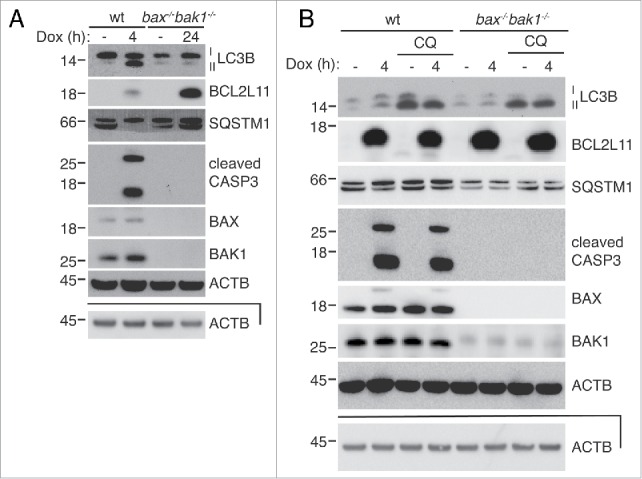
Autophagy induced by overexpression of the BH3-only protein BCL2L11 is strictly dependent on BAX and BAK1. (A) Overexpression of BCL2L11s only induces LC3B lipidation when BAX and BAK1 are present. MEFs bearing a doxycycline-inducible BCL2L11s lentiviral construct were treated with doxycycline for the indicated times and analyzed by western blotting. (B) Western blot of wild-type and bax−/−bak1−/− MEFS after overexpression of BCL2L11s with doxycycline for 4 h with or without 10 μM treatment with the autolysosome inhibitor chloroquine (CQ). The lower ACTB panels are of the same samples, diluted, and run on a separate gel.
Discussion
At least 2 models have been proposed to explain how the prosurvival BCL2 family members inhibit autophagy. Our data support a model where autophagy is induced when the BCL2 family members are inhibited and release BAX and BAK1, which then activate autophagy via a still uncharacterized mechanism.7,8 The alternative model argues that BCL2 and its related family members bind directly to BECN1 to inhibit autophagy, and was supported by claims that BH3 mimetics induce autophagy in the absence of BAX and BAK1.4,10,23,24
The results presented here, however, indicate that the concentrations of BH3 mimetic compounds required to induce BAX- and BAK1-independent LC3B lipidation are too high to reflect on-target activity. For instance, at these high concentrations, a less-active enantiomer of ABT-263 had similar effects on LC3B lipidation as the functional BH3 mimetics at these concentrations. Importantly, in our hands, none of the compounds tested induced autolysosome formation or function in bax−/−bak1−/− cells, even though LC3B lipidation occurred.7 While it is possible that the flow cytometry experiments are not sensitive enough to detect a moderate reduction in flux because the EGFP signal is quite strong, LC3B turnover experiments were sensitive enough to detect autophagic flux in cells where BAX and BAK1 were activated with moderate concentrations of ABT-737. In contrast, no LC3B turnover was observed in BAX- and BAK1-null cells with either moderate or high concentrations of BH3 mimetics, even if they caused an increase in LC3B lipidation. Importantly, when the BH3 mimetics were functionally replaced with the potent BH3-only protein BCL2L11, autophagic flux was strictly BAX and BAK1 dependent.
So how could LC3B lipidation occur without autophagic flux? It is possible that the pathway (or parts of the pathway) leading to LC3B lipidation is activated, while the ULK1 complex and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex may remain unaffected. For instance, cells null for RB1CC1/FIP200, which is part of the ULK1 complex, still can make LC3B-II but cannot induce autophagic flux.25 Similar trends have been reported when levels of the components of the class III PtdIns3K complex are reduced with RNAi.26 The requirement of the different members of the ubiquitin-like pathways would need to be determined to elucidate if LC3B-II formation induced by high concentrations of BH3 mimetic occurs in a canonical manner. Regardless, the LC3B lipidation we observed is independent of the BH3 mimetic activity of the compounds when BAX and BAK1 are absent.
If the BCL2-BECN1 model is correct, the BH3-only proteins, BH3 mimetic compounds, and BAX and BAK1 should all compete for the same binding site of BCL2 and related proteins. However, in studies performed by Pedro et al. in fibroblasts, BECN1 dissociated from BCL2 after LC3B lipidation was observed,10 which is inconsistent with release of BECN1 being the major factor driving autophagy in these cells. This slow dissociation is particularly surprising given the weak binding affinity of the BH3 domain of BECN1 for prosurvival proteins.20,21 Moreover, the BECN1 BH3-prosurvival complexes are significantly less stable than complexes of BH3-only proteins and prosurvival proteins. The latter must be antagonized to induce apoptosis, yet apoptotic death occurs relatively quickly after treatment and we readily detected dissociation of these higher affinity complexes in our immuno-precipitation experiments. One potential explanation is that the effects observed by Pedro et al. were caused by cell death from prolonged treatment with concentrations of compound that were sufficiently high to cause off-target toxicity.
While we observed specific binding of apoptotic BH3 domain-containing proteins to overexpressed BCL2 and/or BCL2L1, including some interactions which are considered very weak (e.g., BAK1 binding to BCL2),27,28 we were unable to demonstrate specific binding of BECN1 to either prosurvival protein under diverse conditions in either mouse or human cell lines. BECN1 is potentially a “sticky” protein and this may also explain the apparent binding of BECN1 to BCL2 seen by Pedro et al., where a negative isotype control was not shown.10
While our data suggest that the prosurvival BCL2 proteins do not regulate autophagy by binding directly to BECN1, BECN1 may still be involved in autophagy induced by BAX and BAK1 activation as part of its canonical role in the PtdIns3K complex. There have also been reports of BECN1-independent autophagy induced by cytotoxic compounds that also activate BAX and BAK1.29-31 It is therefore also possible that BECN1 is not essential for autophagy induced by the BH3 mimetics tested here. Obviously the role of BECN1 in autophagy regulated by the BCL2 family would profit from further study.
It has only been possible to address the direct effects of prosurvival BCL2 family members on autophagy independently of their canonical function of regulating apoptosis through the use of BAX- and BAK1-deleted cell lines.7,10 Use of such lines is important, because many stimulants of autophagy, such as starvation, MTOR inhibition, and chemotherapeutic drugs, can also induce apoptosis. It has also been reported that caspases can inhibit BECN1-dependent autophagy downstream of apoptosis by cleaving this autophagy protein, further complicating the interpretation of results where both autophagy and apoptosis are induced.32 Overall, studying the induction of autophagy in the absence of intrinsic apoptosis will bring new insight into how autophagy is regulated and the fine interplay between cell survival and cell suicide pathways.
Materials and methods
Cell lines, constructs, and compounds
Mouse embryonic fibroblasts (MEFs) are previously described and were immortalized with SV40 large T antigen.7 HCT116 lines were kindly provided by Dr Richard Youle (NIH).33 WEHI-7 cells34 were infected with FUCas9Cherry35 and gRNA against BAX (AGTTTCATCCAGGATCGAGC) and BAK1 (TCATCGCAGCCCACCTTCGG) into FgH1tUTG35 to create a Bax- and Bak1-deleted line. ABT-737, ABT-199, and ABT-263 were obtained from Selleck, while A-900526 was kindly provided by AbbVie.11 A-1331852 (A-1852)14 was made as previously described.36 Chloroquine and bafilomycin A1 were purchased from Sigma (C6628-25G, B1973_10UG). The construct pBABe puro mCherry-EGFP-LC3B is available from Addgene (Plasmid #22418, deposited by Jayanta Debnath). BCL2L11s overexpression was achieved by infecting MEFs with the previously described lentiviral construct pFTRE 3G rtTA Puro/Bims and the addition of doxycycline.7
Western blotting and viability assays with propidium iodide
These assays were performed essentially as described.7 Briefly, MEF, WEHI-7, or HCT116 cells were treated with compounds at the indicated times and concentrations. If viability assays were performed, 10% of cells (both attached and unattached) were resuspended in 10 μg/mL propidium iodide (PI) for flow cytometry analysis, while the rest was lysed in a Triton X-100-based ONYX buffer (20 mM Tris, pH 7.5, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 10% glycerol, 1% Triton X-100 (Sigma-Aldrich, T9284)) including protease inhibitors (Roche, 11697498001) and separated on 10% or 4–12% gradient NuPAGE Bis Tris gels (Invitrogen, WG1202BX10). Nitrocellulose membranes were blotted with antibodies raised against LC3B (D11; Cell Signaling Technology, 3868), SQSTM1 (Cell Signaling Technology, 5114), BECN1 (Cell Signaling Technology, D40C5), BCL2 (3F11; WEHI, in house), MCL1 (19C4; WEHI, in house), BCL2L1 (44; BD Biosciences, 610746), BCL2L11 (3C5; WEHI, in house or 3C5; Enzo Biochem, Inc., ALX-804-527-C100), BAX (49F9-13-13; WEHI, in house), BAK1 (aa23-38; Sigma-Aldrich, B5897), cleaved CASP3 (Cell Signaling Technology, 9661), and ACTB (Sigma, AC-15).
CellTiter-Glo experiments
Metabolic viability experiments were performed using a CellTiter-Glo (Promega, G7570) luminescent cell viability assay. mcl1−/− mouse embryonic fibroblasts (1500 cells/well) were plated overnight in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco, 16000–044) in an opaque 96-well plate. The following day, the medium was removed and replaced with medium containing various concentrations of ABT-737, ABT-263, or the enantiomer of ABT-263 and incubated for 24 h before developing the assay as per the manufacturer's instructions.
Autolysosome formation experiments
Autolysosome formation experiments were performed essentially as previously described.7 bax−/−bak1−/− MEFs infected with pBABe puro mCherry-EGFP-LC3B were treated with compounds in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum or starved in HBSS for 24 h before processing and analysis by flow cytometry. The percentage of cells with high EGFP also positive for mCherry were calculated. Details of gating are detailed in Fig. S2A.
Co-immunoprecipitations
MEFs and HCT116 cells were infected with a doxycycline-inducible mouse BCL2 or BCL2L1 lentiviral vector as previously described.7 8.0 × 106 cells were induced for 24 h with 1 μg/mL doxycycline (Sigma-Aldrich, D3447), and then subsequently treated with compound or vehicle for 4 h. Cells were trypsinized, washed, and then lysed in 250 μL ice-cold modified ONYX buffer (20 mM Tris, pH 7.5, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 10% glycerol) with either 1% CHAPS (Bio Basic, CD0110) or 1% Triton X-100 supplemented with protease inhibitor cocktail and incubated on ice for 30 min before being centrifuged for 5 min at 16000 × g at 4°C. The supernatant fraction was precleared with 50 μL of equilibrated protein-G-sepharose beads (GE Healthcare, 17-0618-06) for 30 min at 4°C before 1 μg of BCL2 (3F11; WEHI, in house), BCL2L1 (1C2; WEHI, in house), CASP2/caspase-2 (BCL2L1 isogenic negative control) (10C6; WEHI, in house) or glutathione S-transferase (GST; isogenic negative control for BCL2) (GT2; WEHI, in house) antibody was added and incubated for 2 h at 4°C on a turning wheel after which fresh beads were added for an additional hour. Samples are gently washed 5 times with 1 mL of lysis buffer (ONYX + CHAPS or Triton X-100) prior to elution with 60 μL of 2 x SDS-loading buffer.
Recombinant protein expression and purification
Recombinant BCL2L1 with a 24-residue C-terminal truncation (BCL2L1ΔC24) was expressed in E. coli as a GST fusion protein, and purified exactly as described previously using glutathione-affinity chromatography followed by size exclusion chromatography.17 The GST tag was removed by PreScission protease cleavage (Genescript, Z02799).
Peptides
Synthetic peptides were purchased from Mimotopes and were all >90% pure. The sequences were: BECN1 BH3: GEASDGGTMENLSRRLKVTGDLFDIMSGQTDVDH, BCL2L11 BH3: DMRPEIWIAQELRRIGDEFNAYYARR, BAD BH3: NLWAAQRYGRELRRMSDEFVDSFKKG; BAK1 BH3: PSSTMGQVGRQLAIIGDDINRRYDSE.
Surface plasma resonance binding assays
Relative binding affinities were determined by solution competition assays using a Biacore 3000 instrument as described previously.37 Briefly, 5 nM purified BCL2L1ΔC24 was incubated with BH3 peptides or ABT-737 for at least 2 h before injection onto a CM5 chip on which either wild-type BCL2L11 BH3 peptide or an inert BCL2L11 BH3 mutant peptide (BCL2L11 4E) was immobilized. Specific binding of BCL2L1ΔC24 to the chip surface in the presence and absence of competitor was quantified by subtracting the signal obtained on the BCL2L11-mutant channel from that obtained on the wild-type BCL2L11 channel. The ability of peptides to prevent protein binding to immobilized BCL2L11 BH3 was expressed as IC50, calculated by nonlinear curve fitting of the data by using GraphPad Prism 6 (GraphPad Software).
Statistical analyses
The mean, standard deviation, and standard error of the mean were calculated using Prism 6 software (GraphPad).
Supplementary Material
Abbreviations
- BAK1
BCL2-antagonist/killer 1
- BAX
BCL2-associated X protein
- BCL2
B-cell CLL/lymphoma 2
- BECN1
Beclin 1, autophagy-related
- DKO
double-knockout
- GST
glutathione S-transferase
- MAP1LC3B
microtubule-associated protein 1 light chain 3
- MCL1
myeloid cell leukemia sequence 1
- MEFs
mouse embryonic fibroblasts
- PI
propidium iodide
- PtdIns3K
phosphatidylinositol 3-kinase
- SQSTM1
sequestosome 1
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are very grateful to Dr Grant Dewson for his helpful suggestions and discussions and Drs David Segal and David Huang for reagents. We thank Dr Grant Dewson and Alex Delbridge for the guide RNAs against BAX and BAK1.
Funding
Funding for this project was provided by National Health and Medical Research Council (NHMRC) Program Grant 1020136 and Project Grant 1049949. LML holds an NHMRC Peter Doherty Early Career Fellowship (1035502), EFL holds an NHMRC Career Development Fellowship (1024620), and DLV a NHMRC Senior Principal Research Fellowship (1020136). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS and from support from the Australian Cancer Research Fund.
References
- [1].Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9:47-59; PMID:18097445; http://dx.doi.org/ 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- [2].Lalaoui N, Lindqvist LM, Sandow JJ, Ekert PG. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol 2015; 39:63-9; PMID:25736836; http://dx.doi.org/ 10.1016/j.semcdb.2015.02.003 [DOI] [PubMed] [Google Scholar]
- [3].Lindqvist LM, Simon AK, Baehrecke EH. Current questions and possible controversies in autophagy. Cell Death Discov 2015; 1:pii: 15036; PMID:26682061; http://dx.doi.org/ 10.1038/cddiscovery.2015.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- [5].Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011; 42:23-35; PMID:21353614; http://dx.doi.org/ 10.1016/j.molcel.2011.02.009 [DOI] [PubMed] [Google Scholar]
- [7].Lindqvist LM, Heinlein M, Huang DC, Vaux DL. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci USA 2014; 111:8512-7; PMID:24912196; http://dx.doi.org/ 10.1073/pnas.1406425111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lindqvist LM, Vaux DL. BCL2 and related prosurvival proteins require BAK1 and BAX to affect autophagy. Autophagy 2014; 10:1474-5; PMID:24991825; http://dx.doi.org/ 10.4161/auto.29639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 2009; 16:1135-45; PMID:19300452; http://dx.doi.org/ 10.1038/cdd.2009.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pedro JM, Wei Y, Sica V, Maiuri MC, Zou Z, Kroemer G, Levine B. BAX and BAK1 are dispensable for ABT-737-induced dissociation of the BCL2-BECN1 complex and autophagy. Autophagy 2015; 11:452-9; PMID:25715028; http://dx.doi.org/ 10.1080/15548627.2015.1017191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al.. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68:3421-8; PMID:18451170; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5836 [DOI] [PubMed] [Google Scholar]
- [12].van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et al.. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10:389-99; PMID:17097561; http://dx.doi.org/ 10.1016/j.ccr.2006.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee EF, Clarke OB, Evangelista M, Feng Z, Speed TP, Tchoubrieva EB, Strasser A, Kalinna BH, Colman PM, Fairlie WD. Discovery and molecular characterization of a Bcl-2-regulated cell death pathway in schistosomes. Proc Natl Acad Sci USA 2011; 108:6999-7003; PMID:21444803; http://dx.doi.org/ 10.1073/pnas.1100652108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, et al.. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 2015; 7:279ra40; PMID:25787766; http://dx.doi.org/ 10.1126/scitranslmed.aaa4642 [DOI] [PubMed] [Google Scholar]
- [15].Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al.. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19:202-8; PMID:23291630; http://dx.doi.org/ 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- [16].Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 2007; 3:374-6; PMID:17438366; http://dx.doi.org/ 10.4161/auto.4237 [DOI] [PubMed] [Google Scholar]
- [17].Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17:393-403; PMID:15694340; http://dx.doi.org/ 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]
- [18].Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al.. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435:677-81; PMID:15902208; http://dx.doi.org/ 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]
- [19].Ku B, Liang C, Jung JU, Oh BH. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res 2011; 21:627-41; PMID:21060336; http://dx.doi.org/ 10.1038/cr.2010.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-xL's target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol 2007; 372:223-35; PMID:17659302; http://dx.doi.org/ 10.1016/j.jmb.2007.06.069 [DOI] [PubMed] [Google Scholar]
- [21].Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 2007; 282:13123-32; PMID:17337444; http://dx.doi.org/ 10.1074/jbc.M700492200 [DOI] [PubMed] [Google Scholar]
- [22].Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem 1998; 273:10777-83; PMID:9553144; http://dx.doi.org/ 10.1074/jbc.273.17.10777 [DOI] [PubMed] [Google Scholar]
- [23].Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/ 10.1038/embor.2008.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al.. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/ 10.1038/sj.emboj.7601689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497-510; PMID:18443221; http://dx.doi.org/ 10.1083/jcb.200712064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005; 19:1294-305; PMID:15901672; http://dx.doi.org/ 10.1101/gad.1304105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 2011; 44:517-31; PMID:22036586; http://dx.doi.org/ 10.1016/j.molcel.2011.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gao P, Bauvy C, Souquere S, Tonelli G, Liu L, Zhu Y, Qiao Z, Bakula D, Proikas-Cezanne T, Pierron G, et al.. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J Biol Chem 2010; 285:25570-81; PMID:20529838; http://dx.doi.org/ 10.1074/jbc.M110.118125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008; 15:1318-29; PMID:18421301; http://dx.doi.org/ 10.1038/cdd.2008.51 [DOI] [PubMed] [Google Scholar]
- [31].Grishchuk Y, Ginet V, Truttmann AC, Clarke PG, Puyal J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011; 7:1115-31; PMID:21646862; http://dx.doi.org/ 10.4161/auto.7.10.16608 [DOI] [PubMed] [Google Scholar]
- [32].Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 2010; 17:268-77; PMID:19713971; http://dx.doi.org/ 10.1038/cdd.2009.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang C, Youle RJ. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1's inhibitory effect on Bak. Oncogene 2012; 31:3177-89; PMID:22056880; http://dx.doi.org/ 10.1038/onc.2011.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Harris AW, Bankhurst AD, Mason S, Warner NL. Differentiated functions expressed by cultured mouse lymphoma cells. II. Theta antigen, surface immunoglobulin and a receptor for antibody on cells of a thymoma cell line. J Immunol 1973; 110:431-8; PMID:4630643 [PubMed] [Google Scholar]
- [35].Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold MJ. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep 2015; 10(8):1422-32; PMID:25732831 [DOI] [PubMed] [Google Scholar]
- [36].WANG LE. (Us) dgu, wang xilu (us), tao zhi-fu (us), brunko milan (us), kunzer aaron r (us), wendt michael d (us), song xiaohong (us), frey robin (us), hansen todd m (us), sullivan gerard m (us), judd andrew (us), souers andrew (us). 8 - carbamoyl - 2 - (2,3- di substituted pyrid - 6 - yl) −1,2,3,4 -tetrahydroisoquinoline derivatives as apoptosis - inducing agents for the treatment of cancer and immune and autoimmune diseases. Abbvie inc. (1 North Waukegan Road, North Chicago, Illinois, 60064, US), 2013. (application number: WO/2013/055897). [Google Scholar]
- [37].Smith BJ, Lee EF, Checco JW, Evangelista M, Gellman SH, Fairlie WD. Structure-guided rational design of alpha/beta-peptide foldamers with high affinity for BCL-2 family prosurvival proteins. Chembiochem 2013; 14:1564-72; PMID:23929624; http://dx.doi.org/ 10.1002/cbic.201300351 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.