

ABSTRACT
Hypoxia induces protective autophagy in glioblastoma cells and new therapeutic avenues that target this process may improve the outcome for glioblastoma patients. Recent studies have suggested that the autophagic process is upregulated in glioblastomas in response to extensive hypoxia. Hypoxia also induces the upregulation of a specific set of proteins and microRNAs (miRNAs) in a variety of cell types. IL6 (interleukin 6), an inflammatory autocrine and paracrine cytokine that is overexpressed in glioblastoma, has been reported to be a biomarker for poor prognosis because of its tumor-promoting effects. Here, we describe a novel tumor-promoting mechanism of IL6, whereby hypoxia-induced IL6 acts as a potent initiator of autophagy in glioblastoma via the phosphorylated (p)-STAT3-MIR155-3p pathway. IL6 and p-STAT3 levels correlated with the abundance of autophagic cells and HIF1A levels in human glioma tissues and with the grade of human glioma, whereas inhibition of exogenous or endogenous IL6 repressed autophagy in glioblastoma cells in vitro. Knockdown of endogenous MIR155-3p inhibited IL6-induced autophagy, and enforced expression of MIR155-3p restored the anti-autophagic activity of IL6 inhibitors. We show that the hypoxia-IL6-p-STAT3-MIR155-3p-CREBRF-CREB3-ATG5 pathway plays a central role in malignant glioma progression, with blockade of the IL6 receptor by tocilizumab demonstrating a certain level of therapeutic efficacy in a xenograft model in vivo, especially in combination with temozolomide. Moreover, tocilizumab inhibits autophagy by promoting tumor apoptosis. Collectively, our findings provide new insight into the molecular mechanisms underlying hypoxia-induced glioma cell autophagy and point toward a possible efficacious adjuvant therapy for glioblastoma patients.
KEYWORDS: ATG5, autophagy, CREB3, CREBRF, glioblastoma, hypoxia, IL6, microRNAs, STAT3, tocilizumab
Introduction
Autophagy, which means “self-eating,” is a process that involves the formation of double-membrane vesicles containing damaged or old organelles (autophagosomes). After fusion with the lysosome (generating an autolysosome), autophagy provides cells with reclaimed essential elements for survival during starvation, hypoxia, immune response and chemoradiotherapy.1 Because cancer cells are overconsumptive, these cells tend to develop ubiquitous autophagy to cope with the relatively infertile tumor microenvironment.2-4 Although the autophagic process in normal cells functions as a mechanism that prevents tumorigenesis through the selective cleanup of damaged organelles and specific oncogene proteins, this process is a double-edged sword—autophagy acts as a cytoprotective mechanism in established, advanced tumor cells that have increasing metabolic demands that need to be met.5,6 Indeed, accumulating evidence demonstrates that autophagy favors tumor development, and this situation is accordingly a topic of oncological interest.7
IL6 (interleukin 6), an inflammatory autocrine and paracrine cytokine overexpressed in glioblastoma multiforme (GBM) and many other malignant tumors,8-13 has been reported to be a biomarker for poor prognosis in patients with various types of tumors, such as GBM.12,13 This observation may be partially explained by the capacity of IL6 to promote proliferation,9,11,14 angiogenesis,15 apoptosis,9 invasion and migration in different malignancies.10 However, the manner in which IL6 contributes to tumor progression by initiating autophagy has yet to be thoroughly investigated.
Because of the rapid growth progression and a relatively inadequate blood supply, hypoxia is a common feature in solid tumors, and tumor hypoxia is an independent prognostic factor associated with poor survival.16 Although numerous studies have suggested that hypoxia activates multiple cellular processes in tumors, such as proliferation,17 angiogenesis,18,19 migration,20 invasion,21 and autophagy,22,23 the mechanisms by which hypoxia induces autophagy remain to be elucidated in detail.
Gliomas are the most malignant type of brain tumor, accounting for more than 70% of all brain tumors.24 GBM is the most common type, with an age-adjusted incidence rate that ranges from 0.59 to 3.69 per 100,000 people.25 There appears to be general agreement that the properties of GBM largely consist of high mortality and recurrence rates,26 uncontrollable invasiveness,27 strong angiogenesis,28 a widespread hypoxic region,16,29,30 increased IL6 expression,8,13 and upregulated autophagic processes.31 Despite the multitude of mechanisms that have been proposed to explain the hypoxia-induced autophagy of tumor cells, there are no studies evaluating the impact of prominently expressed IL6 on this cellular event.
Members of the STAT (signal transducer and activator of transcription) protein family are important transducers of many cytokines and growth factors in virtually all tumors.32 Among the 7 members of the family, STAT3 is the one most commonly constitutively activated in several cancers, including GBM.33 It is well accepted that STAT3 serves as a downstream target of IL6, which induces STAT3 activation through tyrosine 705 phosphorylation and cytoplasmic-to-nuclear shuttling, recognition of STAT3-specific DNA motifs, and transcriptional activation of target genes that are implicated throughout tumor development and involved in proliferation, survival, invasion and angiogenesis.34,35 Recent studies have demonstrated that STAT3 also regulates the expression of several microRNAs (miRNAs). miRNAs comprise a class of noncoding endogenous RNAs that regulate the expression of genes that participate in almost every biological process by binding to the 3′-untranslated region (3′-UTR) of target protein-coding mRNA molecules.36 Moreover, several miRNAs have been well characterized as being involved in autophagy regulation.37 Indeed, there is increasing evidence that miRNAs might be the link between hypoxia-induced IL6 levels and the enhancement of tumor autophagy.
CREBRF/LRF (CREB3 regulatory factor) is a novel cellular protein and a negative regulator of CREB3/Luman/LZIP (cAMP responsive element binding protein 3) that can recruit nuclear CREB3 to discrete foci in the nucleus, promote CREB3 protein degradation and repress CREB3-mediated activation of unfolded protein response element-containing promoters.38 CREB3 is the primary member of the CREB3 family. All CREB3 family members appear to play a role in the unfolded protein response,39 during which endoplasmic reticulum (ER)-resident molecular chaperones and foldases are induced, attenuating translation to reduce the load on the ER.40 Unfolded proteins can also be targeted for proteasomal degradation via ubiquitination.41 CREB family proteins, including CREB1, CREB2 and CREB3, bind to the cAMP-responsive element recognition sequence of cAMP-sensitive genes to regulate transcription. Although it has been reported that CREB1 can upregulate autophagy genes,42,43 and that CREBRF is involved in inducing cell apoptosis through the ER stress pathway,44 there is no evidence to date that the CREBRF-CREB3 pathway is involved in regulating autophagy in tumor cells.
In the present study, we report a novel tumor-promoting mechanism of IL6 in which hypoxia-induced IL6 promotes tumor cell survival by upregulating autophagy in GBM through the p-STAT3-MIR155-3p pathway. We first investigated the considerable initiating effect of IL6 during the hypoxia process, and we found that hypoxic pretreatment of tumor cells induced significant IL6 expression and autophagy activation. More importantly, the application of exogenous IL6 increased autophagic activity, whereas knocking down endogenous IL6 or treatment with IL6 antibodies alleviated hypoxia-induced autophagy. To understand the mechanisms of the autophagy induced by IL6, we screened the entire complement of genomic miRNAs using gene chips (Human miRCURY™ LNA expression array). Analysis of the data revealed dramatic changes in multiple molecules under hypoxia, especially those related to IL6 and autophagy. Based on these results, we selected the molecules downstream of IL6 implicated in the autophagic process for further examination. Finally, we provide evidence that the p-STAT3-MIR155-3p pathway plays a central role in the impact of IL6. Our results suggest potential uses for anti-IL6 therapeutic strategies in adjuvant therapy for glioma patients. In a broader sense, the data also support the application of a monoclonal antibody to block the hypoxia-IL6-p-STAT3-MIR155-3p-CREBRF-CREB3-ATG5 pathway at the source. Several attempts have been made to therapeutically regulate the autophagic-promoting activity of IL6, and we were encouraged by the fact that tocilizumab, a novel monoclonal antibody injectable for rheumatoid arthritis that targets IL6, could achieve this therapeutic effect through specific blockade of the IL6 receptor. We conducted a series of successful tocilizumab therapeutic experiments using glioma cell xenograft mouse models, especially in combination with temozolomide; however, further clinical investigations are needed.
Results
IL6 levels correlated with the density of autophagic cells, HIF1A levels in human glioma tissues and the grade of human glioma
Extensive hypoxic areas have been found in glioma tissues;45 however, the relationship between IL6 and the level of autophagy in the hypoxic areas of tumors remains unclear. In fact, a causal link between IL6 and LC3B levels and hypoxia levels in gliomas has not been reported, and it is also unknown whether IL6 is involved in the upregulation of autophagy in such hypoxic areas. To examine whether the expression of IL6 correlates with autophagy and the level of hypoxia in human glioma, we performed immunohistochemical staining to detect the expression of IL6, STAT3 and p-STAT3 (the primary downstream signaling molecules of IL6), LC3B (a marker of autophagy) and HIF1A in 101 human glioma specimens with different grades and 3 normal brain tissues (Table 1). As shown in Fig. 1A, the expression of IL6, p-STAT3, HIF1A, and LC3B positively correlated with the WHO grade of glioma (Fig. 1C), and Pearson's or Spearman's rho rank correlation tests showed that IL6, p-STAT3, HIF1A, LC3B and grade were positively correlated. Most correlation indexes were approximately 0.7 to 0.8, indicating a strong positive relationship (Fig. 1D). In addition, the expression of the IL6-p-STAT3 pathway strongly correlated with LC3B and HIF1A levels; consistent with this finding, the abundance of autophagic cells was also associated with HIF1A levels (Fig. 1A). Furthermore, immunohistochemical staining of continuous paraffin sections revealed the colocalization of the IL6, p-STAT3, HIF1A, and LC3B proteins in high-grade glioma tissues (Fig. S1), particularly in the hypoxic area around tumor vessels (Fig. 1B, the enlarged view of the lower right green box in Fig. S1). The maximum diffusion distance of the oxygen supply around the tumor vessel in solid tumors was approximately 100 to 150 μm,46 which agreed with our staining results and illustrated the hypoxic regions around tumor vessels (Fig. 1B). Taken together, these results demonstrate that IL6 expression correlates with the density of autophagic cells and HIF1A levels in high-grade (WHO III and IV) gliomas.
Figure 1.

Levels of colocalized IL6, HIF1A, LC3B and p-STAT3 positively correlate with the WHO grade in human glioma tissues. (A) The expression of IL6, HIF1A, p-STAT3, and LC3B positively correlated with the WHO grade of gliomas. Ninety human gliomas and 3 normal brain tissue samples of different WHO grades were processed by immunohistochemistry. STAT3 levels were basically stable. (B) Immunohistochemical staining of continuous paraffin sections revealed the colocalization of IL6, p-STAT3, HIF1A, and LC3B proteins in the hypoxic area around the tumor vessels of a sample of high-grade glioma tissues (WHO III) (enlarged view of the lower right green box in Fig. S1). (C) Quantification of IHC staining of glioma tissues with different WHO grades. IRS, immunoreactive score. (D) Pearson's or Spearman's rho rank correlation tests of IL6, p-STAT3, HIF1A, LC3B and grades.
To further investigate tumor-associated cells in this hypoxic glioma microenvironment and to clarify the cellular sources of IL6, we collected 5 freshly removed, surgical WHO grade IV glioma tissue samples for fast-frozen sectioning, and staining revealed most of the tumor-associated cells in the microenvironment, including T cells (CD3), macrophages (CD68, CD11B), microglia (CD68), M1 tumor-associated macrophages (CD11B, CD16), M2 tumor-associated macrophages (CD206), tumor-associated dendritic cells (CD11C), monocytes and some NK cells (CD16) (Fig. S2A). These tumor-associated cells accounted for approximately 30% of all cells in the glioma tissues, as some of the markers overlapped. Importantly, the IL6-positive cells accounted for over 90% of all cells. As IL6 can be derived from many sources, including glioma cells and macrophages, we double stained these markers and found that the IL6 was primarily secreted from tumor cells (Fig. S2C), with only 25% of the total amount of IL6 being secreted by nontumor immune cells (Fig. S2D). Thus, glioma cells are clearly stimulated by strong IL6 signals in a complex hypoxic microenvironment.
Hypoxia induces autophagy activation and IL6 upregulation in glioblastoma cells
It is generally accepted that hypoxia activates autophagy as an evolutionarily conserved cellular catabolic process.23 To verify this, we performed a GFP-LC3B puncta-formation assay and an LC3B conversion assay. Using GBM cells (U251) stably expressing a GFP-LC3B fusion protein, GFP-LC3B localization was examined by fluorescence microscopy. GFP-LC3B puncta appearing in the cytoplasm reflect the recruitment of the LC3B protein to phagophores, the precursor to autophagosomes. As shown in Fig. 2A, there was a significant increase in GFP-LC3B puncta in hypoxic cells, a result that was confirmed by the quantification of GFP-LC3B foci per cell. Moreover, we detected the conversion of LC3B-I to LC3B-II and the degradation of SQSTM1/p62 (autophagy markers) by western blotting. Consistent with the GFP-LC3B puncta-formation assay, hypoxia led to a significant time-dependent upregulation of LC3B-II and downregulation of SQSTM1 (Fig. 2B). Thus, both assays suggested that hypoxia induces autophagosome accumulation. In addition, treatment of normoxic and hypoxic cells with 3-methyladenine (3-MA) inhibited the autophagy (Fig. 2C),47 as previously observed, whereas treatment of normoxic and hypoxic cells with bafilomycin A1 (BAF) blocked autophagic flux and resulted in an increase in hypoxia-induced autophagy (Fig. 2C).48 To investigate the function of IL6 in hypoxia-induced autophagy, we used ELISA to examine the secretion of IL6 in a GBM cell culture supernatant (U251) under hypoxic stress. As shown in Fig. 2D, the secretion level of IL6 was low under normal culture conditions (21% oxygen), but hypoxia (1% oxygen) treatment induced a significant increase in IL6 secretion.
Figure 2.
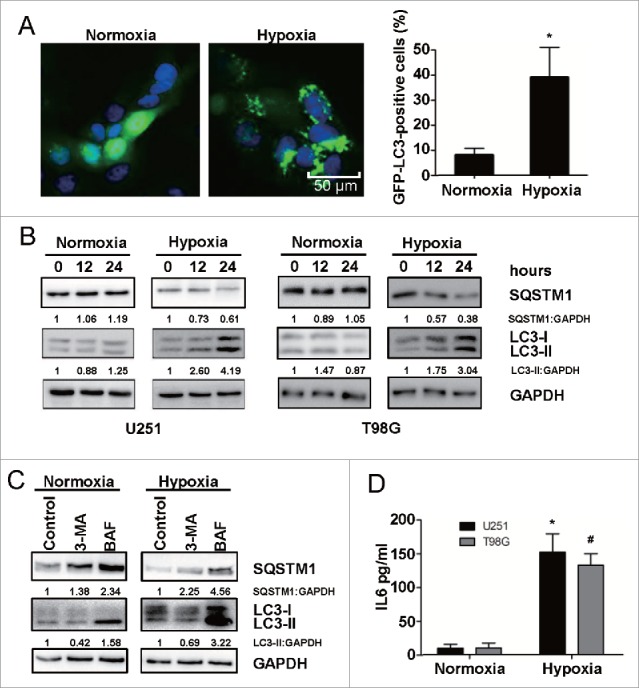
Hypoxia induces autophagy activation and upregulates IL6 in human glioma cells. (A) Hypoxia promotes GFP-LC3B translocation. pSELECT-GFP-LC3B transfection revealed LC3B puncta in U251 cells treated with hypoxia (1% O2, 5% CO2, and 94% N2 at 37°C) for 24 h. Cells were fixed and stained with DAPI for nuclear visualization. Representative images are shown. Scale bar: 50 μm. Quantitative analysis of GFP-LC3B puncta is shown in the right panel. At least 100 high-power fields were examined in each experimental group. The data shown are the mean ± s.d. of 4 independent experiments. *, P < 0.0001; 2-tailed t test. (B) Hypoxia induced LC3B conversion and SQSTM1 degradation in U251 and T98G cells. LC3B and SQSTM1 levels were examined by western blot analysis in GBM cells after hypoxia treatment (1% O2, 5% CO2, and 94% N2 at 37°C) for 12 and 24 h. GAPDH served as the loading control. (C) Western blot analysis showing that 3-methyladenine (3-MA) inhibited autophagy in U251 cells and treatment of normoxic and hypoxic cells with bafilomycin (BAF) blocked autophagic flux. (D) Hypoxia increased the secretion of IL6 in U251 and T98G cell culture supernatants, as revealed by ELISA. The data shown are the mean ± s.d. of 3 independent experiments. * and #, P < 0.001; 2-tailed t test.
Exogenous IL6 triggers autophagy and inhibition of endogenous IL6 represses autophagy in glioblastoma cells
An increasing number of studies have shown that cancer cells, including GBM cells, undergo autophagy in response to hypoxia,45 yet it remains unknown whether hypoxia-induced IL6 is a central mediator of hypoxia-induced autophagy. Thus, we examined whether IL6 could independently induce autophagy in GBM cells by utilizing GFP-LC3B transient transfection, the LC3B conversion assay and transmission electron microscopy to visualize the aggregation of expressed LC3B in U251 cells. We determined the induction of autophagy through the localization of the autophagosome-specific protein LC3B using GFP-LC3B transient transfection. As shown in Fig. 3A, we observed abundant autophagosomes in IL6-treated cells. Moreover, a western blot analysis was performed to examine whether IL6 treatment induced the processing of LC3B-I to LC3B-II, and the level of the latter increased with the duration of IL6 treatment (Fig. 3B). This effect was observed to occur in a dose-dependent manner in IL6 cells treated for 24 h. In contrast, the opposite trend was observed for SQSTM1 (Fig. 3D), the downregulation of which indicates the activation of autophagy.49 Transmission electron microscopy also revealed abundant characteristic autophagosomes in IL6-treated U251 cells (Fig. 3C). To further support the fact that both endogenous IL6 and exogenous IL6 trigger autophagy, we examined LC3B-II and SQSTM1 protein levels after attenuating IL6 activity by small interfering RNA (siRNA) and the use of a recombinant human antibody. The data showed that a blockade of both endogenous and exogenous IL6 repressed autophagy in GBM cells (Fig. 3E, F). Furthermore, to test whether the IL6-induced increase in LC3B-II was due to autophagy induction or the inhibition of autolysosome function, BAF was used to inhibit autophagic flux. As shown in Fig. 3G, although LC3B-II levels increased in BAF-treated cells due to inhibition of LC3B-II lysosomal degradation, the LC3B-II level was even higher in cells additionally treated with IL6. We also found that the expression of SQSTM1 was decreased in IL6-treated cells in the absence of BAF, suggesting that autophagy was activated and that SQSTM1 was degraded via autophagy. The elevated level of SQSTM1 in cells treated with BAF and IL6 indicated that autophagy was blocked by BAF and that SQSTM1 accumulated in GBM cells (Fig. 3G).
Figure 3.

IL6 induces autophagy activation, and inhibition of IL6 represses autophagy in human glioma cells. (A) Exogenous IL6 promotes GFP-LC3B translocation. pSELECT-GFP-LC3B-transfected U251 cells treated with IL6 (20 ng/ml) for 24 h. Scale bar: 50 μm. Quantitative analysis of GFP-LC3B puncta is shown in the right panel. The data shown are the mean ± s.d. of 4 independent experiments. *, P < 0.0001; 2-tailed t test. (B) Exogenous IL6 induced LC3B conversion and STAT3 activation in U251 and T98G cells. LC3B, STAT3 and p-STAT3 levels were examined by western blot analysis in GBM cells after treatment with IL6 (20 ng/ml) for 0, 12, 24 h. GAPDH served as the loading control. (C) Images from transmission electron microscopy showing characteristic autophagosomes (arrows) in U251 cells after treatment with IL6 (20 ng/ml) for 24 h. N, nucleus. At least 50 cells were examined in each experimental group. The data shown are the mean ± s.d. of 3 independent experiments. *, P < 0.0001; 2-tailed t test. (D) Exogenous IL6 induces LC3B conversion and SQSTM1 degradation in GBM cells in a dose-dependent manner after a 24-h treatment. Inhibition of endogenous IL6 (E) and exogenous IL6 (F) represses LC3B conversion and SQSTM1 degradation in GBM cells as measured by western blot analysis. (G) Autophagic flux inhibition of IL6-treated U251 cells with bafilomycin A1 (BAF). ACTB served as the loading control.
Activation of the IL6-p-STAT3 pathway is involved in hypoxia-induced autophagy in glioblastoma cells
As discussed above, IL6 was shown to exert a facilitative influence on the activation of autophagy in GBM cells, and hypoxia could elevate IL6 secretion, suggesting that IL6 may be a crucial mediator of hypoxia-induced autophagy. Using U251 cells transiently transfected with GFP-LC3B, we found that under normoxic conditions, exogenous IL6 induced intense autophagy, similar to hypoxia alone (Fig. 4A, C). Moreover, we observed that a recombinant human IL6 antibody dramatically impaired IL6-induced autophagy and even hypoxia-induced autophagy (Fig. 4). The results of western blot analysis suggested that the IL6 antibody inhibited hypoxia-induced autophagy by blocking the IL6-p-STAT3 pathway (Fig. 4B and D). Additionally, IL6 siRNA against endogenous IL6 also blocked activation of the IL6-p-STAT3 pathway and hypoxia-induced autophagy in glioblastoma cells (Fig. S3).
Figure 4.
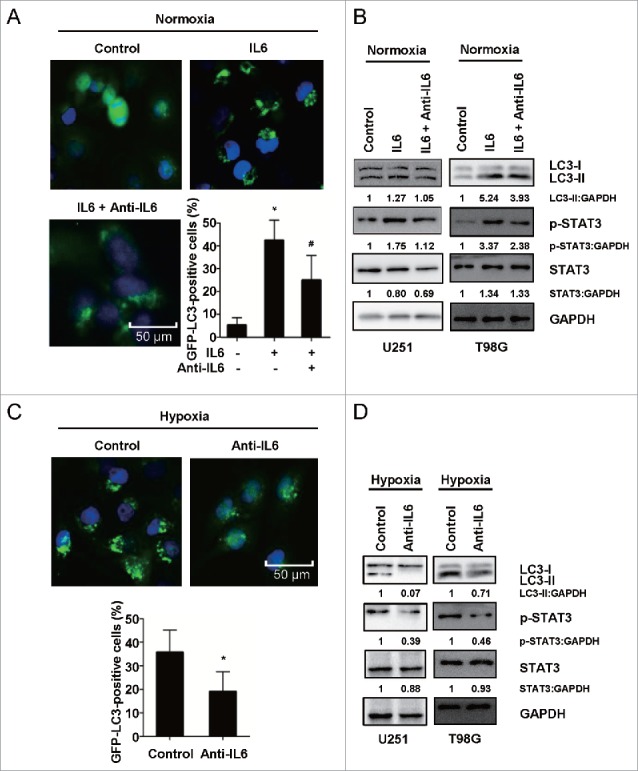
Activation of the IL6-p-STAT3 pathway is involved in hypoxia-induced autophagy in glioblastoma cells. (A) An antibody against exogenous IL6 inhibited GFP-LC3B translocation. pSELECT-GFP-LC3B-transfected U251 cells treated with IL6 (20 ng/ml) and an IL6 antibody (1 μg/ml) for 24 h. Scale bar: 50 μm. Quantitative analysis of GFP-LC3B puncta is shown in the right panel. The data shown are the mean ± s.d. of 4 independent experiments. * and #, P<0.001; one-way ANOVA. (B) An antibody against exogenous IL6 inhibited LC3B conversion and STAT3 activation in U251 and T98G cells. LC3B, STAT3 and p-STAT3 levels were examined by western blot analysis in GBM cells after treatment with IL6 (20 ng/ml) and an IL6 antibody (1 μg/ml) for 24 h. GAPDH served as the loading control. (C) An antibody against exogenous IL6 inhibited GFP-LC3B translocation in hypoxic U251 cells. pSELECT-GFP-LC3B-transfected U251 cells treated with IL6 antibody (1 μg/ml) for 24 h under hypoxic conditions. Scale bar: 50 μm. The quantitative analysis of GFP-LC3B puncta is shown in the right panel. The data shown are the mean ± s.d. of 4 independent experiments. *, P < 0.0001; 2-tailed t test. (D) An antibody against exogenous IL6 inhibited LC3B conversion and STAT3 activation in hypoxic U251 and T98G cells. LC3B, STAT3 and p-STAT3 levels were examined by western blot analysis after treatment of hypoxic GBM cells with an IL6 antibody (1 μg/ml) for 24 h. GAPDH served as the loading control.
MiR155-3p is involved in IL6-induced autophagy in hypoxic glioblastoma cells
Because several miRNAs have been well characterized as modulators of autophagy and hypoxia is an independent autophagy-promoting factor, we used a normoxic and hypoxic U251 cell miRNA microarray to identify hypoxia-induced miRNAs. These data revealed 84 significantly differentially expressed miRNAs, including MIR210, a novel miRNA marker of hypoxia, and MIR155-3p, the homolog of which (MIR155-5p) is a potent inducer of autophagy via MTOR pathway targeting (Fig. 5A and C).37 We further verified the levels of MIR155-3p in hypoxic U251 cells by quantitative real-time PCR, and the validated expression results were consistent with the microarray results. MIR155-3p expression was time dependent in hypoxia-treated U251 cells (Fig. 5B) and dose dependent in IL6-treated cells (Fig. 5D). To further investigate whether MIR155-3p and IL6 are linked, we utilized siRNA and a recombinant human antibody that has been previously demonstrated to interfere with IL6. As shown in Figure 5D and E , suppression of IL6 significantly reduced MIR155-3p expression.
Figure 5.
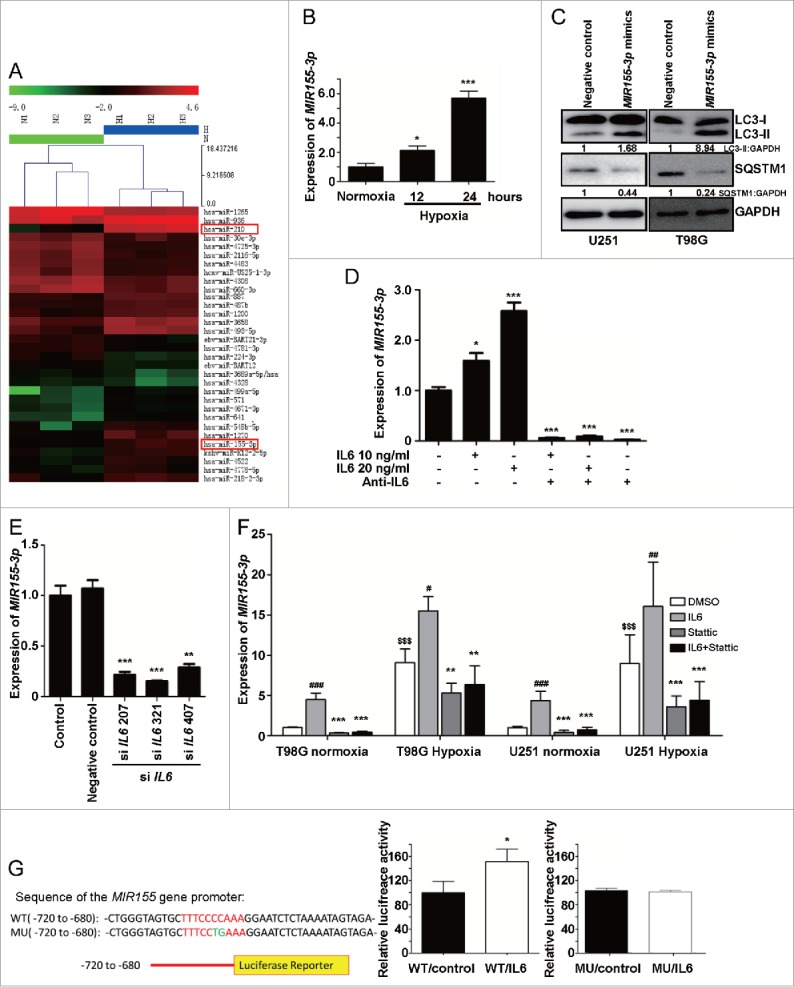
MIR155-3p is upregulated by hypoxia, and IL6 can induce autophagy in glioblastoma cells. (A) The miRCURY™ RNA expression array revealed 84 significantly differentially expressed miRNAs (partial data shown in Fig. 5A) between normoxic and hypoxic U251 cells. The hypoxic miRNA marker MIR210 and the target miRNA MIR155-3p are indicated. (B) The expression levels of MIR155-3p in hypoxic U251 cells (hypoxia treatment for 0, 12, and 24 h) were assessed by quantitative real-time PCR. The data shown are the mean ± s.d. of 5 independent experiments. *, P < 0.05; ***, P < 0.0001; one-way ANOVA. (C) MIR155-3p overexpression induced LC3B conversion and SQSTM1 degradation in U251 and T98G cells at 48 h after MIR155-3p mimic transfection, as shown by western blot analysis. GAPDH served as the loading control. (D) Exogenous IL6 upregulated and an antibody against exogenous IL6 inhibited the expression levels of MIR155-3p, as determined by quantitative real-time PCR. The data shown are the mean ± s.d. of 5 independent experiments. *, P < 0.01; ***, P < 0.0001; one-way ANOVA. (E) Knockdown of endogenous IL6 inhibited the expression of MIR155-3p, as determined by quantitative real-time PCR. (F) The specific p-STAT3 inhibitor Stattic inhibited the function of STAT3. (G) A “5′-TTTCCCCAAA-3′” p-STAT3-binding element is present at −731 in the MIR155-3p promoter. Mutation of the p-STAT3-binding element eliminated the promoting effect of the IL6-p-STAT3 pathway on MIR155-3p expression compared with the wild-type element. The data shown are the mean ± s.d. of 5 independent experiments. * and #, P < 0.01; ** and ##, P < 0.001; ***, ### and $$$, P < 0.0001; * by Student's t-test for Stattic groups versus IL6 groups, # by the Student t test for IL6 groups vs. DMSO control groups, and $ by Student t test for hypoxia groups versus normoxia groups.
To further examine the relationship between p-STAT3 and MIR155-3p, we utilized a specific p-STAT3 inhibitor, Stattic, to inhibit the function of STAT3. We found that Stattic blocked the IL6- and hypoxia-induced upregulation of MIR155-3p in U251 and T98G glioma cells (Fig. 5F). We then searched for p-STAT3-binding elements in the sequence of the MIR155-3p promoter and identified a “5′-TTTCCCCAAA-3′” element at −731. To assess the direct promoting effect of p-STAT3 on the MIR155-3p promoter, p-STAT3-binding element mutation and luciferase reporter assays were performed using U251 glioma cells. Mutation of the p-STAT3-binding element eliminated the promoting effect of the IL6-p-STAT3 pathway on MIR155-3p expression compared with the wild-type element (Fig. 5G). These results suggested that MIR155-3p may be involved in regulating the autophagic process downstream of the IL6-p-STAT3 pathway.
MIR155-3p knockdown antagonizes hypoxia-induced autophagy and the pro-autophagic effects of IL6 on human glioma cells
To investigate whether the downregulation of MIR155-3p could affect hypoxia-mediated glioma cell autophagy, we used a MIR155-3p inhibitor. miRNA inhibitors are synthetic, 2′-O-methyl-modified, single-stranded molecules that interfere with miRNA function by sequestering these molecules via irreversible binding.50,51 The GFP-LC3B puncta-formation assay showed that the MIR155-3p inhibitor significantly suppressed U251 cell autophagy under hypoxic conditions, an effect that was partially rescued by exogenous IL6 (Fig. 6A). The results of western blot analysis clearly indicated that MIR155-3p knockdown blocked the IL6-induced increase in LC3B-II as well as SQSTM1 degradation under hypoxic conditions (Fig. 6B).
Figure 6.
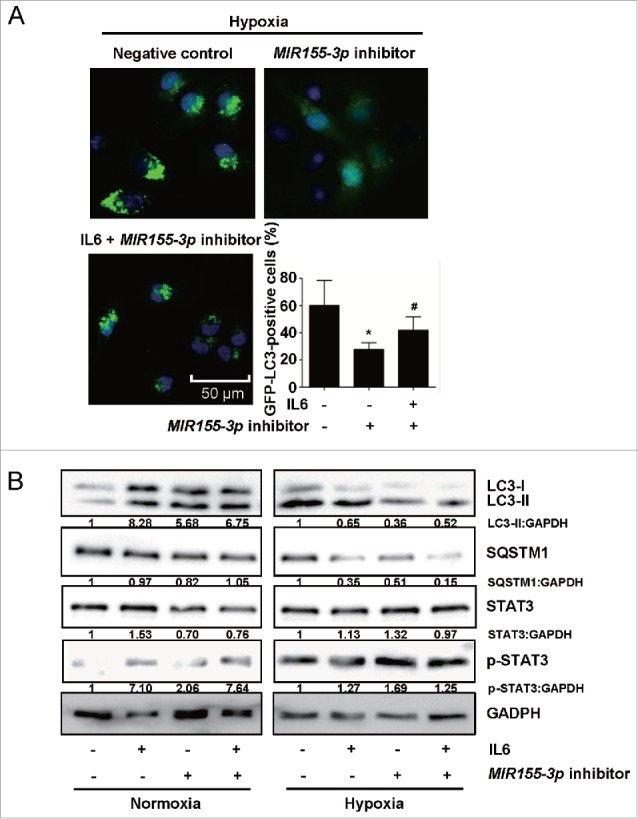
MIR155-3p knockdown antagonizes hypoxia-induced autophagy in human glioma cells. (A) The MIR155-3p inhibitor suppressed GFP-LC3B translocation. pSELECT-GFP-LC3B and MIR155-3p inhibitor-cotransfected U251 cells were treated with IL6 (20 ng/ml) for 24 h. Scale bar: 50 μm. The quantitative analysis of GFP-LC3B puncta is shown in the right panel. The data shown are the mean ± s.d. of 4 independent experiments. * and #, P < 0.001; one-way ANOVA. (B) MIR155-3p inhibition suppresses LC3B conversion and STAT3 activation in U251 and T98G cells. LC3B, SQSTM1, STAT3 and p-STAT3 levels were examined by western blot analysis in GBM cells after treatment with IL6 (20 ng/ml) for 24 h. GAPDH served as the loading control.
MIR155-3p enhances hypoxia-induced autophagy and rescues the anti-autophagic effects of IL6 inhibition on human glioma cells
To further understand the pro-autophagic role of MIR155-3p, we investigated the effects of its overexpression using miRNA mimics, which consist of synthetic, double-stranded, modified RNA molecules that imitate the functions of endogenous miRNAs.52 The GFP-LC3B puncta-formation assay showed that the MIR155-3p mimics further enhanced glioma cell autophagy under hypoxic conditions, partially rescuing the anti-autophagic effects of IL6 siRNA and the recombinant human IL6 antibody (Fig. 7A). Western blot analysis of the LC3B conversion assay showed that overexpression of MIR155-3p had pro-autophagic effects similar to exogenous IL6 and rescued the anti-autophagic effects of the IL6 antibody and siRNA by upregulating LC3B-II levels and downregulating SQSTM1 levels, respectively, particularly under hypoxic conditions (Fig. 7B).
Figure 7.
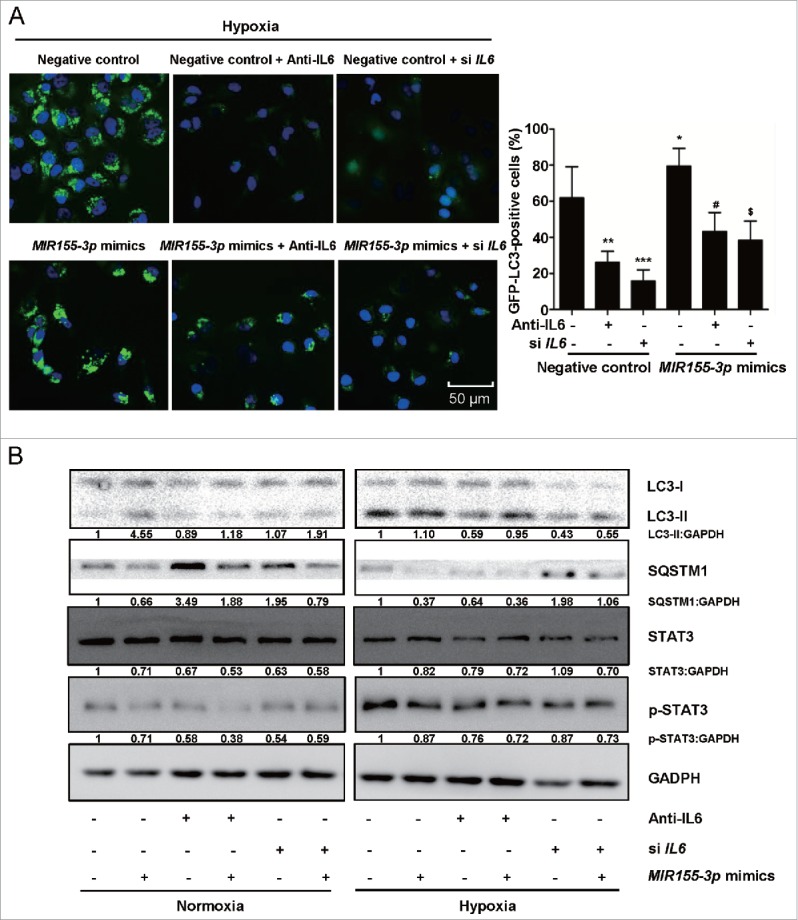
MIR155-3p overexpression enhances hypoxia-induced autophagy and resists the effects of IL6 inhibition on human glioma cells. (A) MIR155-3p mimic promoted GFP-LC3B translocation. pSELECT-GFP-LC3B and MIR155-3p mimic-co-transfected U251 cells treated with the IL6 antibody (1 μg/ml) and siRNAs (Si IL6 321) for 24 h. Scale bar: 50 μm. Quantitative analysis of GFP-LC3B puncta is shown in the right panel. The data shown are the mean ± s.d. of 4 independent experiments. *, # and $, P < 0.01; **, P < 0.001; ***, P < 0.0001; one-way ANOVA. (B) The MIR155-3p mimic induced LC3B conversion and STAT3 activation in U251 and T98G cells. LC3B, SQSTM1, STAT3 and p-STAT3 levels were examined by western blot analysis. GAPDH served as the loading control.
MIR155-3p enhances hypoxia-induced autophagy by directly targeting the CREBRF-CREB3-ATG5 pathway in human glioma cells
Next, we searched for potential MIR155-3p target genes in miRDB (http://mirdb.org/miRDB/index.html) and identified the best potential target gene, CREBRF, with a full target score and 3 perfectly strong binding seed sequences (http://mirdb.org/cgi-bin/search.cgi?searchType=miRNA&full=mirbase&searchBox=MIMAT0004658). To assess the direct inhibitory effect of MIR155-3p on CREBRF gene transcription, 3′-UTR seed mutation and 3′-UTR luciferase assays were performed using U251 glioma cells (Fig. 8A). The luciferase activity in MIR155-3p-transfected cells decreased to approximately half of the activity observed using the control miRNA (Fig. 8B). To examine the inhibitory effect of MIR155-3p at the protein level, we performed western blotting at 48 h after MIR155-3p inhibitor and mimic transfection into U251 and T98G cells and observed significantly decreased CREBRF protein levels in hypoxia-treated U251 and T98G cells. The level of CREBRF protein was decreased significantly in MIR155-3p mimic-transfected glioma cells compared to those transfected with the negative control miRNA but increased significantly in MIR155-3p inhibitor-transfected glioma cells (Fig. 8C). As CREBRF is a negative regulator of CREB3, the CREB3 level showed an opposite trend (Fig. 8C).
Figure 8.

MIR155-3p enhances hypoxia-induced autophagy by directly targeting the CREBRF-CREB3-ATG5 pathway in human glioma cells. (A) The best potential MIR155-3p target gene, CREBRF, in miRDB, and the 3′-UTR seed mutation. (B) The 3′-UTR seed mutation and 3′-UTR luciferase assays in U251 glioma cells. The luciferase activity in MIR155-3p-transfected cells decreased to approximately half of the activity observed using the control miRNA. (C) The inhibitory effect of MIR155-3p at the protein level of CREBRF and CREB3. We performed western blotting at 48 h after MIR155-3p inhibitor and mimic transfection into U251 and T98G cells and observed significantly decreased CREBRF protein levels in hypoxia-treated U251 and T98G cells. As CREBRF is a negative regulator of CREB3, the CREB3 level showed an opposite trend. (D) The CREB3 siRNA knockdown decreases autophagy levels and ATG5 protein levels of U251 and T98G cells. (E) and (F) The key pathway proteins in primary glioma cells by western blotting. (G) The CREB3-induced autophagy-related (ATG) genes in U251 and T98G cells by quantitative real-time PCR. (H) CREB3 siRNA inhibits GFP-LC3B translocation. pSELECT-GFP-LC3B-transfected U251 cells treated with IL6 (20 ng/ml) for 24 h. Scale bar: 50 μm. Quantitative analysis of GFP-LC3B puncta is shown in the right panel. The data shown are the mean ± s.d. of 4 independent experiments. * and #, P < 0.05; ** and ##, P < 0.01; 2-tailed t test. 155i, MIR155-3p inhibitor; 155m, MIR155-3p mimic; NC, negative control.
To further test the relationship between the autophagy level and MIR155-3p-induced CREB3, we utilized a specific CREB3 siRNA for knockdown. As expected, the cells transfected with CREB3 siRNA exhibited a reduction in CREB3 protein and significantly reduced autophagy under conditions of both normoxia and hypoxia (Fig. 8D). To elucidate the mechanisms of CREB3-induced autophagy, several autophagy-related (ATG) genes were examined by quantitative real-time PCR in T98G and U251 cells, revealing a significant decrease in ATG5 after CREB3 knockdown (Fig. 8G) that was confirmed by western blot in both cell lines (Fig. 8D). However, ATG3, which was only decreased in U251 cells, and other ATGs exhibited no change at the protein level (data not shown).
Furthermore, we confirmed the changes in those key pathway proteins in primary glioma cells by western blotting (Fig. 8E and F). In conclusion, it is tempting to postulate that the hypoxia-IL6-p-STAT3-MIR155-3p-CREBRF-CREB3-ATG5 pathway plays a pivotal role in hypoxia-induced autophagy in glioma cells (Fig. 12).
Figure 12.
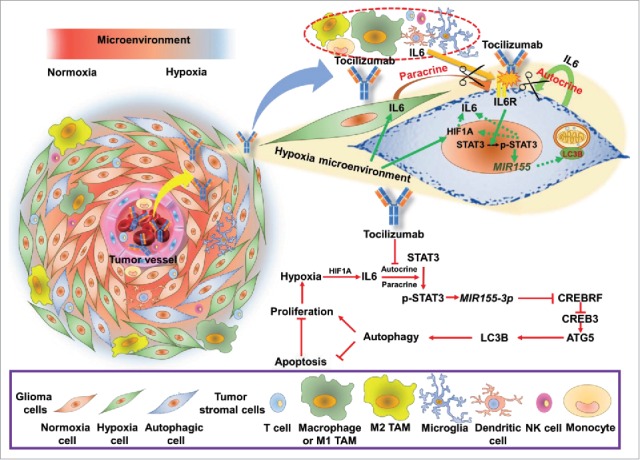
Proposed model and hypothesis of the novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. The hypoxic regions around the tumor vessels (Fig. 1B) and hypoxia-induced IL6 trigger hypoxic glioma cell autophagy through the p-STAT3-MIR155-3p-CREBRF pathway. Because upregulated autophagy inhibits tumor apoptosis and promotes proliferation, we blocked the IL6 receptor using tocilizumab and significantly suppressed tumor growth by shifting from autophagy to apoptosis. TAM, tumor-associated macrophage.
Inhibition of IL6 induces autophagy-enhanced apoptosis in GBM cells
In recent years, accumulating evidence has demonstrated that autophagy may function as a protective mechanism in tumor cells and that therapy-induced apoptosis can be potentiated by autophagy inhibition.53,54 To determine the biological significance of autocrine and paracrine IL6-induced autophagy on apoptotic cell death, the recombinant human IL6 antibody was utilized to prevent autophagy through antibody neutralization. A CCK-8 assay was utilized to determine cell viability, and ANXA5#x2011;FITC-PI and TUNEL staining assays were performed to examine the level of apoptosis in GBM cells. As shown in Figure 9A, the IL6 antibody significantly suppressed GBM cells only under hypoxic conditions. In agreement with this observation, hypoxia-induced apoptotic cell death was augmented in the presence of the IL6 antibody, as demonstrated by ANXA5–x2011;FITC-PtdIns (Fig. 9B) and TUNEL (Fig. 9C) assays. During apoptosis, CASP3, as the final effector molecule, cleaves PARP into fragments of 85 kDa and 25 kDa, and PARP cleavage is one of the most common characteristics of apoptosis.55 Accordingly, we examined cleaved CASP3 and PARP protein levels by western blot analysis, and the data further indicated that application of the IL6 antibody resulted in the activation of apoptotic signaling pathways, including activated CASP3 and PARP (Fig. 9D). This type of crosstalk between autophagy and apoptosis provides a possible avenue for interference via hypoxia-induced autophagy of glioma cells through anti-IL6 adjuvant therapy in the treatment of glioma patients.
Figure 9.
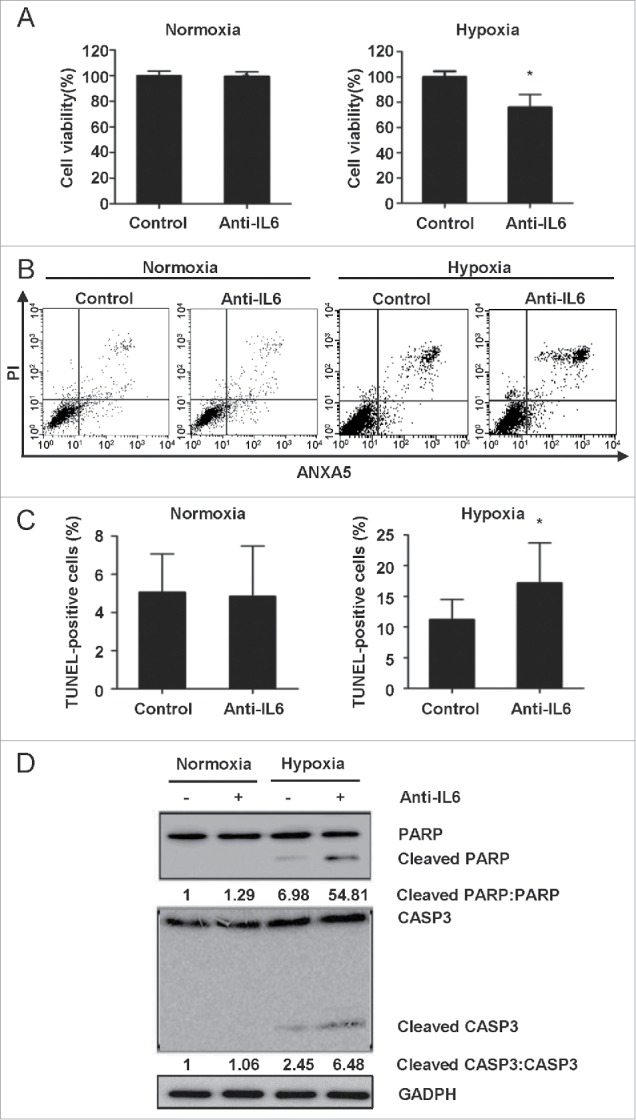
Inhibition of IL6 induces apoptosis in GBM cells. (A) A CCK-8 assay was performed to assess cell viability in hypoxia-treated U251 cells in the presence or absence of an IL6 antibody for 48 h. (B) and (C) ANXA5 FITC-PI and TUNEL staining assays were performed to examine the level of apoptosis in hypoxic GBM cells treated with an IL6 antibody (1 μg/ml). (D) Cleavage of PARP and CASP3 was induced by the IL6 antibody (1 μg/ml) in hypoxic GBM cells. PARP, cleaved PARP, CASP3 and cleaved CASP3 levels were examined by western blot analysis. GAPDH served as the loading control. The data are the mean ± s.d. *, P < 0.01 compared with the control group.
Tocilizumab inhibits tumor growth by antagonizing hypoxia-induced autophagy and promoting apoptosis in a xenograft tumor model
Tocilizumab, a novel monoclonal antibody from Roche, recently became available in China and is mainly used in the treatment of rheumatoid arthritis due to its targeting of IL6 receptors. As discussed above, we found that the IL6 antibody exerted a suppressive effect on glioma cells in vitro by antagonizing hypoxia-induced autophagy and promoting apoptosis, suggesting that tocilizumab may inhibit glioma growth in vivo. Therefore, we injected tocilizumab into the caudal veins of nude mice that bore subcutaneous U251 xenografts. No major side effects were noted throughout the study. At the end of the study, we harvested the subcutaneous tumors and peripheral blood serum for analysis. Relative to the control group, the average tumor volumes of the tocilizumab group were significantly decreased (Fig. 10A and B), and IL6 in the peripheral blood serum was lower according to ELISA (Fig. 10C). To examine whether the treatment mechanism agreed with our hypothesis, we used immunohistochemical staining to detect the expression of IL6, STAT3 and p-STAT3, LC3B, HIF1A, KI67 and cleaved CASP3 in the xenograft specimens. As shown by HE staining (Fig. 10D), the U251 xenografts grew in a more compact manner and were more homogeneous compared with glioma specimens; however, IL6, STAT3 and p-STAT3, LC3B, and HIF1A staining was essentially similar. As expected, the expression levels of IL6 in serum and IL6, p-STAT3 and LC3B in xenograft tissue were significantly decreased in the tocilizumab group (Fig. 10D). Finally, a pronounced decrease in tumor cell proliferation (KI67) and an increase in apoptosis (cleaved CASP3) were noted in the tocilizumab-treated xenografts (Fig. 10E). These findings suggested an unexpected role for anti-IL6 therapy in the treatment of glioma by blocking autophagy and inducing apoptosis in glioma cells.
Figure 10.
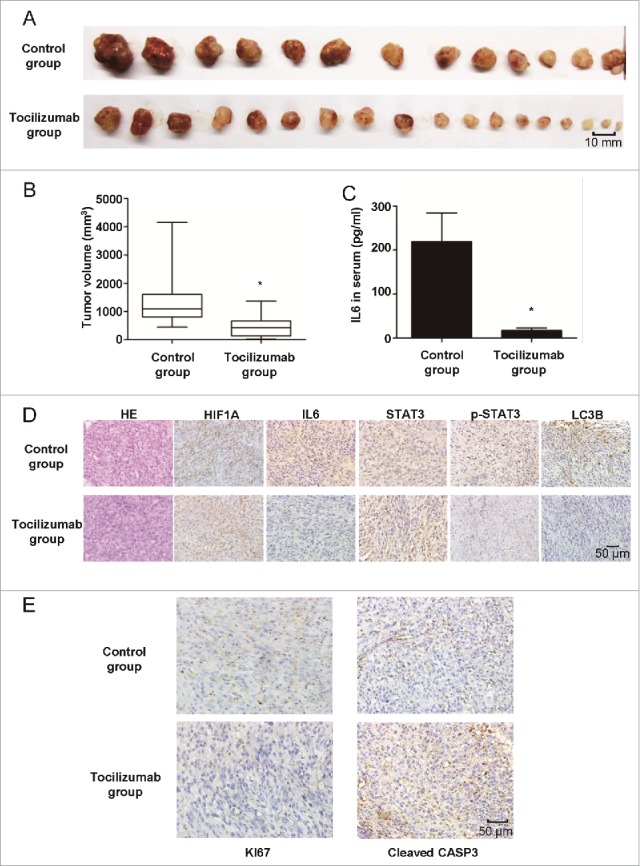
The IL6 monoclonal antibody tocilizumab inhibits the growth of GBM cells in vivo by inhibiting IL6-induced autophagy and promoting apoptosis. (A) and (B) Tocilizumab markedly inhibited tumor growth in U251 cell xenografts, as measured by tumor volume. *, P < 0.001 compared with the control group. (C) IL6 in the peripheral blood serum of the tocilizumab group was maintained at a lower level, as examined by ELISA, relative to the control group. *, P < 0.001 compared with the control group. (D) Tocilizumab reduced the expression of IL6, p-STAT3 and LC3B in all xenograft specimens, as evaluated by immunohistochemical staining. (E) Tocilizumab inhibited the proliferation and induced apoptosis in xenografts. The expression of KI67 and cleaved CASP3 in all xenograft specimens was examined by immunohistochemical staining.
It is well known that autophagy promotes the survival of tumor cells exposed to chemotherapy. However, there are different opinions concerning chemotherapy-induced autophagy and the efficiency of therapy.56,57 Furthermore, it remains unclear whether blockage of chemotherapy-induced autophagy could increase treatment efficiency. Temozolomide (TMZ), an oral chemotherapy drug, is used in the treatment of some brain tumors, as a second-line treatment for astrocytoma and as a first-line treatment for glioblastoma.58 In the present study, we investigated the combination therapy of temozolomide and tocilizumab in our xenograft tumor models and subsequently examined the ability of anti-IL6 therapy to reduce temozolomide-induced autophagy and improve its efficiency in vivo. As anticipated, the combination treatment resulted in a reduction in tumor volume compared with the single-drug groups (Fig. 11A and B). We then detected the expression of IL6, p-STAT3, LC3B, KI67 and cleaved CASP3 in the xenograft specimens by immunohistochemical staining. As expected, xenograft expression of IL6, p-STAT3 and LC3B in the temozolomide group was significantly increased (Fig. 11C), but combination therapy with tocilizumab blocked chemotherapy-induced autophagy and increased treatment efficiency by inducing significant apoptosis (Fig. 11C). To gain further insight into the involvement of the IL6-p-STAT3-MIR155-3p pathway in autophagy in vivo, we tested the effects of IL6 and MIR155-3p knockdown on tumor growth. To verify whether IL6 and MIR155-3p knockdown in glioma cells could block autophagy and induce apoptosis in vivo, adenovirus vector-transfected U251 cells were transplanted into nude mice through subcutaneous injection. The results indicated that knockdown of IL6 and MIR155-3p could inhibit tumor growth in vivo by blocking autophagy and inducing apoptosis in glioma cells, confirming the central role played by the IL6-p-STAT3-MIR155-3p pathway in controlling hypoxia-induced autophagy (Fig. S4).
Figure 11.
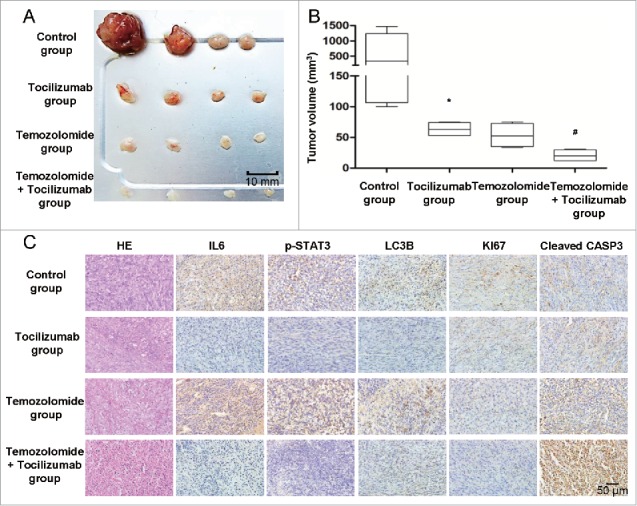
The IL6 monoclonal antibody tocilizumab increases the efficiency of temozolomide for GBM treatment in vivo by inhibiting IL6-induced autophagy and promoting apoptosis. (A) and (B) Tocilizumab and temozolomide combination therapy markedly inhibited tumor growth in U251 cell xenografts, as measured by tumor volume. *, P < 0.05 compared with the control group. #, P < 0.05 compared with the temozolomide group. (C) Tocilizumab reduced the expression of IL6, p-STAT3 and LC3B and induced apoptosis in temozolomide-treated xenograft specimens, as examined by immunohistochemical staining.
These data recapitulated the in vitro observations and showed that anti-IL6 therapy could be utilized for the treatment of glioma patients by antagonizing hypoxia-induced autophagy and promoting the apoptosis of tumor cells in hypoxic regions (Fig. 12).
Discussion
Autophagy promotes tumor survival under hypoxic metabolic stress conditions and mediates resistance to anti-tumor therapies such as radiation and chemotherapy.59,60 Although numerous anti-tumor agents have been reported to trigger cellular autophagy,61 novel adjuvant therapeutic strategies that efficaciously target GBMs are desperately needed to improve the currently unfavorable outcome of glioma patients. Recent accumulating evidence has demonstrated that the inhibition of autophagy triggers apoptosis.47 In the present study, we discovered a novel anti-autophagy therapeutic strategy by targeting hypoxia-induced IL6 in glioma cells. Our hypothesis that hypoxia-induced IL6 is a potent autophagy initiator in GBM via the p-STAT3-MIR155-3p pathway was confirmed by a series of experiments in vitro and in vivo. As shown in Fig. 10, hypoxic regions were distributed around tumor vessels (Fig. 1B), and hypoxia-induced IL6 triggered autophagy in hypoxic glioma cells through the p-STAT3-MIR155-3p pathway. Because upregulated autophagy inhibits tumor apoptosis and promotes proliferation, we blocked the IL6 receptor using tocilizumab, which significantly suppressed tumor growth by shifting from autophagy into apoptosis (Figs. 9 and 10E). This work is significant because it characterizes the pivotal role of the IL6-p-STAT3-MIR155-3p pathway in hypoxia-induced autophagy in glioma cells, and this is the first demonstration of the therapeutic efficacy of IL6 receptor monoclonal antibody injection for the treatment of gliomas in vivo.
The tumor microenvironment plays a critical role in tumor progression, and malignant proliferative tumor cells are deprived of oxygen as the tumor rapidly outgrows its blood supply.62 Tumor hypoxia can strongly induce cells to develop an aggressive and treatment-resistant phenotype that leads to rapid progression and poor prognosis.16 HIF1A, a key transcription factor involved in adaptation to hypoxic stress,63 plays an important role in hypoxia-induced autophagy, activating several autophagy-inducing molecules, such as BNIP3 (BCL2/adenovirus E1B 19kDa interacting protein 3) and IGFBP3 (insulin like growth factor binding protein 3).45,64 In addition, HIF1A-independent AMPK activates more severe hypoxia,22 and AMPK can contribute to autophagy via an MTOR inhibition mechanism.65,66 Nevertheless, similar to our previous demonstrations of high IL6 expression levels in gliomas and many other tumors,9-11,14,15,67 we found in the current study that IL6 also plays a role in hypoxia-induced autophagy.
IL6, a biomarker that indicates poor prognosis when overexpressed in many malignant tumors,9-13 promotes tumor progression by autocrine and paracrine mechanisms and binds to its receptor to activate downstream STAT3.34 Both phosphorylated and nonphosphorylated STAT3 can positively regulate the expression of IL6 and further promote a feed-forward loop.68-70 Furthermore, IL6 is a cytokine that is not only involved in inflammation and infection responses but also in the regulation of metabolic, regenerative, and neural processes as well as tumor progression.71 There are several lines of evidence suggesting that IL6 is a pleiotropic cytokine with significant functions in regulating the immune system.72 For instance, as a potent pro-inflammatory cytokine, IL6 plays a pivotal role in host defense against pathogens and acute stress. However, increased or deregulated expression of IL6 significantly contributes to the pathogenesis of various human diseases, including tumors.72 IL6R (interleukin 6 receptor) exists in 2 forms, a membrane-bound and soluble form;73 after binding IL6, the IL6-IL6R complex activates JAK1 (Janus kinase 1), JAK2 (Janus kinase 2), and TYK2 (tyrosine kinase 2) via IL6ST/gp130 (interleukin 6 signal transducer) signaling, leading to the phosphorylation of STAT3. STAT3 is an important regulator of a number of anti-apoptotic genes, and its activity is associated with tumor growth, survival, angiogenesis, and metastatic processes,74 especially in glioblastoma progression.33 However, the review articles cited have only summarized the current role of IL6-STAT3-regulated processes in promoting GBM progression with regard to proliferation, migration,75 anti-apoptosis, angiogenesis,76 relapse, and immune escape,77,78 but without the important aspect of autophagy. However, recent reports have shown that STAT3 affects autophagy via the transcriptional regulation of several autophagy-related genes, such as HIF1A and other genes, and microRNAs with autophagy-modulating targets.79 Our study first clearly demonstrated that the hypoxia-IL6-STAT3-MIR155-3p-CREBRF-CREB3 pathway mediates IL6-induced autophagy in glioma cells under hypoxic conditions. These observations reveal a novel positive feedback mechanism of the IL6-STAT3-HIF1A pathway that is promoted by hypoxia-induced autophagy and provide new insight into the contribution of IL6 to tumor progression by initiating protective autophagy. Because multiple upstream activators converge on the STAT3 pathway, targeting only one of these activators of the STAT3 pathway is unlikely to be sufficient for therapy. For tumor types that express high levels of IL6, such as GBM,80 our in vivo experimental results suggest that tocilizumab may be a potential therapy for glioma patients, at least as an adjuvant therapy combined with current chemotherapy.
Membrane-bound IL6R stimulates a classical signaling pathway, whereas soluble IL6R acts in a trans-signaling pathway.75 The classical and trans-signaling pathways are thought to contribute to the anti-inflammatory and pro-inflammatory activities of IL6, respectively,72 and the proliferation and survival of tumor cells are promoted by IL6 through both paracrine and autocrine mechanisms. After activation by IL6, various types of cells, such as tumor-associated macrophages,81 myeloid-derived suppressor cells,77,78 and endothelial cells,72 are involved in the development of a suppressive immunomicroenvironment and a metastatic tumor microenvironment.82 Several IL6-associated cytokines have growth factor activity, and inflammatory mediators such as HMGB1, IL23, and IL17 can promote tumor growth by activating the IL6-STAT3 pathway in a mouse model of melanoma.83 In addition, IL6 modulates the balance between regulatory T cells and T helper 17 cells in the tumor microenvironment and promotes the generation of cancer stem cells from noncancer stem cells.84
miRNAs are increasingly reported to be involved in the modulation of different autophagic stages.37 For example, ULK1 (unc-51 like autophagy activating kinase 1) is targeted by MIR20A and MIR106;85 BECN1/beclin 1 by MIR30A, MIR376B and MIR519A;86-89 RAB5A (RAB5A, member RAS oncogene family) by MIR101 and MIR630;89,90 RB1CC1/FIP200 (RB1 inducible coiled-coil 1) by MIR224;91 and ATGs by MIR30A, MIR181A, MIR374A, MIR630, MIR376B, MIR204, MIR224, MIR375, MIR519A, MIR885, and MIR-101.91-93 Almost all of these miRNAs are negative regulators of autophagy. MIR155-5p and MIR7 are rare miRNA inducers of autophagy.94 Mature miRNAs are processed from longer 70-nucleotide pre-miRNAs by RNASE3-like enzymes in the cytoplasm; pre-miRNAs are long duplex hairpins that contain several bulges or mispairings and are capped by an apical loop of variable size.95,96 Previous studies have suggested that the 5′ arm of the pre-miRNA stem-loop sequence will become the unique, mature miRNA, whereas the 3′ arm will be rapidly degraded.97 Nevertheless, growing evidence now suggests that miRNAs encoded from the 3′ arm (−3p) exist simultaneously with those encoded by the 5′ arm (−5p) and target a completely different set of mRNAs because of their disparate sequences, even though they originate from the same precursor.98 Although MIR155-5p has been previously reported to be an effective inducer of autophagy by directly targeting RHEB, RICTOR, and RPS6KB2 of the MTOR pathway in CNE and HeLa cells,37 MIR155-3p, a completely different mature miRNA of the MIR155 pre-miRNA, is also produced, though its exact role in tumors has not yet been elucidated. Our data first identified MIR155-3p, which was found in a screen of our hypoxia microarray, as a central player in the hypoxia-induced IL6 pro-autophagic pathway. The direct regulation of MIR155 pre-miRNAs by STAT3 has also been confirmed by Li et al.99 Unlike MIR155-5p, the TargetScan 6.2 algorithm cannot currently be utilized to determine the putative targets of MIR155-3p. However, one would speculate that this miRNA could potentially regulate the expression of thousands of mRNA targets.100 Nonetheless, the miRDB database can be used for MIR155-3p target prediction, and we found 248 potential targets of MIR155-3p. Our results indicated that the top predicted target, CREBRF, is a direct target of MIR155-3p and mediates the autophagic-promoting effect of MIR155-3p by regulating CREB3 and, in turn, ATG5. Therefore, our theoretical hypoxia-IL6-p-STAT3-MIR155-3p-autophagy pathway appears to play crucial roles in GBM malignant progression, and STAT3, which is centrally located, may have feed-forward control and transcriptionally regulate MIR155-3p (Fig. 12).
More than a half a century after its discovery, novel roles for the IL6 signaling pathway in tumor formation and progression have been consistently uncovered, including roles in regulating the proliferation, migration, activation, morphology, and metabolic state of tumor cells. However, in preclinical models of wide metastatic disease, IL6 inhibitors do not lead to disease regression.101 Tocilizumab has recently been approved for the treatment of rheumatoid arthritis by the FDA and China's SFDA, but there are no apparent plans to use this agent for malignancies. Tocilizumab treatment in patients with rheumatoid arthritis results in better clinical outcomes,102,103 and other related studies have shown that serum IL6 levels and serum MIF (macrophage migration inhibitory factor [glycosylation-inhibiting factor]) levels are significantly decreased with tocilizumab treatment.104,105 Regardless, our in vivo findings in a GBM mouse model provide insight that may be useful for the development of efficacious anti-IL6 therapies by tocilizumab injection. There are at least 2 possible explanations for this unexpected therapeutic effect on gliomas: the unique biological characteristics of glioma cells mentioned above and, most importantly, the unique immune microenvironment of the tumor. In addition, the decreased serum MIF level after tocilizumab injection might be another reason due to the tumor-promoting effects of MIF in gliomas.106,107
It is well established that the central nervous system is not devoid of immune cells and does not possess only ineffective immune cells; instead, immunosuppressive regulation of the immune balance occurs in gliomas,108-110 similar to that which occurs in the regulation of HLA-related molecules,111-114 the secretion of immunosuppressive factors, and the recruitment of immunosuppressive cells.115 The population of type M2 glioma-associated macrophages (GAMs) is thought to largely contribute to the induction of the immunosuppressive glioma microenvironment. Multiple immunosuppressive factors are secreted by glioma cells, including IL6, which could polarize GAMs toward tumor-promoting M2 GAMs.116 In contrast, M2 GAMs deteriorate this microenvironment by releasing IL6,117 which also stimulates glioma growth, neovascularization, and invasiveness.117,118 Furthermore, myeloid-derived suppressor cells are another remarkable immunosuppressive cell type in gliomas that are also induced by tumor-derived IL6.119 Although the subcutaneous gliomas in our studies are substantially different than in situ tumors, we detected an abundance of M2 tumor associated macrophage infiltration via immunohistochemical assays. Therefore, the observed therapeutic effect of tocilizumab may partially be attributed to blockade of the immunosuppressive effects of IL6.
In conclusion, this study demonstrated that hypoxia-induced IL6 is a potent autophagy inducer in GBM via the p-STAT3-MIR155-3p-CREBRF-CREB3-ATG5 pathway and that IL6-induced autophagy could protect GBM cells from apoptotic death. We investigated the ability of tocilizumab to block this pathway at the source, and our positive results suggest potential uses of this drug for glioma patients in anti-IL6 therapeutic strategies, such as adjuvant therapy combined with TMZ. However, modifications of the immunosuppressive microenvironment in the presence of tocilizumab remain to be defined, and evaluation of the therapeutic efficiency of tocilizumab combined with other common strategies would be helpful for advancing clinical investigations.
Materials and methods
Tissue samples and cell lines
The human glioma cell lines U251 and T98G were purchased from the Chinese Academy of Sciences Cell Bank and identified by the STR site detection assay (Microread Genetics Co, Beijing). One hundred and one human glioma tissue samples including 44 low-grade gliomas (8 grade I tumors and 36 grade II tumors), 57 high-grade gliomas (21 grade III tumors and 36 grade IV tumors), and 3 normal brain tissues from decompression operation were obtained from the Department of Neurosurgery of Qilu Hospital of Shandong University from October 2013 to June 2015. The glioma specimens were verified and classified according to the WHO classification standard of tumors by 2 experienced clinical pathologists. Our study was approved by the Institutional Review Board of Shandong University. Written informed consent was obtained from all patients, and the hospital ethics committee approved the experiments.
Reagents, cell culture and hypoxia treatment
All reagents in the study are as follows: 3-methyladenine (ApexBio Technology, A8353), bafilomycin A1 (Enzo Life Sciences, BML-CM110-0100), recombinant human IL6 (ProSpec, CYT-213), anti-human IL6 (ProSpec, ANT-109), tocilizumab/Actemra® (Roche Pharma, S20130020), temozolomide (Sigma-Aldrich, T2577). All cells were cultured in DMEM (Invitrogen, SP10001) supplemented with 10% fetal bovine serum (Gibco, 10099-141) and maintained at 37°C with 5% CO2 in a humidified chamber. Hypoxic conditions were induced by incubating the cells in a modular incubator chamber flushed with a gas mixture containing 1% O2, 5% CO2, and 94% N2 at 37°C.
Immunohistochemical staining
Human glioma tissue samples or solid tumors removed from sacrificed mice were fixed with 4% formaldehyde. Paraffin-embedded tumor tissues were sectioned to 5-μm thickness and mounted on positively charged microscope slides, and 1 mM EDTA (pH 8.0; Beyotime Biotechnology, P0085) for HIF1A or citrate solution (pH 6.0; Beyotime Biotechnology, P0083) for other antigens was used for antigen retrieval. Endogenous peroxidase activity was quenched by incubating the slides in methanol containing 3% hydrogen peroxide, followed by washing in phosphate-buffered saline (PBS; Beyotime Biotechnology, ST476) for 6 min. The sections were incubated for 2 h at room temperature with normal goat serum (ZSGB-BIO, ZLI-9021) and subsequently incubated at 4°C overnight with primary antibodies (1:200, HIF1A [Abcam, ab82832]; 1:400, IL6 [Abcam, ab6672, ab9324]; 1:300, STAT3 [Abcam, ab5073]; 1:100, p-STAT3 [Abcam, ab76315]; 1:400, LC3B [Abcam, ab48394]; 1:100, KI67 [Cell Signaling Technology, 9027]; and 1:300, cleaved CASP3 [Cell Signaling Technology, 9664]). The sections were then rinsed with PBS and incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies (ZSGB-BIO, PV-9000), followed by reaction with diaminobenzidine (ZSGB-BIO, ZLI-9033) and counterstaining with Mayer's hematoxylin (Beyotime Biotechnology, C0107). HE staining was performed free of charge by the pathology department of Qilu Hospital of Shandong University. Evaluation of the staining reaction was performed in accordance with the immunoreactive score = staining intensity × percentage of positive cells.120,121 A staining intensity value of 0 was negative; 1, weak; 2, moderate; and 3, strong. A percentage of positive cells value of 0 was negative; 1, 10% positive cells; 2, 11-50% positive cells; 3, 51-80% positive cells; and 4, more than 80% positive cells. Five visual fields from different areas of each tumor were used for the immunoreactive score evaluation.
Immunofluorescence staining
U251 cells were plated on glass slides in 24-well culture plates at a concentration of 2×105 cells/well for 24 h and subsequently treated with reagents for an additional 48 h in serum-free DMEM. Thereafter, the cells were fixed with a 4% formaldehyde solution in PBS, permeabilized with 0.5% Triton X-100 (Beyotime Biotechnology, ST795) in PBS, stained with the primary antibody overnight, and labeled with anti-mouse or anti-rabbit IgG conjugated with FITC (Santa Cruz Biotechnology, sc-358949, sc-2012). The cells were counterstained with DAPI and observed under an Olympus BX61 fluorescence microscope. Images were scanned using a DP71 CCD (charge-coupled device) digital camera.
GFP-LC3 stable cell lines and quantitative GFP-LC3 analyses
A U251 GFP-LC3 stable cell line was established by transient transfection of the pSELECT-GFP-LC3 lentiviral vector (Invivogen, VXI0541). GFP-LC3 puncta formation under normoxic or hypoxic conditions was determined by capturing images using a DP71 CCD digital camera microscope (Olympus). To quantify autophagic cells after drug treatment, we counted the number of autophagic cells as determined by the presence of GFP-LC3 puncta (≥20 puncta = a positive cell) in 100 high-power fields.
Western blot analysis
After the desired treatment, cells were washed twice with cold PBS and harvested with a rubber scraper. Cell pellets were lysed and kept on ice for at least 30 min in a buffer (Beyotime Biotechnology, P0013) containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Nonidet P-40 (Beyotime Biotechnology, P0013F), 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (Beyotime Biotechnology, ST506). The lysates were cleared by centrifugation, and the supernatant fractions were collected. Cell lysates were then separated by SDS-PAGE (Beyotime Biotechnology, P0012A) and subjected to western blot analysis with primary antibodies and horseradish peroxidase-conjugated secondary antibodies. Antibodies against the following were used for western blotting: SQSTM1 (Cell Signaling Technology, 5114S), LC3B (Abcam, ab63817), GAPDH (Cell Signaling Technology, 3683S), STAT3 (Abcam, ab68153), p-STAT3 (Abcam, ab76315), ACTB (Cell Signaling Technology, 12262S), PARP (Abcam, ab32138), cleaved PARP (Abcam, ab32064), CASP3 (Cell Signaling Technology, 9668T), cleaved CASP3 (Cell Signaling Technology, 9664S), KI67/PCNA (Cell Signaling Technology, 2586S), CREBRF (Santa Cruz Biotechnology, sc-133747), CREB3 (Abcam, ab42454), and ATG5 (Abcam, ab108327). GAPDH served as the loading control in all experiments involving hypoxic treatment due to the upregulation of ACTB under hypoxia.
ELISA
GBM culture supernatant was obtained from plates of treated cells, and blood serum samples were collected during the in vivo experiment. ELISAs were processed using a Human IL6 Valukine ELISA Kit (R&D Systems, VAL102) according to the manufacturer's instructions. The absorbance at 450 nm was measured using a microplate reader.
Transmission electron microscopy
Cells were fixed with 3% glutaraldehyde in PBS for 2 h, washed 5 times with 0.1 M cacodylate buffer, and postfixed with 1% OsO4 in 0.1 M cacodylate buffer containing 0.1% CaCl2 for 1.5 h at 4°C. The samples were then stained with 1% Millipore-filtered uranyl acetate, dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in LX-112 medium (Ladd Research Industries, 21210). After polymerization of the resin at 60°C for 48 h, ultrathin sections were cut with a Leica Ultracut microtome (Leica). Sections were stained with 4% uranyl acetate and lead citrate, and images were obtained using a JEM-100cxII electron microscope (JEM).
Small interfering RNA, miR inhibitor/mimics and adenovirus vector transfection
IL6 and negative control siRNAs were synthesized by GenePharma (China). The MIR155-3p inhibitor/mimics and negative control sequences were synthesized by Rio-Bio (China). The MIR155-3p inhibitor, IL6 siRNA, CREB3 siRNA and negative control adenovirus vectors were also synthesized by Rio-Bio (China). The sequences of siRNAs were as follows: Human IL6-207, 5′-CCC AGG AGA AGA UUC CAA ATT-3′ (sense) and 5′-TTG GGU CCU CUU CUA AGG UUU-3′ (antisense); IL6-321, 5′-GGA GAC AUG UAA CAA GAG UTT-3′ (sense) and 5′-TTC CUC UGU ACA UUG UUC UCA-3′ (antisense); IL6-417, 5′-CUU CCA AUC UGG AUU CAA UTT-3′ (sense) and 5′-TTG AAG GUU AGA CCU AAG UUA-3′ (antisense); CREB3-1299, 5′-GCA GUC AGA AGU GCC GAA ATT-3′ (sense) and 5′-TTC GUC AGU CUU CAC GGC UUU-3′ (antisense). The sequences of MIR155-3p inhibitor/mimics were as follows: MIR155-3p inhibitor, 5′- UGU UAA UGC UAA UAU GUA GGA G −3′; MIR155-3p mimics, 5′- CUC CUA CAU AUU AGC AUU AAC A −3′ (sense) and 5′-UUA AUG CUA AUA UGU AGG AGU U-3′ (antisense). The sequences of negative control were as follows: 5′-UUC UCC GAA CGU GUC ACG UTT-3′ (sense) and 5′-ACG UGA CAC GUU CGG AGA ATT-3′ (antisense). The miRNA inhibitor negative control was as follows: 5′-CAG UAC UUU UGU GUA GUA CAA-3′. The siRNAs or inhibitor/mimics were transfected into U251 and T98G cells for 48 h using Lipofectamine 2000 (Invitrogen, 11668-027) according to the protocol of the manufacturer. The MIR155-3p inhibitor, IL6 siRNA and negative control adenovirus vectors were transfected into U251 cells for 72 h according to the protocol of the manufacturer.
Cell viability assay
The CCK-8 assay (Dojindo Molecular Technologies, CK04) was used to test cell viability. Tumor cells in medium containing 10% fetal bovine serum were seeded into 96-well, flat-bottomed plates at 5×103 cells/well and incubated at 37°C overnight. After the desired treatment, the cells were incubated for an additional 4 h with 100 μl of serum-free DMEM and 10 μl of CCK-8 at 37°C. The absorbance at 450 nm was measured using a microplate reader.
ANXA5-fluorescein isothiocyanate (FITC) assay
To assess the degree of apoptosis of different groups of GBM cells, the extent of ANXA5-FITC-propidium iodide (PI) staining was determined by flow cytometry using the ANXA5/PtdIns staining kit (Beyotime Biotechnology, C1063). Samples were measured using an Epics XL-MCL flow cytometer (Beckman Coulter, Brea, CA, USA) and analyzed with WinMDI 2.8 software.
TUNEL assay
GBM cells were plated on glass slides in 24-well culture plates at 2×105 cells/well for 24 h and subsequently treated with drugs for an additional 48 h in serum-free DMEM. Glass slides with GBM cells and paraffin-embedded tumor sections were stained by the TUNEL technique using a TACS®2 TdT-Fluor in situ apoptosis detection kit (Trevigen, 4822-96-K) according to the instructions of the manufacturer. TUNEL-positive cells were counted from at least 100 random fields under a fluorescence microscope.
Human miRCURY™ LNA Array analysis
U251 cells were cultured under normoxic and hypoxic conditions (3 samples were prepared for each condition to serve as biological replicates), and then total RNA was extracted for microarray analysis. Total RNA was harvested using TRIzol (Invitrogen, IS-10007) and the miRNeasy mini kit (QIAGEN, 217004) according to the manufacturers' instructions. The RNA quantity was measured using a NanoDrop 1000, and the samples were labeled with the miRCURY™ Hy3™/Hy5™ Power labeling kit and hybridized onto the miRCURY™ LNA Array (v.18.0). After washing, the slides were scanned using the Axon GenePix 4000B microarray scanner. Scanned images were then imported into GenePix Pro 6.0 software (Axon) for grid alignment and data extraction. After normalization, significantly differentially expressed miRNAs were identified through Volcano Plot filtering. Finally, hierarchical clustering was performed to assess distinguishable gene expression profiles among samples.
RNA extraction and real-time quantitative PCR
Total RNA was extracted using TRIzol according to the manufacturer's protocol. Then, total RNA (50 ng) was reverse-transcribed with miR stem-loop RT primers or with U6 RT primers using a ReverTra Ace qPCR RT kit (TOYOBO, FSQ-101) according to the manufacturer's protocol to generate cDNA. Real-time PCR was performed using a SYBR Premix Ex Taq™ Kit (TAKARA, RR420A) with each primer. The reactions were performed using a LightCycler 2.0 Instrument (Roche). U6 expression was used as the endogenous control. The absolute expression levels were calculated as concentration ratios using a Roche LightCycler® 2.0 system.
Bioinformatic prediction and luciferase reporter assay
Common MIR155-3p targets predicted by computer-aided algorithms were obtained using the online miRNA prediction tool of miRBase. The reporter constructs containing pGL3-CREBRF and pGL3-mutCREBRF, with a mutated target seed sequence, were obtained from Bio-Asia (Jinan, China). Mutation of the p-STAT3 binding element (Bio-Asia, Jinan, China) and luciferase reporter assays were also performed using U251 glioma cells, which were cotransfected with luciferase reporters along with test substances or vehicle using Lipofectamine 2000. Forty-eight h after the cells were transfected, luciferase assays were performed using a luciferase assay kit according to the manufacturer's instructions.
Tumor xenograft model
The experiments conformed to the Animal Management Rule of the Chinese Ministry of Health (documentation 55, 2001), and the experimental protocol was approved by the Animal Care and Use Committee of Shandong University. Fifty BALB/c nude (nu/nu) female mice were purchased from Vital River Laboratories in batches. U251 cells (5×106 cells in 50 μl of serum-free DMEM) were inoculated subcutaneously into the right groin of the first batch of 5 5-wk-old female mice after acclimatization for a wk. Tumor volumes were calculated as (L ×W2)/2, where L is the length in millimeters and W is the width in millimeters. After 30 d, 3 of these mice developed a visible tumor mass, and the maximal tumor was resected using aseptic technique. The tumor tissue was rapidly cut into pieces of the same size, and each was transplanted subcutaneously into the right groin of a second batch of 45 female mice. When the tumors reached a mean volume of 90-120 mm3 (only 35 mice developed uniform tumors), the animals were randomized into 2 groups (15 in the control group and 20 in the treatment group). In vivo experiments were then performed to investigate the effect of tocilizumab injection: tocilizumab was injected into the caudal vein (8 mg/kg/month in normal saline), and an equivalent volume of normal saline was injected into the control group. Tumors were dissected at 30 d after transplantation and frozen in liquid nitrogen or fixed in formalin (2 mice were lost from each group due to suffocation), and blood serum samples were collected. Combination therapy tests of temozolomide plus tocilizumab were performed with the same xenograft tumor models. In the experiment, 20 mice were randomly divided into 4 groups. In the tocilizumab and combination groups, tocilizumab was injected into the caudal veins of 5 mice (8 mg/kg/mo in normal saline), with an equivalent volume of normal saline injected into 5 mice of the control and temozolomide groups. Intragastric administration of temozolomide (50 mg/kg) for 5 d in the temozolomide and combination groups was performed at the same time. Tumors were dissected at 30 d after transplantation and frozen in liquid nitrogen or fixed in formalin (1 mouse was lost from each group due to suffocation during intragastric administration). For in vivo IL6 and MIR155-3p knockdown tests, adenovirus vectors were transfected into U251 cells, and the same concentration of cell suspension was transplanted into nude mice (45 mice in 3 groups) through subcutaneous injection. Tumors were dissected at 28 d after transplantation and frozen in liquid nitrogen or fixed in formalin (due to individual differences, the negative control group and MIR155-3p knockdown group both included 5 mice without xenografts, and the IL6 knockdown group included 4 mice without xenografts).
Statistical analysis
Data analyses were conducted with SPSS 16.0 (SPSS, IL, USA) and GraphPad-Prism5 (GraphPad, CA, USA). Descriptive statistics including means ± SD, Student's t test, non-parametric Kruskal-Wallis tests for multiple comparison, the Mann-Whitney Test for 2-group comparison, Kaplan-Meier plots, log-rank tests, one-way ANOVAs, Pearson's correlation test and Spearman's correlation test were used to analyze significant differences; *, P < 0.05; **, P < 0.01; and ***, P < 0.001 were considered statistically significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- 3′-UTR
3′-untranslated region
- AMPK
adenosine 5′-monophosphate-activated protein kinase
- ATG3
autophagy-related 3
- ATG5
autophagy-related 5
- BAF
bafilomycin A1
- CASP3
caspase 3
- CCK-8
cell counting kit-8
- CD
clusters of differentiation
- CREB3
cAMP responsive element binding protein 3
- CREBRF
CREB3 regulatory factor
- ELISA
enzyme-linked immunosorbent assay
- ER
endoplasmic reticulum
- GAM
glioma-associated macrophage
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GBM
glioblastoma
- GFP
green fluorescent protein
- HIF1A
hypoxia inducible factor 1 α subunit
- IL6
interleukin 6
- IL6R
interleukin 6 receptor
- JAK1
Janus kinase 1
- M1
macrophages type 1
- M2
macrophages type 2
- MAP1LC3B/ LC3B
microtubule associated protein 1 light chain protein 3 β
- MIF
macrophage migration inhibitory factor (glycosylation-inhibiting factor)
- MIR/miR/miRNA
microRNA
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- PARP
poly(ADP-ribose) polymerase; p-STAT3, phosphorylated signal transducer and activator of transcription 3
- siRNA
small interfering RNA; SQSTM1/p62, sequestosome 1
- STAT3
signal transducer and activator of transcription 3
- TMZ
temozolomide
- TUNEL
deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- WHO
world health organization
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Professor Xun Qu and Professor Jian Wang for helpful comments and advice on this work and Pamela F, a Senior Editor at American Journal Experts, for language advice.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81101594; 81372719; 81172403; 81300510; 81402077; 81571284; 91542115) and Taishan Scholars of Shandong Province of China (No. ts201511093).
References
- [1].Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990-5; PMID:15528435; http://dx.doi.org/ 10.1126/science.1099993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol 2014; 85:830-8; PMID:24574520; http://dx.doi.org/ 10.1124/mol.114.091850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res 2014; 74:647-51; PMID:24459182; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2966 [DOI] [PubMed] [Google Scholar]
- [4].Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell 2013; 155:1216-9; PMID:24315093; http://dx.doi.org/ 10.1016/j.cell.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol 2010; 32:431-6; PMID:20814791; http://dx.doi.org/ 10.1007/s00281-010-0220-1 [DOI] [PubMed] [Google Scholar]
- [6].Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells 2010; 15:923-33; PMID:20670274; http://dx.doi.org/ 10.1111/j.1365-2443.2010.01433.x [DOI] [PubMed] [Google Scholar]
- [7].Aredia F, Guaman OLM, Giansanti V, Scovassi AI. Autophagy and cancer. Cells 2012; 1:520-34; PMID:24710488; http://dx.doi.org/ 10.3390/cells1030520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N. Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res 1990; 50:6683-8; PMID:2208133 [PubMed] [Google Scholar]
- [9].Cavarretta IT, Neuwirt H, Untergasser G, Moser PL, Zaki MH, Steiner H, Rumpold H, Fuchs D, Hobisch A, Nemeth JA, et al.. The antiapoptotic effect of IL-6 autocrine loop in a cellular model of advanced prostate cancer is mediated by Mcl-1. Oncogene 2007; 26:2822-32; PMID:17072336; http://dx.doi.org/ 10.1038/sj.onc.1210097 [DOI] [PubMed] [Google Scholar]
- [10].Badache A, Hynes NE. Interleukin 6 inhibits proliferation and, in cooperation with an epidermal growth factor receptor autocrine loop, increases migration of T47D breast cancer cells. Cancer Res 2001; 61:383-91; PMID:11196191 [PubMed] [Google Scholar]
- [11].Chang KT, Tsai CM, Chiou YC, Chiu CH, Jeng KS, Huang CY. IL-6 induces neuroendocrine dedifferentiation and cell proliferation in non-small cell lung cancer cells. Am J Physiol Lung Cell Mol Physiol 2005; 289:L446-53; PMID:15894558; http://dx.doi.org/ 10.1152/ajplung.00089.2005 [DOI] [PubMed] [Google Scholar]
- [12].Chang CY, Li MC, Liao SL, Huang YL, Shen CC, Pan HC. Prognostic and clinical implication of IL-6 expression in glioblastoma multiforme. J Clin Neurosci 2005; 12:930-3; PMID:16326273; http://dx.doi.org/ 10.1016/j.jocn.2004.11.017 [DOI] [PubMed] [Google Scholar]
- [13].Tchirkov A, Khalil T, Chautard E, Mokhtari K, Veronese L, Irthum B, Vago P, Kémény JL, Verrelle P. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer 2007; 96:474-6; PMID:17224923; http://dx.doi.org/ 10.1038/sj.bjc.6603586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Goswami S, Gupta A, Sharma SK. Interleukin-6-mediated autocrine growth promotion in human glioblastoma multiforme cell line U87MG. J Neurochem 1998; 71:1837-45; PMID:9798907; http://dx.doi.org/ 10.1046/j.1471-4159.1998.71051837.x [DOI] [PubMed] [Google Scholar]
- [15].Liu Q, Li G, Li R, Shen J, He Q, Deng L, Zhang C, Zhang J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J Neurooncol 2010; 100:165-76; PMID:20361349; http://dx.doi.org/ 10.1007/s11060-010-0158-0 [DOI] [PubMed] [Google Scholar]
- [16].Vaupel P. Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. Oncologist 2008; 13(Suppl 3):21-6; PMID:18458121; http://dx.doi.org/ 10.1634/theoncologist.13-S3-21 [DOI] [PubMed] [Google Scholar]
- [17].Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer 2002; 2:38-47; PMID:11902584; http://dx.doi.org/ 10.1038/nrc704 [DOI] [PubMed] [Google Scholar]
- [18].Mongiardi MP. Angiogenesis and hypoxia in glioblastoma: a focus on cancer stem cells. CNS Neurol Disord Drug Targets 2012; 11:878-83; PMID:23131159; http://dx.doi.org/ 10.2174/1871527311201070878 [DOI] [PubMed] [Google Scholar]
- [19].Jensen RL. Brain tumor hypoxia: tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J Neurooncol 2009; 92:317-35; PMID:19357959; http://dx.doi.org/ 10.1007/s11060-009-9827-2 [DOI] [PubMed] [Google Scholar]
- [20].Ji RC. Hypoxia and lymphangiogenesis in tumor microenvironment and metastasis. Cancer Lett 2014; 346:6-16; PMID:24333723; http://dx.doi.org/ 10.1016/j.canlet.2013.12.001 [DOI] [PubMed] [Google Scholar]
- [21].Elstner A, Holtkamp N, von DA. Involvement of Hif-1 in desferrioxamine-induced invasion of glioblastoma cells. Clin Exp Metastasis 2007; 24:57-66; PMID:17357815; http://dx.doi.org/ 10.1007/s10585-007-9057-y [DOI] [PubMed] [Google Scholar]
- [22].Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival. Curr Opin Cell Biol 2010; 22:177-80; PMID:20022734; http://dx.doi.org/ 10.1016/j.ceb.2009.11.015 [DOI] [PubMed] [Google Scholar]
- [23].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ohgaki H. Epidemiology of brain tumors. Methods Mol Biol 2009; 472:323-42; PMID:19107440; http://dx.doi.org/ 10.1007/978-1-60327-492-0_14 [DOI] [PubMed] [Google Scholar]
- [25].Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan JS. Epidemiology of gliomas. Cancer Treat Res 2015; 163:1-14; PMID:25468222; http://dx.doi.org/ 10.1007/978-3-319-12048-5_1 [DOI] [PubMed] [Google Scholar]
- [26].Davis ME, Stoiber AM. Glioblastoma multiforme: enhancing survival and quality of life. Clin J Oncol Nurs 2011; 15:291-7; PMID:21624864; http://dx.doi.org/ 10.1188/11.CJON.291-297 [DOI] [PubMed] [Google Scholar]
- [27].Sayegh ET, Kaur G, Bloch O, Parsa AT. Systematic review of protein biomarkers of invasive behavior in glioblastoma. Mol Neurobiol 2014; 49:1212-44; PMID:24271659; http://dx.doi.org/ 10.1007/s12035-013-8593-5 [DOI] [PubMed] [Google Scholar]
- [28].McNamara MG, Mason WP. Antiangiogenic therapies in glioblastoma multiforme. Expert Rev Anticancer Ther 2012; 12:643-54; PMID:22594899; http://dx.doi.org/ 10.1586/era.12.35 [DOI] [PubMed] [Google Scholar]
- [29].Raza SM, Fuller GN, Rhee CH, Huang S, Hess K, Zhang W, Sawaya R. Identification of necrosis-associated genes in glioblastoma by cDNA microarray analysis. Clin Cancer Res 2004; 10:212-21; PMID:14734472; http://dx.doi.org/ 10.1158/1078-0432.CCR-0155-3 [DOI] [PubMed] [Google Scholar]
- [30].Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 2004; 64:920-7; PMID:14871821; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-2073 [DOI] [PubMed] [Google Scholar]
- [31].Yuan G, Yan SF, Xue H, Zhang P, Sun JT, Li G. Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J Biol Chem 2014; 289:10607-19; PMID:24599950; http://dx.doi.org/ 10.1074/jbc.M113.528760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci 2004; 117:1281-3; PMID:15020666; http://dx.doi.org/ 10.1242/jcs.00963 [DOI] [PubMed] [Google Scholar]
- [33].Luwor RB, Stylli SS, Kaye AH. The role of Stat3 in glioblastoma multiforme. J Clin Neurosci 2013; 20:907-11; PMID:23688441; http://dx.doi.org/ 10.1016/j.jocn.2013.03.006 [DOI] [PubMed] [Google Scholar]
- [34].Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ, Mansell A, Jenkins BJ. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol 2011; 186:1199-208; PMID:21148800; http://dx.doi.org/ 10.4049/jimmunol.1002971 [DOI] [PubMed] [Google Scholar]
- [35].Groner B, Lucks P, Borghouts C. The function of Stat3 in tumor cells and their microenvironment. Semin Cell Dev Biol 2008; 19:341-50; PMID:18621135; http://dx.doi.org/ 10.1016/j.semcdb.2008.06.005 [DOI] [PubMed] [Google Scholar]
- [36].Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu X, Shen Y, Huang TT. Interplay between microRNAs and the STAT3 signaling pathway in human cancers. Physiol Genomics 2013; 45:1206-14; PMID:24192393; http://dx.doi.org/ 10.1152/physiolgenomics.00122.2013 [DOI] [PubMed] [Google Scholar]
- [37].Wan G, Xie W, Liu Z, Xu W, Lao Y, Huang N, Cui K, Liao M, He J, Jiang Y, et al.. Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy 2014; 10:70-9; PMID:24262949; http://dx.doi.org/ 10.4161/auto.26534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Audas TE, Li Y, Liang G, Lu R. A novel protein, Luman/CREB3 recruitment factor, inhibits Luman activation of the unfolded protein response. Mol Cell Biol 2008; 28:3952-66; PMID:18391022; http://dx.doi.org/ 10.1128/MCB.01439-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lu R, Yang P, O'Hare P, Misra V. Luman, a new member of the CREB/ATF family, binds to herpes simplex virus VP16-associated host cellular factor. Mol Cell Biol 1997; 17:5117-26; PMID:9271389; http://dx.doi.org/ 10.1128/MCB.17.9.5117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000; 101:451-4; PMID:10850487; http://dx.doi.org/ 10.1016/S0092-8674(00)80855-7 [DOI] [PubMed] [Google Scholar]
- [41].Sifers RN. Cell biology. Protein degradation unlocked. Science 2003; 299:1330-1; PMID:12610289; http://dx.doi.org/ 10.1126/science.1082718 [DOI] [PubMed] [Google Scholar]
- [42].Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al.. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516:108-11; PMID:25383523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu YL, Lai F, Wilmott JS, Yan XG, Liu XY, Luan Q, Guo ST, Jiang CC, Tseng HY, Scolyer RA, et al.. Noxa upregulation by oncogenic activation of MEK/ERK through CREB promotes autophagy in human melanoma cells. Oncotarget 2014; 5:11237-51; PMID:25365078; http://dx.doi.org/ 10.18632/oncotarget.2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yang Y, Lin P, Chen F, Wang A, Lan X, Song Y, Jin Y. Luman recruiting factor regulates endoplasmic reticulum stress in mouse ovarian granulosa cell apoptosis. Theriogenology 2013; 79:633-9.e1-3; PMID:23270862; http://dx.doi.org/ 10.1016/j.theriogenology.2012.11.017 [DOI] [PubMed] [Google Scholar]
- [45].Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 2012; 72:1773-83; PMID:22447568; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 1998; 58:1408-16; PMID:9537241 [PubMed] [Google Scholar]
- [47].Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025-40; PMID:15657430; http://dx.doi.org/ 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- [50].Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA 2004; 10:544-50; PMID:14970398; http://dx.doi.org/ 10.1261/rna.5235104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol 2004; 2:E98; PMID:15024405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet 2007; 8:173-84; PMID:17304245; http://dx.doi.org/ 10.1038/nrg2006 [DOI] [PubMed] [Google Scholar]
- [53].Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009; 46:210-40; PMID:19552522; http://dx.doi.org/ 10.1080/10408360903044068 [DOI] [PubMed] [Google Scholar]
- [54].Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta 2009; 1793:1516-23; PMID:19167434; http://dx.doi.org/ 10.1016/j.bbamcr.2008.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A, Wu Q, Zhang J, Hong Y. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci 2014; 10:1072-83; PMID:25285039; http://dx.doi.org/ 10.7150/ijbs.9719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang ZS, Wang J, Shen YB, Guo CC, Sai KE, Chen FR, Mei X, Han FU, Chen ZP. Dihydroartemisinin increases temozolomide efficacy in glioma cells by inducing autophagy. Oncol Lett 2015; 10:379-83; PMID:26171034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zou Y, Wang Q, Wang W. MutL homolog 1 contributes to temozolomide-induced autophagy via ataxia-telangiectasia mutated in glioma. Mol Med Rep 2015; 11:4591-6; PMID:25646660 [DOI] [PubMed] [Google Scholar]
- [58].Zanotto-Filho A, Braganhol E, Klafke K, Figueiro F, Terra SR, Paludo FJ, Morrone M, Bristot IJ, Battastini AM, Forcelini CM, et al.. Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett 2015; 358:220-31; PMID:25542083; http://dx.doi.org/ 10.1016/j.canlet.2014.12.044 [DOI] [PubMed] [Google Scholar]
- [59].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Marino ML, Pellegrini P, Di LG, Djavaheri-Mergny M, Brnjic S, Zhang X, Hägg M, Linder S, Fais S, Codogno P, et al.. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem 2012; 287:30664-76; PMID:22761435; http://dx.doi.org/ 10.1074/jbc.M112.339127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hu YL, Jahangiri A, De Lay M, Aghi MK. Hypoxia-induced tumor cell autophagy mediates resistance to anti-angiogenic therapy. Autophagy 2012; 8:979-81; PMID:22714142; http://dx.doi.org/ 10.4161/auto.20232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Eng JW, Kokolus KM, Reed CB, Hylander BL, Ma WW, Repasky EA. A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol Immunother 2014; 63:1115-28; PMID:25307152; http://dx.doi.org/ 10.1007/s00262-014-1617-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Unwith S, Zhao H, Hennah L, Ma D. The potential role of HIF on tumour progression and dissemination. Int J Cancer 2015; 136:2491-503; PMID:24729302; http://dx.doi.org/ 10.1002/ijc.28889 [DOI] [PubMed] [Google Scholar]
- [64].Hsieh DJ, Kuo WW, Lai YP, Shibu MA, Shen CY, Pai P, Yeh YL, Lin JY, Viswanadha VP, Huang CY. 17beta-Estradiol and/or Estrogen Receptor beta Attenuate the Autophagic and Apoptotic Effects Induced by Prolonged Hypoxia Through HIF-1alpha-Mediated BNIP3 and IGFBP-3 Signaling Blockage. Cell Physiol Biochem 2015; 36:274-84; PMID:25967966; http://dx.doi.org/ 10.1159/000374070 [DOI] [PubMed] [Google Scholar]
- [65].Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-26; PMID:18439900; http://dx.doi.org/ 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/ 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bao B, Ali S, Ahmad A, Azmi AS, Li Y, Banerjee S, Kong D, Sethi S, Aboukameel A, Padhye SB, et al.. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment. PLoS One 2012; 7:e50165; PMID:23272057; http://dx.doi.org/ 10.1371/journal.pone.0050165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, Wels J, Theilen T, Granitto S, Zhang X, et al.. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013; 15:848-62; PMID:23814496; http://dx.doi.org/ 10.1593/neo.13706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 2007; 21:1396-408; PMID:17510282; http://dx.doi.org/ 10.1101/gad.1553707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A 2013; 110:16975-80; PMID:24082147; http://dx.doi.org/ 10.1073/pnas.1315862110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011; 1813:878-88; PMID:21296109; http://dx.doi.org/ 10.1016/j.bbamcr.2011.01.034 [DOI] [PubMed] [Google Scholar]
- [72].Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, Yao Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther 2014; 141:125-39; PMID:24076269; http://dx.doi.org/ 10.1016/j.pharmthera.2013.09.004 [DOI] [PubMed] [Google Scholar]
- [73].Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor: a paradigm for cytokines. Science 1992; 258:593-7; PMID:1411569; http://dx.doi.org/ 10.1126/science.1411569 [DOI] [PubMed] [Google Scholar]
- [74].Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/ 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Santer FR, Malinowska K, Culig Z, Cavarretta IT. Interleukin-6 trans-signalling differentially regulates proliferation, migration, adhesion and maspin expression in human prostate cancer cells. Endocr Relat Cancer 2010; 17:241-53; PMID:19966016; http://dx.doi.org/ 10.1677/ERC-09-0200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med 2008; 14:109-19; PMID:18261959; http://dx.doi.org/ 10.1016/j.molmed.2007.12.007 [DOI] [PubMed] [Google Scholar]
- [77].Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 2010; 22:238-44; PMID:20171075; http://dx.doi.org/ 10.1016/j.coi.2010.01.021 [DOI] [PubMed] [Google Scholar]
- [79].You L, Wang Z, Li H, Shou J, Jing Z, Xie J, Sui X, Pan H, Han W. The role of STAT3 in autophagy. Autophagy 2015; 11:729-39; PMID:25951043; http://dx.doi.org/ 10.1080/15548627.2015.1017192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rolhion C, Penault-Llorca F, Kemeny JL, Lemaire JJ, Jullien C, Labit-Bouvier C, Finat-Duclos F, Verrelle P. Interleukin-6 overexpression as a marker of malignancy in human gliomas. J Neurosurg 2001; 94:97-101; PMID:11147905; http://dx.doi.org/ 10.3171/jns.2001.94.1.0097 [DOI] [PubMed] [Google Scholar]
- [81].Jackaman C, Nelson DJ. Are macrophages, myeloid derived suppressor cells and neutrophils mediators of local suppression in healthy and cancerous tissues in aging hosts. Exp Gerontol 2014; 54:53-7; PMID:24291067; http://dx.doi.org/ 10.1016/j.exger.2013.11.009 [DOI] [PubMed] [Google Scholar]
- [82].Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 2012; 22:275-81; PMID:22313874; http://dx.doi.org/ 10.1016/j.semcancer.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tang Q, Li J, Zhu H, Li P, Zou Z, Xiao Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in murine models of melanoma. Mediators Inflamm 2013; 2013:713859; PMID:24453427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, Lee YJ. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 2013; 25:961-9; PMID:23333246; http://dx.doi.org/ 10.1016/j.cellsig.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wu H, Wang F, Hu S, Yin C, Li X, Zhao S, Wang J, Yan X. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal 2012; 24:2179-86; PMID:22781751; http://dx.doi.org/ 10.1016/j.cellsig.2012.07.001 [DOI] [PubMed] [Google Scholar]
- [86].Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X, Chen X, Zhang CY, Zhang Q, Zen K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem 2012; 287:4148-56; PMID:22157765; http://dx.doi.org/ 10.1074/jbc.M111.307405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, Liu CG, Yang JM. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009; 5:816-23; PMID:19535919; http://dx.doi.org/ 10.4161/auto.9064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Korkmaz G, le SC, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012; 8:165-76; PMID:22248718; http://dx.doi.org/ 10.4161/auto.8.2.18351 [DOI] [PubMed] [Google Scholar]
- [89].Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-DeltaNp63alpha-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle 2012; 11:1247-59; PMID:22356768; http://dx.doi.org/ 10.4161/cc.11.6.19670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, Jäättelä M, Lund AH. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30:4628-41; PMID:21915098; http://dx.doi.org/ 10.1038/emboj.2011.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Guo X, Xue H, Guo X, Gao X, Xu S, Yan S, Han X, Li T, Shen J, Li G.. MiR224-3p inhibits hypoxia-induced autophagy by targeting autophagy-related genes in human glioblastoma cells. Oncotarget 2015; 39:41620–37; PMID:26536662; http://dx.doi.org/ 10.18632/oncotarget.5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fu LL, Wen X, Bao JK, Liu B. MicroRNA-modulated autophagic signaling networks in cancer. Int J Biochem Cell Biol 2012; 44:733-6; PMID:22342941; http://dx.doi.org/ 10.1016/j.biocel.2012.02.004 [DOI] [PubMed] [Google Scholar]
- [93].Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis 2012; 33:2018-25; PMID:22902544; http://dx.doi.org/ 10.1093/carcin/bgs266 [DOI] [PubMed] [Google Scholar]
- [94].Fang Y, Xue JL, Shen Q, Chen J, Tian L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology 2012; 55:1852-62; PMID:22234835; http://dx.doi.org/ 10.1002/hep.25576 [DOI] [PubMed] [Google Scholar]
- [95].Ritchie W, Legendre M, Gautheret D. RNA stem-loops: to be or not to be cleaved by RNAse III. RNA 2007; 13:457-62; PMID:17299129; http://dx.doi.org/ 10.1261/rna.366507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Roy-Chaudhuri B, Valdmanis PN, Zhang Y, Wang Q, Luo QJ, Kay MA. Regulation of microRNA-mediated gene silencing by microRNA precursors. Nat Struct Mol Biol 2014; 21:825-32; PMID:25086740; http://dx.doi.org/ 10.1038/nsmb.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al.. A uniform system for microRNA annotation. RNA 2003; 9:277-9; PMID:12592000; http://dx.doi.org/ 10.1261/rna.2183803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011; 39:D152-7; PMID:21037258; http://dx.doi.org/ 10.1093/nar/gkq1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Li P, Grgurevic S, Liu Z, Harris D, Rozovski U, Calin GA, Keating MJ, Estrov Z. Signal transducer and activator of transcription-3 induces microRNA-155 expression in chronic lymphocytic leukemia. PLoS One 2013; 8:e64678; PMID:23750211; http://dx.doi.org/ 10.1371/journal.pone.0064678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene 2013; 532:1-12; PMID:23246696; http://dx.doi.org/ 10.1016/j.gene.2012.12.009 [DOI] [PubMed] [Google Scholar]
- [101].Chang Q, Daly L, Bromberg J. The IL-6 feed-forward loop: a driver of tumorigenesis. Semin Immunol 2014; 26:48-53; PMID:24613573; http://dx.doi.org/ 10.1016/j.smim.2014.01.007 [DOI] [PubMed] [Google Scholar]
- [102].Kojima T, Yabe Y, Kaneko A, Hirano Y, Ishikawa H, Hayashi M, Miyake H, Takagi H, Kato T, Terabe K, et al.. Monitoring C-reactive protein levels to predict favourable clinical outcomes from tocilizumab treatment in patients with rheumatoid arthritis. Mod Rheumatol 2013; 23:977-85; PMID:23099471; http://dx.doi.org/ 10.3109/s10165-012-0782-y [DOI] [PubMed] [Google Scholar]
- [103].Bay-Jensen AC, Platt A, Byrjalsen I, Vergnoud P, Christiansen C, Karsdal MA. Effect of tocilizumab combined with methotrexate on circulating biomarkers of synovium, cartilage, and bone in the LITHE study. Semin Arthritis Rheum 2014; 43:470-8; PMID:23932312; http://dx.doi.org/ 10.1016/j.semarthrit.2013.07.008 [DOI] [PubMed] [Google Scholar]
- [104].Shimamoto K, Ito T, Ozaki Y, Amuro H, Tanaka A, Nishizawa T, Son Y, Inaba M, Nomura S. Serum interleukin 6 before and after therapy with tocilizumab is a principal biomarker in patients with rheumatoid arthritis. J Rheumatol 2013; 40:1074-81; PMID:23637318; http://dx.doi.org/ 10.3899/jrheum.121389 [DOI] [PubMed] [Google Scholar]
- [105].Kasama T, Isojima S, Umemura M, Tsukamoto H, Tokunaga T, Furuya H, Yanai R, Takahashi R, Nakamura M, Inagaki K. Serum macrophage migration inhibitory factor levels are correlated with response to tocilizumab therapy in patients with rheumatoid arthritis. Rheumatol Int 2014; 34:429-33; PMID:23670804; http://dx.doi.org/ 10.1007/s00296-013-2778-0 [DOI] [PubMed] [Google Scholar]
- [106].Mittelbronn M, Platten M, Zeiner P, Dombrowski Y, Frank B, Zachskorn C, Harter PN, Weller M, Wischhusen J. Macrophage migration inhibitory factor (MIF) expression in human malignant gliomas contributes to immune escape and tumour progression. Acta Neuropathol 2011; 122:353-65; PMID:21773885; http://dx.doi.org/ 10.1007/s00401-011-0858-3 [DOI] [PubMed] [Google Scholar]
- [107].Wang XB, Tian XY, Li Y, Li B, Li Z. Elevated expression of macrophage migration inhibitory factor correlates with tumor recurrence and poor prognosis of patients with gliomas. J Neurooncol 2012; 106:43-51; PMID:21725855; http://dx.doi.org/ 10.1007/s11060-011-0640-3 [DOI] [PubMed] [Google Scholar]
- [108].Harris MG, Hulseberg P, Ling C, Karman J, Clarkson BD, Harding JS, Zhang M, Sandor A, Christensen K, Nagy A, et al.. Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci Rep 2014; 4:4422; PMID:24651727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Weller M. More on the immune privilege of glioblastoma. Neuro Oncol 2012; 14:527-8; PMID:22539528; http://dx.doi.org/ 10.1093/neuonc/nos110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Galea I, Bechmann I, Perry VH. What is immune privilege (not). Trends Immunol 2007; 28:12-8; PMID:17129764; http://dx.doi.org/ 10.1016/j.it.2006.11.004 [DOI] [PubMed] [Google Scholar]
- [111].Mehling M, Simon P, Mittelbronn M, Meyermann R, Ferrone S, Weller M, Wiendl H. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism. Acta Neuropathol 2007; 114:111-9; PMID:17541610; http://dx.doi.org/ 10.1007/s00401-007-0231-8 [DOI] [PubMed] [Google Scholar]
- [112].Rossi ML, Esiri MM, Jones NR, Coakham HB, Moss TH, Cruz-Sanchez FF, Carey MP. Characterization of the mononuclear cell infiltrate and HLA-Dr expression in 19 oligodendrogliomas. Surg Neurol 1991; 36:119-25; PMID:1891756; http://dx.doi.org/ 10.1016/0090-3019(91)90229-3 [DOI] [PubMed] [Google Scholar]
- [113].Rebmann V, Regel J, Stolke D, Grosse-Wilde H. Secretion of sHLA-G molecules in malignancies. Semin Cancer Biol 2003; 13:371-7; PMID:14708717; http://dx.doi.org/ 10.1016/S1044-579X(03)00028-2 [DOI] [PubMed] [Google Scholar]
- [114].Wiendl H, Mitsdoerffer M, Hofmeister V, Wischhusen J, Bornemann A, Meyermann R, Weiss EH, Melms A, Weller M. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol 2002; 168:4772-80; PMID:11971028; http://dx.doi.org/ 10.4049/jimmunol.168.9.4772 [DOI] [PubMed] [Google Scholar]
- [115].Albesiano E, Han JE, Lim M. Mechanisms of local immunoresistance in glioma. Neurosurg Clin N Am 2010; 21:17-29; PMID:19944963; http://dx.doi.org/ 10.1016/j.nec.2009.08.008 [DOI] [PubMed] [Google Scholar]
- [116].Wei J, Gabrusiewicz K, Heimberger A. The controversial role of microglia in malignant gliomas. Clin Dev Immunol 2013; 2013:285246; PMID:23983766; http://dx.doi.org/ 10.1155/2013/285246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012; 33:312-9; PMID:22159219; http://dx.doi.org/ 10.1093/carcin/bgr289 [DOI] [PubMed] [Google Scholar]
- [118].Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 2008; 216:15-24; PMID:18553315; http://dx.doi.org/ 10.1002/path.2370 [DOI] [PubMed] [Google Scholar]
- [119].Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol 2006; 16:53-65; PMID:16168663; http://dx.doi.org/ 10.1016/j.semcancer.2005.07.005 [DOI] [PubMed] [Google Scholar]
- [120].Kieback DG, Press MF, Atkinson EN, Edwards GL, Mobus VJ, Runnebaum IB, Kreienberg R, Jones LA. Prognostic significance of estrogen receptor expression in ovarian cancer. Immunoreactive Score (IRS) vs. Composition Adjusted Receptor Level (CARL). Anticancer Res 1993; 13:2489-96; PMID:8135488 [PubMed] [Google Scholar]
- [121].Friedrichs K, Gluba S, Eidtmann H, Jonat W. Overexpression of p53 and prognosis in breast cancer. Cancer 1993; 72:3641-7; PMID:8252480; http://dx.doi.org/ 10.1002/1097-0142(19931215)72:12%3c3641::AID-CNCR2820721215%3e3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.