

Abstract
Bone metastases occur in up to 70% of advanced breast cancer. For most patients with breast cancer, bone metastases are predominantly osteolytic. Interactions between tumor cells and stromal cells in the bone microenvironment drive osteolytic bone metastasis, a process that requires the activation of osteoclasts, cells that break down bone. Here, we report that ABL kinases promoted metastasis of breast cancer cells to bone by regulating the crosstalk between tumor and the bone microenvironment. ABL kinases protected tumor cells from apoptosis induced by TRAIL (TNF-related apoptosis-inducing ligand), activated the transcription factor STAT5, and promoted osteolysis through the STAT5-dependent expression of genes encoding the osteoclast activating factors interleukin 6 (IL6) and matrix metalloproteinase-1 (MMP1). Furthermore, ABL kinases increased the abundance of the Hippo pathway mediator TAZ and the expression of TAZ-dependent target genes that promote bone metastasis. Knockdown of ABL kinases or treatment with ABL-specific allosteric inhibitor impaired osteolytic metastasis of breast cancer cells in mice. These findings revealed a role for ABL kinases in regulating tumor-bone interactions and provide a rationale for targeting both tumor and the bone microenvironment with ABL-specific inhibitors.
Introduction
The ABL family of non-receptor tyrosine kinases, ABL1 (also known as c-Abl) and ABL2 (also known as Arg), links diverse extracellular stimuli to signaling pathways that control cell growth, survival, adhesion, migration and invasion (1–3). ABL tyrosine kinases play an oncogenic role in human leukemias (4, 5) and promote the progression of solid tumors (5, 6). ABL kinases elicit pro-tumorigenic or anti-tumorigenic effects in breast cancer cells and promote cancer cell invasion (7–10). However, whether ABL kinases have a role in the regulation of cellular processes critical for metastasis, other than invasion, has not yet been evaluated. Here we uncovered a critical role for the ABL kinases in the regulation of breast cancer metastasis to bone.
Bone metastases occur in up to 70% of patients with advanced breast cancer and are associated with high mortality and morbidity (11, 12). While the mechanisms that drive tumor cell homing, invasion and colonization to the bone are poorly understood, it is increasingly apparent that bone metastasis requires interactions between tumor and stromal cells in the bone microenvironment (13). When breast cancer cells invade into the bone microenvironment, they produce molecules that activate osteoclastic bone resorption, leading to the release of growth factors stored in the bone matrix to promote tumor growth. Currently, there are no available therapies to cure breast cancer metastasis. Thus, there is a need to identify molecules that could be targeted simultaneously in tumor and bone to disrupt the tumor-stromal cells interactions that drive metastasis.
Here we report that increased expression of ABL1 and ABL2 correlated with enhanced breast cancer metastasis and decreased metastasis-free survival. Using metastasis models that bypass invasion and intravasation, we uncovered roles for the ABL kinases in the regulation of breast cancer cell survival and colonization in the bone microenvironment. Further, we identified a role for ABL kinases in promoting the expression of multiple pro-bone-metastasis genes such as AXL (which encodes a receptor tyrosine kinase), IL6 (which encodes interleukin-6), MMP1 (which encodes matrix metalloproteinase 1) and TNC (which encodes tenascin-C) through TAZ- and STAT5-mediated signaling. Moreover, we found that treatment with a selective allosteric inhibitor of the ABL kinases or simultaneous depletion of both ABL kinases in breast cancer cells impaired breast cancer bone metastases and decreased osteoclast activation in vitro and osteolysis in vivo.
Results
Increased expression of ABL kinase-encoding genes correlates with breast cancer metastasis
To evaluate whether altered expression of the ABL genes is associated with breast cancer progression and metastasis we examined the expression of ABL1 and ABL2 in normal and invasive breast tumor specimens using published TCGA datasets (14–16). ABL2 DNA and RNA abundance was significantly increased in breast tumor specimens (Fig. 1, A and B). To further evaluate the importance of enhanced ABL abundance in the context of metastasis, we analyzed an integrative database assembled from 22 publicly available datasets containing information on metastasis-related relapse (17). We found that increased ABL2 mRNA abundance correlated with metastasis across all subtypes of breast cancer, primarily the basal type (Fig. 1, C and D), whereas high ABL1 mRNA abundance significantly correlated with metastasis in HER2-enriched breast cancer but not in other breast cancer subtypes (Fig. 1E). Furthermore, high ABL1 mRNA was associated with bone metastasis in a microarray dataset reporting organ-specific metastasis (Fig. 1F) (18). Collectively these findings support a link between increased expression of the ABL genes and increased breast cancer metastasis.
Fig. 1.
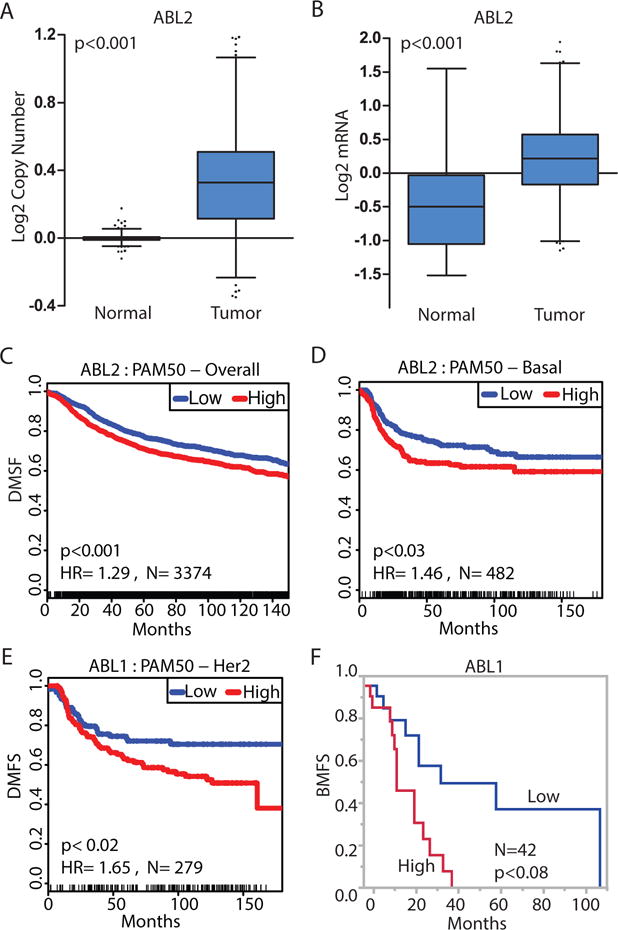
Increased expression of ABL genes in invasive breast cancer is associated with metastasis.
(A) ABL2 copy number in 813 normal samples compared with 789 invasive breast tumor samples in the TCGA database. (B) ABL2 mRNA abundance in 61 normal samples compared with 532 invasive breast tumor samples in the TCGA database. Results shown in (A) and (B) are based on data generated by the TCGA Research Network (http://cancergenome.nih.gov/); whiskers represent 1th and 99th percentile. (C–D) Kaplan-Meier representation of the probability of cumulative overall distant metastasis-free survival (DMFS) in 2830 breast cancer cases (C), or 482 basal breast cancer cases (D) according to ABL2 expression. (E) Kaplan-Meier representation of the probability of cumulative overall distant metastasis-free survival in 279 HER2 enriched breast cancer cases according to ABL1 expression. (F) Kaplan-Meier representation of the probability of cumulative bone metastasis-free survival (BMFS) in 42 breast cancer cases according to ABL1 expression. P values (log rank test) and hazard ratio (HR) are shown in the graph.
ABL family protein kinases are required for bone metastasis
To directly evaluate the relationship between ABL family kinases and metastasis, we analyzed ABL1 and ABL2 protein abundance in MDA-MB-231-derived breast cancer cell lines with different organ metastasis tropisms (19). The MDA-MB-231-derived 1833 cell line, which is characterized by enhanced bone-specific metastasis compared to the parental cell line or cell lines with increased tropism to lung and brain, showed increased abundance of ABL1 and ABL2 (fig. S1A). To examine the functional role of ABL kinases in these cells, endogenous ABL kinases were depleted with previously characterized shRNAs specific against ABL1 and ABL2 (20). The lentiviral-encoded shRNAs decreased the abundance of ABL1 and ABL2 by 80% at day 2 after viral transduction, but ABL1 abundance was slightly increased by day 21 after viral transduction (fig. S1B). Depletion of ABL kinases did not affect cell growth in monolayers or colony formation in matrigel (fig. S1, C, D and E), but decreased cell invasion in both 1833 triple-negative and HER2-positive SKBR3 breast cancer cells (fig. S2, A, B, C, and D). Conversely, overexpression of constitutively active forms of ABL1 (ABL1PP) and ABL2 (ABL2PP) enhanced the invasiveness of the parental MDA-MB-231 cells (fig. S2, E, F, and G).
ABL kinases regulate cancer cell invasion (21), but it is unclear whether they play a role in the regulation of subsequent steps of the metastatic cascade. To investigate whether inhibition of ABL kinases interferes with metastatic processes other than invasion, we depleted ABL1 and ABL2 (ABL1/ABL2) in two bone metastatic breast cancer cell lines 1833 and SCP28 (22), and evaluated the metastatic potential of these cells following intra-cardiac injection into immune-deficient mice. This mouse model bypasses the initial invasion step and allows for analysis of subsequent steps in the metastatic cascade. The 1833 and SCP28 breast cancer cells were engineered to express reporters with luciferase and green fluorescent protein (GFP) to monitor metastatic progression by bioluminescence imaging (Fig. 2A). We found that ABL kinase knockdown increased the survival of tumor-bearing mice (Fig. 2B) and markedly inhibited bone metastases by 1833 and SCP28 breast cancer cells as measured by bioluminescence imaging (Fig. 2, C, D, E and F) and haematoxylin and eosin (HE) staining (Fig. 2, G and H). Decreased metastasis by ABL-deficient breast cancer cells was accompanied by a significant reduction in the extent of hind-limb osteolytic lesions, as determined by X-ray and μCT imaging (Fig. 2, I and J).
Fig. 2.
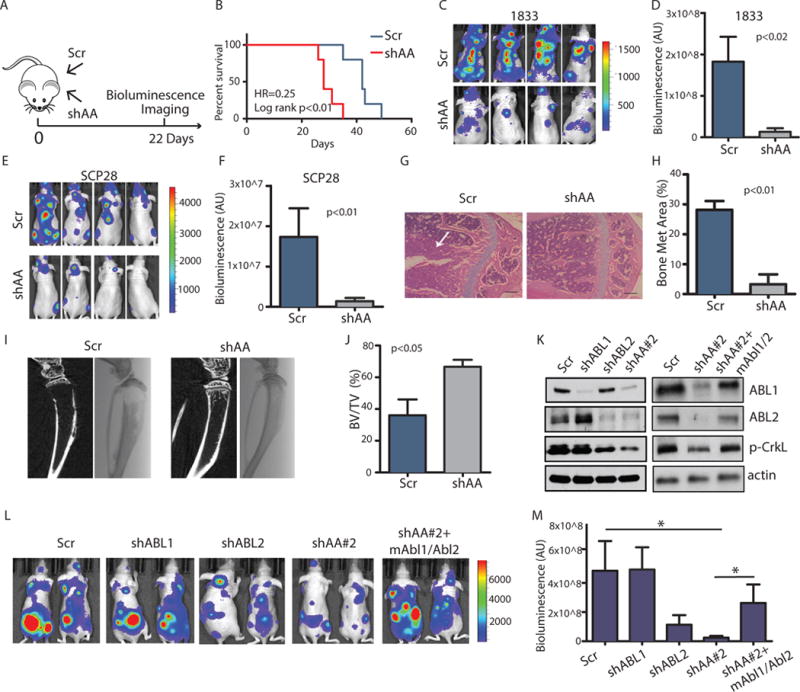
Knockdown of ABL kinases decreases breast cancer bone metastasis.
(A) The experimental design. (B) Survival of mice after intracardiac injection of 1833 (1×105) breast cancer cells transduced with control shRNA (Scr) or shRNAs against ABL1 and ABL2 (shAA). N=10 mice/per group. (C–F) Bioluminescent images (C, E) of bone metastasis from representative mice at day 22 after inoculation with 1833 cells (N=10 mice/group) or day 35 after inoculation with SCP28 cells (N=8 mice/group). Quantification of bone metastases (D, F). (G–H) Representative HE staining (G) and quantification of HE-stained tumor area of bone lesions. Arrows indicate tumor. N=3 mice/group. Scale bar=200μM. Met, metastatic. (I–J) Representative Xray and μCT reconstruction (I) and quantification of bone volume/total volume from μCT analysis of the mouse tibias (J). N=3 mice/group. (K) Representative immunoblots of 1833 cells transfected with control shRNA (Scr), shRNA against ABL1 (shABL1), ABL2 (shABL2), and shRNA #2 against both ABL1 and ABL2 (shAA#2), and ABL1/ABL2 knockdown cells with overexpression of mouse Abl1/Abl2 (shAA+mAbl1/Abl2). N=3 blots. p, phosphorylated. (L) Bioluminescent images of bone metastases from representative mice at day 18 after inoculation. N=8 mice/group. (M) Quantification of (L). * p<0.05, calculated using One-Way ANOVA followed by Tukey’s HSD.
To evaluate whether ABL1 and ABL2 are individually responsible for promoting metastasis, we employed specific shRNAs to silence either ABL1 or ABL2 in breast cancer cells. We found that ~90% knockdown of ABL1 alone resulted in enhanced ABL2 expression, and did not produce a significant decrease in the phosphorylation of CrKL, a reporter for the activation state of the ABL kinases (Fig. 2K) and did not inhibit metastasis (Fig. 2, L and M). Double knockdown of ABL1 and ABL2 was required to decrease the phosphorylation of CrKL by more than 90%, which indicates inactivation of the endogenous ABL kinases (Fig. 2K). While knockdown of ABL2 alone decreased metastasis, knockdown of both ABL1 and ABL2 was required to significantly decrease metastasis (Fig. 2, L and M). To further validate that the decreased metastasis induced by double knockdown of ABL1 and ABL2 was not due to off target effects of the lentivirus-encoded shRNAs, we employed a second set of ABL shRNAs (shAA#2) and carried out rescue experiments by expressing mouse Abl1 and Abl2 constructs (mAbl1/Abl2) that are resistant to shRNAs against human ABL1 and ABL2. We found that expression of murine Abl1 and Abl2 kinases in the knockdown cells reversed the decreased metastasis (Fig. 2, K, L, and M). Loss of ABL1 and ABL2 in the lung metastatic 4175 breast cancer cell line did not significantly reduce metastasis (fig. S3). These findings reveal a function for ABL kinases in the regulation of breast cancer bone metastasis and tumor-induced osteolysis in vivo.
Allosteric inhibition of the ABL kinases impairs breast cancer bone metastasis
To date few studies have directly evaluated the biological consequences of targeting the ABL kinases with selective inhibitors in solid tumors, including breast cancer in vivo. The ATP-competitive kinase inhibitors imatinib (STI571; trade name Gleevec), dasatinib, and nilotinib inhibit multiple tyrosine kinases in addition to ABL1 and ABL2 (5). Moreover, these ATP-competitive inhibitors induce the formation of B-RAF and C-RAF dimers, leading to ERK activation in diverse cancer cell types (23). Therefore, we wished to use a different approach to evaluate whether ABL kinases play a role in breast cancer metastasis using the allosteric inhibitor GNF5, which targets the unique ABL myristate-binding site and functions as non-ATP-site and selective inhibitor of the ABL kinases (24, 25). Notably, GNF5 does not activate the RAF-ERK pathway in breast cancer cells (23) (fig. S4).
Treatment of tumor-bearing mice with GNF5 starting on day eight following intra-cardiac injection of breast cancer cells (Fig. 3A) resulted in a significant increase in survival (Fig. 3B) and a decrease in bone metastasis burden as measured by bioluminescent imaging (Fig. 3, C and D). Similarly, histological analyses revealed a decrease in bone tumor burden in mice treated with the allosteric inhibitor of the ABL kinases (Fig. 3, E and F). Notably, we found that bone destruction was decreased and the ratio of bone volume to total volume was increased in tumor-bearing mice treated with GNF5 (Fig. 3, G and H). These results demonstrate that ABL kinase activity is required for osteolytic metastasis in breast cancer and suggest that pharmacological inhibition of the ABL kinases may be an effective treatment for bone metastasis.
Fig. 3.
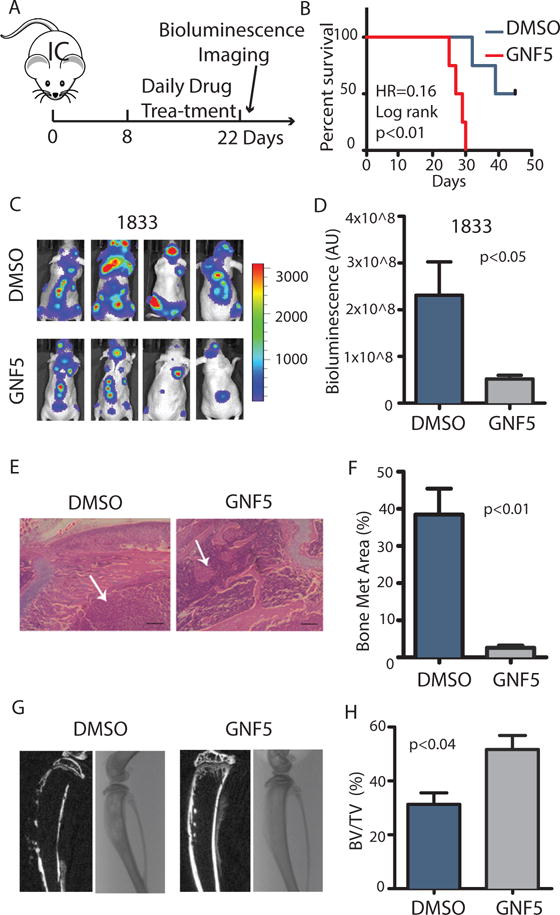
Allosteric inhibition of ABL kinases decreases breast cancer bone metastasis.
(A) The experimental design. (B) Survival of mice after intracardiac injection of 1833 (1×105) breast cancer cells and treatment with either DMSO control or the allosteric ABL inhibitor GNF5. N=10 mice/group. (C) Bioluminescent images of representative mice at day 22 after inoculation. (D) Quantification of bone metastases. N=10 mice/group. (E–F) Representative HE staining (E) and quantification (F) of HE-stained tumor area of bone lesions. Arrows indicate tumor. N=3 mice/group. HE: Scale bar=200μM. (G–H) Representative Xray and μCT reconstruction (G) of mouse tibias and quantification (H) of bone volume/total volume. N=3 mice/group.
ABL kinases are required for tumor cell survival and tumor-induced osteolysis in the bone microenvironment
To directly examine whether ABL kinases play a role in regulating the colonization and survival of breast cancer cells in the bone microenvironment, we injected control or ABL1/ABL2 knockdown breast cancer cells directly into the tibia of immune-deficient mice. Depletion of the ABL kinases reduced tumor expansion in the tibia as measured by both bioluminescent imaging (Fig. 4, A and B) and histological staining (Fig. 4, C and D). Moreover, 3D-μCT reconstruction of the tibia revealed that mice injected with control cells had a significantly higher degree of osteolysis with a decreased ratio of bone volume to total volume compared to mice injected with ABL1/ABL2-knockdown breast cancer cells (Fig. 4, E and F). Depletion of the ABL kinases did not affect breast cancer cell proliferation or colony formation in vitro (fig. S1). Therefore, these findings suggest that ABL1/ABL2-dependent expansion of breast cancer cells is mediated by factors present in the bone microenvironment.
Fig. 4.
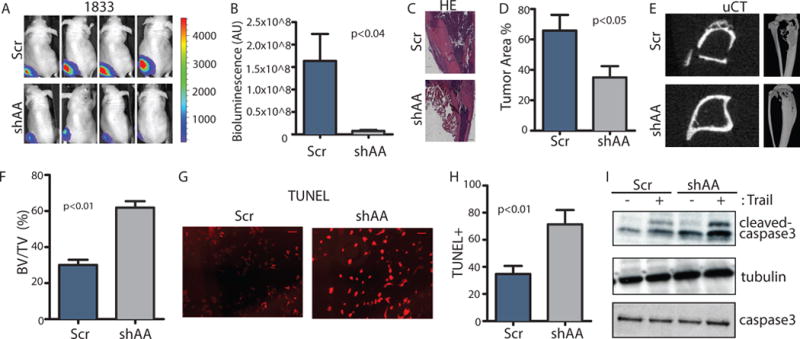
ABL kinases are required for tumor survival and tumor-induced osteolysis in the bone microenvironment.
(A–B) 1×105 control or ABL1/ABL2 knockdown 1833 cells were injected directly into the tibias of the mice. Representative bioluminescent images (A) taken at day 21 after inoculation and quantification (B) of bone lesions are shown. N=5 mice/group. (C–D) Representative HE staining (C) and quantification (D) of HE-stained tumor area of mouse tibias from each group. N=3 mice/group. Scale bar=500μM. (E–F) Representative 3D μCT reconstruction of mouse tibias (E) and quantification (F) of bone volume/total volume (BV/TV) from μCT analysis. N=3 mice (G–H) Representative images (G) of TUNEL staining of cells treated with TRAIL and the indicated shRNA and quantification of the percent of TUNEL-positive cells (H). N=3 biological replicates. Scale bar=100μM. (I) Immunobloting was performed using the indicated antibodies on whole-cell lysates from cells incubated or not with TRAIL. N=3 blots.
Bone metastasis requires reciprocal interactions between tumor cells, stromal cells, and bone cells (12, 26). Several soluble factors released by stromal cells within the bone microenvironment promote tumor growth and survival (27). These factors include Chemokine C-X-C Motif Ligand 12 (CXCL12), a chemokine produced by bone marrow mesenchymal cells that functions as a chemo-attractant and survival factor for cells bearing the Chemokine C-X-C Motif Receptor 4 (CXCR4), and the insulin-like growth factor 1 (IGF-1), a factor that is stored in the bone matrix and released during osteolysis (28). We and others have previously shown that ABL kinases are activated by the binding of CXCL12 and IGF-1 to their cognate receptors (6, 21). Thus, we evaluated whether loss of ABL kinases could affect activation of AKT-mediated survival by these factors. We found that CXCL12 and IGF-1 induced activation of AKT was independent of ABL1 and ABL2 in 1833 breast cancer cells (fig. S5, A and B). In contrast, we observed that ABL kinases protected breast cancer cells from TRAIL-induced cell death (Fig. 4, G, H, and I). TRAIL is a pro-apoptotic member of the tumor necrosis factor family, that induces apoptosis by binding to cell death receptors DR4 and DR5 (29). TRAIL and DR5 are present in clinical breast cancer bone metastases specimens, and DR4 and DR5 are present in 80% of patient bone tumor biopsies (28, 30). TRAIL enhanced apoptosis as measured by cleavage of caspase-3 (Fig. 4I), and knockdown of ABL kinases increased the sensitivity of 1833 breast cancer cells to the pro-apoptotic effects of TRAIL (Fig. 4, G, H and I). These data suggest that ABL kinases promote breast cancer metastasis to the bone in part by increasing tumor cell survival within the bone microenvironment.
Depletion of ABL kinases impairs tumor-induced osteoclast activation in part by decreased IL6 secretion
Osteoclast activation plays a central role in the progression of breast cancer bone metastasis. To directly examine whether ABL kinases regulate tumor-induced osteoclast activation, we employed an in vitro osteoclastogenesis assay (Fig. 5A). Mouse primary bone marrow cells were treated with conditioned media from control or ABL1/ABL2 knockdown breast cancer cells, and then stained for TRAP (an osteoclast marker). Bone marrow cells cultured with conditioned medium derived from ABL1/ABL2 knockdown 1833 and SKBR3 breast cancer cells had decreased numbers of TRAP+ cells compared to the control groups (Fig. 5, B and C; and fig. S6, A and B). These data suggest that inactivation of the ABL kinases in breast cancer cells may impair secretion of soluble factor(s) required for osteoclast activation. Because bone marrow contains a heterogeneous population of cells, factors secreted in an ABL1/ABL2-dependent manner might interact directly with osteoclast progenitors and promote their differentiation, or might instead function to regulate osteoclasts indirectly by modulating the activity of osteoblasts (22). To test the first possibility, we carried out the osteoclastogenesis assay using the RAW264.7 murine pre-osteoclast cell line. Conditioned media derived from ABL1/ABL2 knockdown 1833 breast cancer cells did not impair tumor-induced RAW264.7 pre-osteoclast differentiation (Fig. 5D). These findings suggest that ABL kinases regulate osteoclast maturation indirectly, possibly by modulating osteoblast function.
Fig. 5.
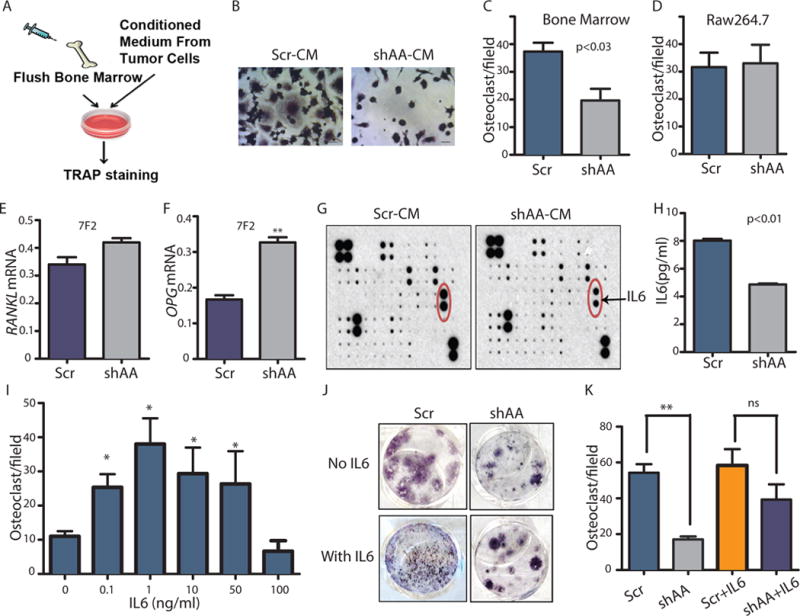
Depletion of ABL kinases impairs tumor-induced osteoclast activation in part by decreasing IL6 secretion.
(A) The in vitro osteoclastogenesis assay. (B) TRAP staining of bone marrow cells treated with conditioned medium (CM) from 1833 breast cancer cells. Scale bar=50μM. (C) Quantification of TRAP+ cells in (B). (D) Quantification of TRAP+ cells derived from RAW 264.7 cells; ns= non-significant. (E–F) RANKL (E) and OPG (F) expression was detected by RT-PCR of the osteoblast cell line 7F2 treated with CM harvested from the indicated 1833 cells. (G) Identification of differentially expressed cytokines in the CM of 1833 cells using a cytokine antibody array. N=2 biological replicates. (H) ELISA quantification of IL6 in CM of the 1833 cells. (I) Quantification of TRAP+ bone marrow-derived osteoclasts incubated with the indicated doses of IL6. * indicates significantly different from 0; p value was calculated using One-Way ANOVA followed by Tukey’s HSD. (J) TRAP staining of bone marrow treated with CM from 1833 cells with or without added IL6. (K) Quantification of TRAP+ osteoclasts in (J). * p<0.05; **p<0.01 p value was calculated using Two-Way ANOVA followed by Tukey’s HSD.
N=3 biological replicates unless otherwise indicated.
Osteoblasts modulate osteoclast activity through secretion of the TNF family member RANKL. Binding of RANKL to the RANK receptor on the surface of osteoclasts activates a pathway essential for osteoclast differentiation. Osteoprotegerin (OPG), a soluble decoy receptor for RANKL, is also produced by osteoblasts and antagonizes the activity of RANKL (31). Several tumor-derived bone metastasis factors can increase RANKL production or decrease OPG secretion by osteoblasts, thereby promoting osteoclast differentiation and activation (12, 32). To evaluate whether ABL kinases might regulate the secretion of osteoblast-derived RANKL or OPG leading to osteoclast differentiation, we analyzed RANKL and OPG mRNA abundance in the murine osteoblast cell line 7F2 in response to conditioned medium from control and ABL1/ABL2 knockdown breast cancer cells. While conditioned medium from ABL1/ABL2-depleted breast cancer cells did not affect RANKL abundance in osteoblasts compared with the cells treated with control conditioned medium (Fig. 5E), we found that conditioned medium from breast cancer cells lacking ABL kinases increased OPG abundance in the osteoblast cell line (Fig. 5F). These findings suggest that ABL kinases promote tumor-induced osteoclast activation in part by increasing OPG abundance in osteoblasts.
To identify tumor-secreted cytokines regulated by the ABL kinases that promote breast cancer metastasis to the bone, we used a human cytokine antibody array to identify changes in cytokine concentrations in the conditioned medium from control and ABL1/ABL2 knockdown cells. We found that IL6 concentrations were decreased in the conditioned medium derived from ABL1/ABL2 knockdown cells compared to that from control cells (Fig. 5G), results that were validated by ELISA (Fig. 5H). IL6 is a multifunctional cytokine with pleiotropic functions that include inducing osteoclast activation, enhancing bone resorption and increasing metastasis (33–35). Inhibition of the IL6 receptor directly blocks osteoclast formation in vitro and in vivo (36). Thus, we evaluated whether addition of IL6 could rescue in part defective osteoclastogenesis induced by conditioned medium from breast cancer cells depleted of the ABL kinases. We added the optimal dose of IL6 required to promote maturation of osteoclasts (Fig. 5I) to conditioned medium derived from ABL1/ABL2 knockdown breast cancer cells. Addition of IL6 to reconstituted CM from ABL1/ABL2-depleted breast cancer cells partially restored osteoclast activation (Fig. 5, J and K). IL6 induced RANKL expression but suppressed OPG expression in the 7F2 osteoblast cell line (fig. S7, A and B). Thus, depletion of the ABL kinases in breast cancer cells and the accompanying decrease of IL6 secretion results in enhanced OPG expression and reduced overall RANKL/OPG ratio, thereby decreasing osteoclast differentiation and bone destruction.
Next generation sequencing reveals ABL1/ABL2- regulated genes in breast cancer cells
To gain insight into the signaling pathway(s) required for ABL1/ABL2-dependent bone metastasis, we evaluated the consequences of single or double inactivation of ABL1 and ABL2 on the transcriptome of breast cancer cells using next generation sequencing (RNAseq) analysis (Fig. 6 and fig. S8A). We found that 180 genes showed significantly decreased expression and 40 genes showed significantly increased expression in ABL1/ABL2 knockdown cells (fig. S8B and Table S1). Principle component analysis revealed that transcripts altered in breast cancer cells depleted of ABL1, ABL2, or both ABL1 and ABL2 were clustered and were distinct from the control group (fig. S8C). This analysis indicated that the transcriptomes of single and double knockdown cells were similar to each other, but different from that of the control cells, supporting the quality and validity of the RNAseq analysis. Comparison of the transcripts revealed that breast cancer cells with knockdown for ABL2 alone shared a similar gene expression pattern with that of the ABL1/ABL2 double knockdown cells (Fig. 6A and fig. S8D). Notably, analysis of cell lysates from ABL2-depleted and ABL1/ABL2-double knockdown breast cancer cells showed a greater reduction of the phosphorylation of CrkL compared to cells with knockdown for ABL1 alone (Fig. 2K). Thus, the altered gene expression profiles correlate with decreased ABL kinase activity in breast cancer cells.
Fig. 6.
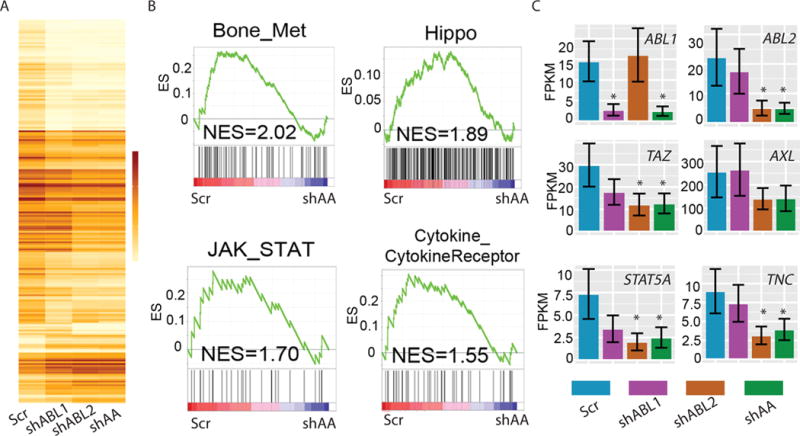
ABL kinases regulate the expression of genes in the JAK/STAT and Hippo pathway signatures in metastatic breast cancer cells.
(A) CummeRbund Heatmap of genes that were differentially expressed in control and single and double ABL1 and ABL2 knockdown cells. (B) GSEA analysis of the indicated gene signatures in ABL1/ABL2 knockdown cells (shAA) compared with control cells (Scr). NES, Normalized Enrichment Score (C) Expression of the indicated genes in control, ABL1 or ABL2 single knockdown and ABL1/ABL2 double knockdown cells quantified using Cufflinks CuffDiff. * indicates significantly (p<0.05 after Benjamini-Hochberg Correction for multiple-testing) different from Scr. Error bars represent Standard Deviation (SD). N=3 biological replicates for (A) and (C).
ABL Kinases Signal to TAZ and STAT5 to Promote Breast Cancer Bone Metastasis
To identify the pathways affected by inactivation of the ABL kinases in metastatic breast cancer cells, we carried out Gene Set Enrichment Analysis (GSEA) using multiple databases (37). In addition to the GSEA oncogenic signature database, we employed the KEGG database and published breast cancer metastasis datasets. We found that a gene signature consisting of 273 genes important for breast cancer bone metastasis showed decreased expression in ABL1/ABL2 knockdown cells (Fig. 6B) (38). Further, inactivation of the ABL kinases resulted in decreased expression of the genes in the Hippo, JAK/STAT, and Cytokine/Cytokine Receptor pathway signatures (Fig. 6B). To identify key molecular mediators of the ABL kinases implicated in the regulation of the ABL1/ABL2-dependent pathways, we analyzed the expression of individual genes for transcripts altered by loss of the ABL kinases. Among transcripts that were decreased in ABL1/ABL2 knockdown cells were TAZ (also known as WWTR1, WW domain containing transcription regulator protein 1), which encodes a transcriptional co-activator in the Hippo pathway, and STAT5A, which encodes a transcription factor (Fig. 6C).
TAZ and the related YAP1 proteins are components of the Hippo pathway and have been implicated in breast cancer progression and metastasis (39, 40). We found that knockdown of the ABL kinases decreased the mRNA expression of TAZ (Fig. 6C), and reduced the protein abundance of TAZ and its downstream target AXL (Fig. 7A, fig S9, A, B). AXL encodes a receptor tyrosine kinase that promotes breast cancer bone metastasis in mouse models (41). Knockdown of ABL2, but not of ABL1, reduced TAZ abundance to a similar extent as ABL1/ABL2 double knockdown (fig. S9A), suggesting that ABL2 has a predominant role in regulating TAZ abundance. Overexpression of ABL1 and ABL2 in both 1833 and parental MDA-MB-231 breast cancer cells increased TAZ abundance (fig. S9C). Further, immunofluorescence staining analysis indicated that the TAZ protein predominantly (~90%) localized in the nuclei of 1833 breast cancer cells (fig. S10A), an effect decreased by double-knockdown of ABL1 and ABL2 (fig. S10B).
Fig. 7.
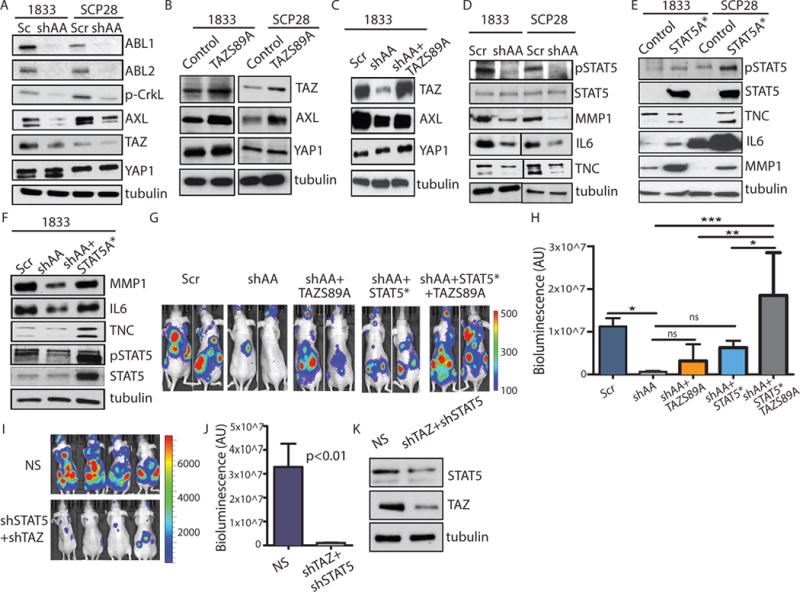
ABL kinases are required for TAZ and STAT5 signaling in breast cancer cells.
(A–C) Immunoblots using the indicated antibodies were performed on whole-cell lysates of 1833 and SCP28 cells. (D) Immunoblots were performed on whole-cell lysates (pSTAT5, STAT5, tubulin), or conditioned medium (MMP1, IL6, TNC). (E) Immunoblots were performed on whole-cell lysates (pSTAT5, STAT5, tubulin), or conditioned medium (MMP1, IL6, TNC) of parental 1833 and SCP28 cells. (F) Immunoblotting using the indicated antibodies were performed on whole-cell lysates (pSTAT5, STAT5, tubulin), or conditioned medium (MMP1, IL6, TNC) of 1833 cells. For (A–F), N=3 blots. (G–H) Bioluminescent images (G) of representative mice at day 25 after intracardiac injection of 1833 cells. Quantification (H) of bone metastasis. N=5 mice/group. * p<0.05; **p<0.01; ***p<0.001. p value was calculated using One-Way ANOVA followed by Tukey’s HSD. (I–J) Bioluminescent images (I) and quantification (J) of bone metastasis from representative mice at day 25 after intracardiac injection of 1833 cells transfected with control (NS) or shRNAs against STAT5 and TAZ (shSTAT5/shTAZ). N=8 mice/group. (K) Immunoblots were performed on whole cell lysate. N=3 blots.
Similarly, inhibiting ABL kinase activity with the allosteric inhibitor GNF5 decreased TAZ protein abundance (fig. S11). TAZ protein abundance was not decreased by GNF5 treatment in breast cancer cells expressing murine Abl2-E505K, a mutant that is resistant to the GNF5 allosteric inhibitor (fig. S11). Moreover, ABL2 mRNA expression positively correlated with TAZ mRNA expression in a TCGA dataset of 971 invasive breast cancer patients (fig. S12). To evaluate whether loss of ABL kinases affected TAZ activity, we performed ChIP analysis using primers for TAZ targets identified by ChIP-Seq analysis (42). We found that depletion of the ABL kinases decreased TAZ binding to some of its target genes (fig. S13). While ABL1 has been reported to phosphorylate YAP1 in response to DNA damage (43), we found that ABL1/ABL2 knockdown did not substantially alter YAP1 protein abundance (Fig. 7A), nuclear localization (fig. S14, A and B), phosphorylation of YAP at Tyr357 (fig. S14C) or binding to some of its downstream targets (fig. S13). However, we cannot rule out the possibility that ABL kinases might regulate YAP1-mediated expression of other target genes in breast cancer cells. Regardless, our data support a role for ABL kinases in the regulation of TAZ protein abundance and activity in breast cancer cells. Moreover, we found that expression of a constitutively active TAZ S89A mutant in ABL1/ABL2 knockdown breast cancer cells (1833 and SCP28) restored the abundance of its target AXL (Fig. 7, B and C). Together these data revealed a functional link between the ABL kinases and TAZ signaling leading to increased AXL abundance in breast cancer cells, and identified a potentially druggable pathway for the treatment of breast cancer bone metastasis.
We found that inactivation of the ABL kinases in breast cancer cells also decreased STAT5A mRNA and downstream expression of STAT5 target genes, including Tenascin C (TNC) (Fig. 6D). STAT5 belongs to a family of transcription factors that regulate cytokine-induced gene expression and is constitutively activated in several human cancers including breast cancer, where it promotes expression of genes encoding cell survival factors (44). STAT5 is also activated by the oncogenic BCR-ABL tyrosine kinase and contributes to the transformation of leukemia cells (45). STAT5 promotes metastasis of human prostate cancer cells (46), and has been implicated in the resistance of metastatic breast cancer cells to targeted therapies (47). Moreover, ablation of a STAT5A allele reduces tumor incidence in a mouse model of breast cancer in which mammary epithelial cells express T antigen (48). We found that depletion of ABL kinases in breast cancer cells decreased STAT5A mRNA expression (Fig. 6D), without decreasing total STAT5 protein abundance as measured by western blotting with antibodies that detect both STAT5A and STAT5B (Fig. 7D, and fig. S9, A and B). However, depletion of ABL kinases decreased the phosphorylation of STAT5 (Fig. 7D, and fig. S9, A and B). Conversely, overexpression of ABL kinases, predominantly ABL1, in both 1833 and parental MDA-MB-231 breast cancer cells increased STAT5 phosphorylation (fig. S9C). Further, we found that double-knockdown of ABL1 and ABL2 decreased the abundance of various secreted proteins, including IL6, TNC, and MMP1 (Fig. 7D). Both MMP1 and IL6 have been linked to the regulation of osteoclast activation (32, 33), and depletion of TNC decreases breast cancer metastasis (49). Expression of a constitutively active STAT5A mutant (STAT5A*) reversed the reduction in MMP1, IL6, and TNC abundance induced by depletion of both ABL kinases in breast cancer cells (Fig. 7, E and F and fig. S15). These findings support a role for STAT5 in regulating the ABL1/ABL2-dependent secretome.
To evaluate whether TAZ and STAT5 pathways promote breast cancer bone metastasis downstream of the ABL kinases, we expressed the constitutively active mutants of TAZ S89A and STAT5* in ABL1/ABL2 knockdown cells. Expression of either TAZ S89A or STAT5* alone in ABL1/ABL2-depleted breast cancer cells only partially rescued bone metastasis, and expression of both STAT5* and TAZ S89A was required to fully rescue the impaired bone metastasis by ABL1/ABL2-depleted cells (Fig. 7, G and H). To evaluate whether depletion of TAZ and STAT5 in breast cancer could phenocopy the reduced bone metastasis caused by depletion of ABL1 and ABL2, we performed intracardiac injection of control or TAZ/STAT5 double knockdown 1833 breast cancer cells, and found that cells depleted of TAZ and STAT5 exhibited markedly impaired metastasis that phenocopied the inhibitory effects of ABL1/ABL2 knockdown (Fig. 7, I, J and K). TAZ and STAT5 might regulate each other (fig. S16). TAZ knockdown with two distinct shRNAs decreased the phosphorylation of STAT5 and to a lesser extent total STAT5 protein abundance (fig. S16A). Moreover, knockdown of STAT5 with two different shRNAs slightly decreased TAZ protein abundance (fig. S16B). However, these reciprocal decreases were much lower than those induced by knockdown of the ABL kinases (Fig. 7). Future studies will evaluate the pathways that mediate the crosstalk between the TAZ and STAT5 pathways. Together our findings suggest that ABL kinases activate TAZ and STAT5 pathways and that co-activation of their downstream targets promote the bone metastasis of breast cancer cells in mouse models. Using a TCGA dataset with 971 invasive breast cancer patients, we found that patients with alterations in the expression of ABL2 and eight validated downstream targets (TAZ, AXL, CTGF, STAT5A, STAT5B, TNC, IL6, and MMP1) exhibited decreased disease-free survival (Fig. 8A).
Fig. 8.
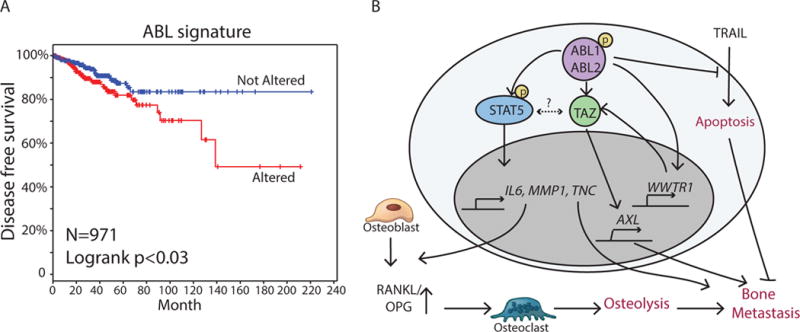
ABL kinases activate TAZ and STAT5 pathways to promote breast cancer bone metastasis.
(A) Kaplan-Meier representation of the probability of cumulative overall disease free survival in TCGA dataset with 971 invasive breast cancer patients according to whether the ABL signature (ABL2, TAZ, AXL, CTGF, STAT5A, STAT5B, TNC, IL6, MMP1) was altered or not. P value was derived by log rank test.
(B) Model for the role of ABL kinases in the regulation of breast cancer bone metastasis.
Discussion
In this work, we uncovered a role for the ABL kinases in promoting breast cancer bone metastasis through the regulation of distinct pathways required for tumor colonization and survival in the bone microenvironment. We showed that ABL kinase activity was required for osteolytic metastasis of breast cancer cells and that depletion or pharmacological inhibition of these kinases impaired breast cancer metastasis to bone. We demonstrated that ABL kinases promoted tumor-induced osteolysis in part through the osteoclast-activating cytokine IL6, increased serum concentrations of which are associated with poor clinical outcome in breast cancer patients (50). IL6 can induce osteoclast activation indirectly by altering the expression of RANKL and OPG in osteoblasts (51). Depletion of ABL kinases in breast cancer cells decreased IL6 concentrations and was accompanied by increased OPG expression in osteoblasts. We found that the addition of IL6 partially enabled medium conditioned by ABL1/ABL2 knockdown breast cancer cells to activate osteoclasts, and suppressed OPG expression in the 7F2 osteoblast cell line. These findings support a role for ABL-mediated IL6 secretion by breast cancer cells in osteoclast activation through decreased OPG expression in osteoblasts. IL6 has pleiotropic roles during tumor progression and metastasis that include increased tumor cell survival, expansion of breast cancer stem cells and resistance to targeted therapies (52). IL6 may also function downstream of the ABL kinases to protect breast cancer cells from TRAIL-induced cell death.
Depletion of ABL kinases in breast cancer cells also decreased the abundance of MMP1, a protease that cleaves fibrillar collagens and promotes the proteolytic release of bound growth factors (32). High serum concentrations of MMP1 correlate with bone metastasis in breast cancer patients (32). Through its various targets, MMP1 promotes not only tumor invasion but also breast cancer colonization to the bone by mechanisms that include the release of membrane-bound EGF-like growth factors from tumor cells, leading to activation of EGFR signaling and suppression of OPG expression in osteoblasts, which in turn promotes the differentiation and activation of osteoclasts required for bone destruction and enhanced tumor growth in the bone microenvironment (32). Thus, decreased osteoclast activation and colonization of breast cancer cells lacking ABL kinases may be mediated in part by reduced MMP1 abundance.
Mechanistically, we found that STAT5 was required for the production of the secreted factors MMP1 IL6, and TNC downstream of ABL kinases. Similar to IL6 and MMP1, TNC is abundant in some breast tumors and promotes metastasis in mouse models (49). We showed that inactivation of the ABL kinases in breast cancer cells resulted in decreased expression of genes in the JAK/STAT and Cytokine/Cytokine Receptor pathway signatures, which may be due to decreased STAT5A mRNA expression as well as reduced STAT5 phosphorylation in ABL1/ABL2-depleted breast cancer cells. Expression of a constitutively active version of STAT5A in ABL1/ABL2 knockdown cells restored the production of secreted factors (IL6, MMP1, and TNC) and partially rescued the ability of breast cancer cells to promote bone metastasis. These findings support a role for the ABL kinase-STAT5 signaling axis in breast cancer metastasis.
In addition to inhibiting STAT5 signaling, we found that depletion of ABL kinases decreased the expression of the Hippo pathway mediator TAZ and downstream target genes in triple-negative and HER2+ breast cancer cells. Inactivation of ABL kinases inhibited expression of the TAZ target gene AXL, which shows increased expression in several human cancers and correlates with poor prognosis, increased invasiveness and metastasis, and enhanced drug resistance (53, 54). AXL mediates resistance to TRAIL-induced cell death in esophageal adenocarcinoma (55); it remains to be determined whether ABL-mediated protection from TRAIL-induced apoptosis in breast cancer cells is mediated by AXL or other targets. Co-expression of both activated TAZ and STAT5 pathways was required to fully rescue the bone metastatic phenotype of ABL1/ABL2 knockdown breast cancer cells. These data, together with the finding that knockdown of both TAZ and STAT5 phenocopied the decrease in breast cancer cell metastasis caused by ABL1/ABL2 depletion, supports a model for a requirement of ABL-dependent TAZ and STAT5 signaling networks in promoting breast cancer metastasis (Fig. 8B). Our data raise the possibility that inhibition of ABL kinases can increase apoptosis of breast cancer cells and block osteoclast activation that is required for osteolytic metastasis. Some Phase I and II trials have been carried out using non-selective ATP-competitive inhibitors such as imatinib to treat patients with advanced breast cancer. However, patients enrolled in these trials were not evaluated for ABL abundance and/or activation (56, 57). The allosteric inhibitors specific for ABL kinases (which are currently in clinical trials) provide a potentially useful tool for selectively targeting ABL kinases in metastatic breast cancer types with an increase in the ABL pathway signature (58). We found that allosteric inhibition of the ABL kinases effectively impaired breast cancer bone metastasis and blocked tumor-induced osteolysis in mouse models. Future studies will test and compare the efficacy of imatinib and allosteric compounds in mouse models of breast cancer. We will also test whether ABL-directed therapy can shrink established bone metastasis in the mouse models. Together, our data suggest that clinical studies may be warranted to evaluate the therapeutic potential of ABL allosteric inhibitors and to determine whether combination therapies that incorporate these compounds are effective in treating metastatic breast cancer. It is also striking that multiple downstream targets of the ABL kinases in breast cancer cells (TAZ, AXL, STAT5 and IL6) have been implicated in therapy resistance. It remains to be determined whether activated ABL kinases play a role in the development of intrinsic and/or acquired resistance in breast cancer cells.
Materials and Methods
Cell Culture
The human breast carcinoma cell line, MDA-MB-231 was purchased from ATCC. The 1833 (bone metastasis), 4175 (lung metastasis), and BrM2a (brain metastasis) sub-lines were derived from the parental cell line MDA-MB-231 (19) and were generous gifts from Dr. Joan Massague (Memorial Sloan-Kettering Cancer Center). The SCP28 (bone metastasis) sub-line was generously provided by Dr. Yibin Kang (Princeton University). MDA-MB-231, their derivative sub-lines and genetically modified versions were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies), antibiotics, and appropriate selection drugs for transfected plasmids. HEK293T cells, a packaging cell line for lentivirus production, and the pre-osteoclast cell line RAW264.7 (ATCC) were maintained in DMEM supplemented with 10% FBS, and antibiotics. The murine osteoblast cell line 7F2 (ATCC) was cultured in alpha-MEM with 10% FBS. The human mammary epithelial cell line (HuMEC) was maintained in Human Basal Serum Free Medium (Life Technologies) with HUMEC Kit (Life Technologies). The human breast cancer cell line SKBR3 was purchased from Duke University Cell Culture Facility, and was maintained in McCoy’s 5 medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies) and antibiotics. All cultures were maintained at 37 degree in humidified air containing 5% CO2.
Antibodies
Antibodies used for Western blotting included cleaved caspase-3, phospho-CrkL(Y207), phospho-Akt (Ser473), Akt, TAZ, YAP1, phospho-STAT5 (Tyr694), STAT5 and ERBB2 from Cell Signaling; β-tubulin and actin from Sigma-Aldrich; ABL2 (9H5) from Santa Cruz; ABL1(8E9) from BD Biosciences; IL6, TNC and phospho-YAP1(Y357) from Abcam; MMP1 from CALBIOCHEM. Antibodies used for Immunofluorescence staining included YAP1 from Cell Signaling and TAZ from BD Biosciences. Antibodies used for CHIP assays were TAZ (V386) and YAP1 (D8H1X) from Cell Signaling.
Tumor Xenografts and Analysis
Procedures involving mice were approved and performed following the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Duke University Division of Laboratory Animal Resources (DLAR). Age-matched female athymic NCr nu/nu mice (5–6 weeks) were used for xenograft experiments. For intra-cardiac injections, cells were harvested from subconfluent culture plates, washed with PBS, and resuspended at 106/mL (1833) or 5×106/mL (SCP28) in PBS; 0.1mL of the suspended cells were injected into the left cardiac ventricle using 30G needles. Mice were anesthetized with isoflurane before injection and imaged by bioluminescence imaging. For intra-tibia injections, mice were anesthetized using a mixture of ketamine (100mg/kg) and xylazine (10mg/kg). The injection site was cleaned with 70% alcohol wipe. Single-cell suspensions (1×105 cells) in a final volume of 10ul were injected into the upper half of the tibia medullary cavity, as felt by a lack of resistance when pushing cells into the cavity. Bioluminescence imaging was used to confirm successful cancer cell innoculation and progression of metastatic bone lesions. The allosteric inhibitor GNF5 was synthesized by Duke University Small Molecule Synthesis Facility. For drug treatment, mice were dosed with GNF5 in DMSO/Peanut Oil (1:9) at 50 mg/kg by intraperitoneal injection daily.
Bone μCT Analysis
Hindlimb bones were excised, fixed in 10% neutral-buffered formalin and imaged using a μCT scanner (Skyscam 1176, Bruker Corp) at 17 μm resolution and 180° scanning with a rotation step of 0.7° per image, 242ms exposure time, 55kV photon energy and 455 μA current. The images were reconstructed using NReconServer and bone volume was analyzed by CT Analysis Software (CTAn, Bruker Corp).
In Vitro Osteoclastogenesis Assay
Bone marrow cells were flushed out from femora and tibia of 6-week-old C57BL/6 mice and plated in basal culture medium (α-MEM supplemented with 10% FBS and antibiotics) overnight. Osteoclastogenesis assay and TRAP staining were conducted as described previously (32). Tumor cells were plated at 2 × 105 per well in 12-well plates to obtain conditioned media for incubation with either bone marrow cells or RAW264.7 cells. RAW264.7 pre-osteoclast cells were plated at 4 × 105 per well in 24-well plates overnight. RAW264.7 medium was replaced by conditioned medium harvested from tumor cells and supplemented with recombinant murine sRANKL (50ng/mL). Medium was changed every 3 days, and TRAP staining was performed on day 6 per manufacturer’s instructions (Sigma).
RNAseq Analysis
For RNAseq analysis, 3×106 breast cancer cells were plated in 10-cm Petri dish in triplicate in complete medium for 24 hours. Cells were harvested and RNA was isolated using the RNeasy kit (Qiagen); 1 μg total RNA input was used for each sample. The libraries were sequenced on an Illumina HiSeq 2000 sequencing system using 50-bp single-ended reads. RNAseq data was mapped to reference genome (HG19) using Bowtie/Tophat. Reads were counted and differential expression between distinct experimental groups was quantified using Cuffdiff. Significant genes were extracted using R CummeRbund.
Viral Transduction
Retroviral or lentiviral constructs were transfected into HEK293T cells with their packaging vectors indicated below using FuGENE6 reagent (Promega). pMX-puro-STAT5A* (kindly provided by Toshio Kitamura, University of Tokyo): pCMV-Gag-Pol and pCMV-VSV-G; Plenti-EF-FH-TAZ-S89A: psPAX2 and pVSV-G; PLKO-NS and shTAZ lentiviral construct (kindly provided by Corinne Linardic, Duke University): pCMV-Rev, pCVM-VSVG; pMDL. Retroviral or lentiviral supernatants were collected and filtered 24 h and 48 h post-transfection. 1833 cells were incubated 48 h with retroviruses or lentiviruses medium in the presence of 8μg/mL Polybrene. Cells were cultured at least 3 days in 3μg/mL Puromycin or 5μg/mL Blasticidin for selection. Lentiviral shRNA-mediated knockdown of ABL1/ABL2 and expression of mouse Abl1/Abl2 were conducted as described previously (20, 21, 59). Lentiviral shRNA-mediated knockdown of STAT5A and STAT5B was conducted per manufacturer’s instruction (Dharmacon RHS4531-EG6776, EG6777).
Immunoblotting
Cells were lysed in radio-immunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors. Cell debris was removed by microcentrifugation, and protein was quantified. Alternatively, equal cell numbers were plated onto 6-well plates and 24 hrs later, conditioned medium was harvested and concentrated using Amicon Ultra centrifugal filters (Millipore). Equal amounts of protein or conditioned media were separated by SDS/PAGE and transferred to nitrocellulose membranes and probed with the indicated antibodies.
Real-Time RT-PCR
RNA was isolated from cancer cells using the RNeasy RNA isolation kit (Qiagen), and cDNA was synthesized using Oligo(dT) primer and M-MLV reverse transcriptase (Invitrogen). Real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad). Primers used were as follows- mouse RANKL (forward): 5′-TTG CAC ACC TCA CCA TCA, (reverse): 5′-TAC GCT TCC CGA TGT TTC; mouse OPG (forward): 5′-CAC TCG AAC CTC ACC ACA, (reverse): 5′-CAA GTG CTT GAG GGC ATA; mouse GAPDH (forward): 5′-CTC ATG ACC ACA GTC CAT GC, (reverse): 5′-ACA CAT TGG GGG TAG GAA CA; human ABL1 (forward): 5′-GGC TGT GAG TAC CTT GCT GC, (reverse): 5′-GGC GCT CAT CTT CAT TCA GGC; human ABL2 (forward): 5′-CCA GCT ACT CCC GAG GCT G, (reverse): 5′-CTT GAT CCC ACA GGG TGA AG; human GAPDH (forward): 5′-GGC TCT CCA GAA CAT CAT CCC, (reverse): 5′-GGG TGT CGC TGT TGA AGT CAG; human MMP1 (forward): 5′-GGT CTC TGA GGG TCA AGC AG, (reverse): 5′-AGT TCA TGA GCA ACA CG; human IL6 (forward): AGA CAG CCA CTC ACC TCT TC, (reverse): 5′-TTT CAC CAG GCA AGT CTC CT; human TNC (forward): 5′-CCC TAC GGG TTC ACA GTT TC, (reverse): 5′-TTC CGG TTC GGC TTC TGT AAC; human AXL (forward): 5′-ATC AGA CCT TCG TGT CCC AG, (reverse): 5′-ATG TCT TGT TCA GCC CTG GA; human WWTR1 (forward): 5′-GGC TGG GAG ATG ACC TTC AC, (reverse): 5′ -AGG CAC TGG TGT GGA ACT GAC; human YAP1 (forward): 5′ -ATG AAC TCG GCT TCA GCC AT, (reverse): 5′ - ACC ATC CTG CTC CAG TGT TG. Analysis was performed using a Bio-Rad CFX384 real-time machine and CFX Manager software. PCR assays were performed in triplicate. Expression of each gene was normalized to that of the GAPDH gene.
Histological Analysis
Hindlimb bones were excised, fixed in 10% neutral-buffered formalin, decalcified, and embedded in paraffin for hematoxylin (H&E) staining (28).
TUNEL Staining
For TUNEL staining, 2×104 cells were seeded onto each single chamber of a 4-chamber side in complete medium. The next day, the medium was replaced with serum-free media containing TRAIL (2ng/mL). After 3 days, cells were fixed using 4% PFA and permeablized with 0.1% Triton X-100. TUNEL staining was performed following the manufacturer’s protocol (Roche Applied Science).
ELISA and Cytokine Array
Conditioned medium was collected after 24 h incubation from confluent cells and was applied to the Human Cytokine Antibody Array C1000 (Raybiotech) or IL6 ELISA kit (R&D) following the manufacturer’s instructions.
Invasion Assay
Invasion was evaluated by plating 25,000 cells in the upper chambers of 8.0 μm pore size matrigel chambers (BD Biosciences) in serum-free medium. Cells were allowed to invade for up to 48 h in the presence of serum-containing medium in the bottom chamber.
Afterwards, the remaining cells, medium, and matrigel were removed from the upper chambers, and cells on the undersurface of the membrane were fixed, stained with DiffQuik (Dade Behring), and quantified by microscopy.
In Vitro Cell Growth Assays
For 2D-cell growth, 3000 cells were seeded onto each well of a 96-well plate. Cell growth was measured daily from day 1 to 5 using CellTiter-Glo (Promega) following the manufacturer’s protocol. For 3D-cell growth, 75μl of matrigel (BD Bioscience) was plated onto each well of a 96-well plate. A single cell suspension (50μl) containing 1500 cells was mixed with matrigel 1:1 and plated on top of the matrigel base onto wells of a 96-well plate; 50μl of complete medium was added and cells were cultured for 14 days. Colonies were analyzed and counted using microscope.
CHIP-QPCR Analysis
CHIP-QPCR was performed using Cell Signaling SimpleChIP Plus Enzymatic Chromatin IP Kit (#9005) according to manufacturer’s instructions. SimpleChIP human CTGF promoter primers were from Cell Signaling (#14927). Validated TAZ/YAP primers, as well as previously validated primers for AJUBA, AMOTL2, and WTIP were used for QPCR analysis (42). Antibodies used for CHIP assays were anti-TAZ (V386) and anti-YAP1 (D8H1X) from Cell Signaling. Primers used were as follows- AXL (forward): 5′-CAG CCT CCT CCT CAC AGA CA; AXL (reverse): 5′-GAG CCC TGA TCA TTC CAC TG; AJUBA (forward): 5′-AAG GAA AGA GTG TGG GGG TAG G; AJUBA (reverse): 5′-ACG CTG GGA ACA AAG TCA CG; AMOTL2 (forward): 5′- TGC CAG GAA TGT GAG AGT TTC; AMOTL2 (reverse): 5′-AGG AGG GAG CGG GAG AAG; WTIP (forward): 5′-GCA GCG CCG TCT CCT TCC T; WTIP (reverse): 5′-GCG GCG GAG GAA TGT AAG CTC.
Mutagenesis
Abl2 E505K mutagenesis was conducted on pBabe-puro-mAbl2 construct using Q5 Site-Directed Mutagenesis Kit (NEB E0554S) according to the manufacturer’s instructions. Primers used were as follows: Abl2 (forward): 5′-CAT CTC TGA AAA GGT AGC TCA G; Abl2 (reverse): 5′-CTG GAG TCA TGG AAC ATT G.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6, JMP Pro and R 3.2. Comparisons of two groups were performed using Student’s t tests (two-tailed). Comparisons involving multiple groups were evaluated using one-way or two-way ANOVA, followed by Tukey’s HSD. For all tests, p<0.05 was considered statistically significant. For all figures, p value was calculated using Student’s t test unless otherwise indicated. Data shown represent averages ± SEM unless otherwise indicated.
Supplementary Material
Fig. S1. ABL protein abundance is increased in breast cancer cells with enhanced bone metastatic activity and ABL1/ABL2 depletion does not affect proliferation in vitro.
Fig. S2. ABL kinases promote breast cancer cell invasion.
Fig. S3. Depletion of ABL kinases does not inhibit metastasis of breast cancer cells, which show tropism to the lung.
Fig. S4. Treatment of breast cancer cells with imatinib but not GNF5 promotes ERK activation.
Fig. S5. CXCL12- and IGF1-mediated survival pathways are independent of ABL kinases.
Fig. S6. Depletion of ABL kinases in SKBR3 breast cancer cells decreases tumor-induced osteoclast activation.
Fig. S7. IL6 affects RANKL and OPG expression in osteoblasts.
Fig. S8. Quality control and global statistics of RNAseq analysis for transcriptome comparison of control versus ABL1/ABL2 knockdown breast cancer cells.
Fig. S9. ABL kinases increase TAZ protein abundance and STAT5 phosphorylation.
Fig. S10. Depletion of ABL kinases reduces the abundance of TAZ in the nucleus.
Fig. S11. Allosteric inhibition of ABL kinases activity decreases TAZ protein abundance.
Fig. S12. ABL2 mRNA expression positively correlates with TAZ mRNA expression in invasive breast cancer patients.
Fig. S13. Depletion of ABL kinases decreases the binding of TAZ to target genes.
Fig. S14. Depletion of ABL kinases does not affect YAP1 protein abundance, localization, or tyrosine phosphorylation in breast cancer cells.
Fig. S15. Expression of a constitutively active STAT5 mutant increases mRNA expression of MMP1, IL6, and TNC.
Fig. S16. Potential interdependence of TAZ and STAT5 in breast cancer cells.
Table S1. Differentially expressed genes in control and ABL1/ABL2 knockdown breast cancer cells.
Acknowledgments
We thank Dr. Joan Massague (Memorial Sloan-Kettering, NY) and Dr. Yibin Kang (Princeton University, NJ) for the breast cancer cell lines and Toshio Kitamura (University of Tokyo) and Corinne Linardic (Duke University) for the viral vectors. We are grateful to Donald P. McDonnell (Duke University School of Medicine, NC) for support with the analysis of breast cancer datasets. We thank Dr. Theodore Slotkin (Duke University School of Medicine, NC) for support with statistical analysis. We thank Dr. Farshid Guilack (Duke University School of Medicine, NC), Dr. Emily Riggs (Duke University School of Medicine, NC), Duke University Light Microscopy Core Facility, and Duke Cancer Center Flow Cytometry Shared Resource for technical assistance. We thank Duke University Department of Pathology for evaluation of histology specimens. We thank Dr. Pameeka Smith-Pearson for supplemental data support.
Funding: This work was supported by National Institutes of Health (NIH) Grant R01 CA155160 (to A.M.P.) and the National Center for Advancing Translational Sciences of the NIH under Award Number UL1TR001117 (to A.M.P.), and NIH Grant CA174643 (to D.P.M).
Footnotes
Author contributions: J. W. and A.M.P. conceived the study, generated the hypotheses, designed the experiments and analyzed the data. J.W. performed all of the experiments. C.R. provided technical expertise and contributed to the mouse experiments. J. S. J. carried out a meta-analysis of previously published microarray data of breast cancer patients. J. W. and A.M.P. wrote the manuscript.
Competing interests: The authors have no conflict of interest to declare.
Data and materials availability: The Gene Expression Omnibus accession number for RNAseq data reported in this paper is GSE69125.
References and Notes
- 1.Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J Cell Sci. 2009;122:3441–3454. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 2010;3:re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- 4.Wong S, Witte ON. The BCR-ABL story: bench to bedside and back. Annual review of immunology. 2004;22:247–306. doi: 10.1146/annurev.immunol.22.012703.104753. [DOI] [PubMed] [Google Scholar]
- 5.Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013;13:559–571. doi: 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganguly SS, Plattner R. Activation of abl family kinases in solid tumors. Genes & cancer. 2012;3:414–425. doi: 10.1177/1947601912458586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanchard Z, Mullins N, Ellipeddi P, Lage JM, McKinney S, El-Etriby R, Zhang X, Isokpehi R, Hernandez B, Elshamy WM. Geminin overexpression promotes imatinib sensitive breast cancer: a novel treatment approach for aggressive breast cancers, including a subset of triple negative. PLoS One. 2014;9:e95663. doi: 10.1371/journal.pone.0095663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gil-Henn H, Patsialou A, Wang Y, Warren M, Condeelis J, Koleske A. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene. 2012 doi: 10.1038/onc.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sirvent A, Boureux A, Simon V, Leroy C, Roche S. The tyrosine kinase Abl is required for Src-transforming activity in mouse fibroblasts and human breast cancer cells. Oncogene. 2007;26:7313–7323. doi: 10.1038/sj.onc.1210543. [DOI] [PubMed] [Google Scholar]
- 10.Srinivasan D, Sims JT, Plattner R. Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival. Oncogene. 2008;27:1095–1105. doi: 10.1038/sj.onc.1210714. [DOI] [PubMed] [Google Scholar]
- 11.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waning DL, Guise TA. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin Cancer Res. 2014;20:3071–3077. doi: 10.1158/1078-0432.CCR-13-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cicek M, Oursler MJ. Breast cancer bone metastasis and current small therapeutics. Cancer Metastasis Rev. 2006;25:635–644. doi: 10.1007/s10555-006-9035-x. [DOI] [PubMed] [Google Scholar]
- 14.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson ER, Chang CY, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends in endocrinology and metabolism: TEM. 2014;25:649–655. doi: 10.1016/j.tem.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bos PD, Zhang XHF, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 20.Chislock EM, Ring C, Pendergast AM. Abl kinases are required for vascular function, Tie2 expression, and angiopoietin-1‚Äìmediated survival. Proceedings of the National Academy of Sciences. 2013;110:12432–12437. doi: 10.1073/pnas.1304188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith-Pearson PS, Greuber EK, Yogalingam G, Pendergast AM. Abl kinases are required for invadopodia formation and chemokine-induced invasion. Journal of Biological Chemistry. 2010;285:40201–40211. doi: 10.1074/jbc.M110.147330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19:192–205. doi: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Packer LM, Rana S, Hayward R, O’Hare T, Eide CA, Rebocho A, Heidorn S, Zabriskie MS, Niculescu-Duvaz I, Druker BJ, Springer C, Marais R. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–727. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, Couch B, Koleske AJ, Smithgall TE, Gray NS. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. The Journal of biological chemistry. 2009;284:29005–29014. doi: 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 27.Ell B, Kang Y. SnapShot: Bone Metastasis. Cell. 2012;151:690–690 e691. doi: 10.1016/j.cell.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 30.Mitsiades N, Poulaki V, Mitsiades C, Tsokos M. Ewing’s sarcoma family tumors are sensitive to tumor necrosis factor-related apoptosis-inducing ligand and express death receptor 4 and death receptor 5. Cancer Res. 2001;61:2704–2712. [PubMed] [Google Scholar]
- 31.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Wang Q, Hu G, Van Poznak C, Fleisher M, Reiss M, Massague J, Kang Y. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes & development. 2009;23:1882–1894. doi: 10.1101/gad.1824809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, Koishihara Y, Ohsugi Y, Kumaki K, Taga T, et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci U S A. 1993;90:11924–11928. doi: 10.1073/pnas.90.24.11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Udagawa N, Takahashi N, Katagiri T, Tamura T, Wada S, Findlay DM, Martin TJ, Hirota H, Taga T, Kishimoto T, Suda T. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. The Journal of experimental medicine. 1995;182:1461–1468. doi: 10.1084/jem.182.5.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell research. 2005;15:57–62. doi: 10.1038/sj.cr.7290266. [DOI] [PubMed] [Google Scholar]
- 36.Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis and rheumatism. 2009;60:2747–2756. doi: 10.1002/art.24781. [DOI] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vashisht S, Bagler G. An approach for the identification of targets specific to bone metastasis using cancer genes interactome and gene ontology analysis. PLoS One. 2012;7:e49401. doi: 10.1371/journal.pone.0049401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, Bicciato S, Piccolo S. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- 40.Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S. Role of TAZ as mediator of Wnt signaling. Cell. 2012;151:1443–1456. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 41.Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT, Micklem DR, Akslen LA, Glackin C, Lorens JB. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107:1124–1129. doi: 10.1073/pnas.0909333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Molecular cell. 2008;29:350–361. doi: 10.1016/j.molcel.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 44.Ferbeyre G, Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochimica et biophysica acta. 2011;1815:104–114. doi: 10.1016/j.bbcan.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 45.de Groot RP, Raaijmakers JA, Lammers JW, Jove R, Koenderman L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood. 1999;94:1108–1112. [PubMed] [Google Scholar]
- 46.Gu L, Vogiatzi P, Puhr M, Dagvadorj A, Lutz J, Ryder A, Addya S, Fortina P, Cooper C, Leiby B, Dasgupta A, Hyslop T, Bubendorf L, Alanen K, Mirtti T, Nevalainen MT. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocrine-related cancer. 2010;17:481–493. doi: 10.1677/ERC-09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811. doi: 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 48.Ren S, Cai HR, Li M, Furth PA. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4335–4339. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- 49.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massague J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature medicine. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salgado R, Junius S, Benoy I, Van Dam P, Vermeulen P, Van Marck E, Huget P, Dirix LY. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. International journal of cancer Journal international du cancer. 2003;103:642–646. doi: 10.1002/ijc.10833. [DOI] [PubMed] [Google Scholar]
- 51.Ara T, Declerck YA. Interleukin-6 in bone metastasis and cancer progression. European journal of cancer. 2010;46:1223–1231. doi: 10.1016/j.ejca.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK, Chung S, Luther T, Paholak HJ, Liu S, Hassan KA, Zen Q, Clouthier SG, Wicha MS. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Molecular cell. 2012;47:570–584. doi: 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuen HF, McCrudden CM, Huang YH, Tham JM, Zhang X, Zeng Q, Zhang SD, Hong W. TAZ expression as a prognostic indicator in colorectal cancer. PLoS One. 2013;8:e54211. doi: 10.1371/journal.pone.0054211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546–9563. doi: 10.18632/oncotarget.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong J, Belkhiri A. AXL mediates TRAIL resistance in esophageal adenocarcinoma. Neoplasia. 2013;15:296–304. doi: 10.1593/neo.122044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Modi S, Seidman AD, Dickler M, Moasser M, D’Andrea G, Moynahan ME, Menell J, Panageas KS, Tan LK, Norton L, Hudis CA. A phase II trial of imatinib mesylate monotherapy in patients with metastatic breast cancer. Breast cancer research and treatment. 2005;90:157–163. doi: 10.1007/s10549-004-3974-0. [DOI] [PubMed] [Google Scholar]
- 57.Cristofanilli M, Morandi P, Krishnamurthy S, Reuben JM, Lee BN, Francis D, Booser DJ, Green MC, Arun BK, Pusztai L, Lopez A, Islam R, Valero V, Hortobagyi GN. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2008;19:1713–1719. doi: 10.1093/annonc/mdn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Pendergast AM. The Emerging Role of ABL Kinases in Solid Tumors. Trends in cancer. 2015;1:110–123. doi: 10.1016/j.trecan.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chislock EM, Pendergast AM. Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS One. 2013;8:e85231. doi: 10.1371/journal.pone.0085231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ABL protein abundance is increased in breast cancer cells with enhanced bone metastatic activity and ABL1/ABL2 depletion does not affect proliferation in vitro.
Fig. S2. ABL kinases promote breast cancer cell invasion.
Fig. S3. Depletion of ABL kinases does not inhibit metastasis of breast cancer cells, which show tropism to the lung.
Fig. S4. Treatment of breast cancer cells with imatinib but not GNF5 promotes ERK activation.
Fig. S5. CXCL12- and IGF1-mediated survival pathways are independent of ABL kinases.
Fig. S6. Depletion of ABL kinases in SKBR3 breast cancer cells decreases tumor-induced osteoclast activation.
Fig. S7. IL6 affects RANKL and OPG expression in osteoblasts.
Fig. S8. Quality control and global statistics of RNAseq analysis for transcriptome comparison of control versus ABL1/ABL2 knockdown breast cancer cells.
Fig. S9. ABL kinases increase TAZ protein abundance and STAT5 phosphorylation.
Fig. S10. Depletion of ABL kinases reduces the abundance of TAZ in the nucleus.
Fig. S11. Allosteric inhibition of ABL kinases activity decreases TAZ protein abundance.
Fig. S12. ABL2 mRNA expression positively correlates with TAZ mRNA expression in invasive breast cancer patients.
Fig. S13. Depletion of ABL kinases decreases the binding of TAZ to target genes.
Fig. S14. Depletion of ABL kinases does not affect YAP1 protein abundance, localization, or tyrosine phosphorylation in breast cancer cells.
Fig. S15. Expression of a constitutively active STAT5 mutant increases mRNA expression of MMP1, IL6, and TNC.
Fig. S16. Potential interdependence of TAZ and STAT5 in breast cancer cells.
Table S1. Differentially expressed genes in control and ABL1/ABL2 knockdown breast cancer cells.