

SUMMARY
The pontocerebellar hypoplasias (PCHs) are a heterogeneous group of autosomal recessive disorders characterized by hypoplasia of the ventral pons and cerebellum, with variable cerebral involvement and severe psychomotor retardation. Eight different subtypes (PCH1-8) have been reported up to now. PCH2 is the most common type, generally caused by homozygous mutations in the TSEN54 gene and characterized by cerebellar hypoplasia that affects the hemispheres more severely than the vermis, progressive cerebral atrophy, microcephaly, dyskinesia, seizures and death in early childhood. We present two cousins with PCH2. Both patients presented with exaggerated startle response in the newborn period. Here we discuss the clinical and neuroradiological findings of PCH2, and its differentiation from familial startle disease or hereditary hyperekplexia.
Keywords: pontocerebellar hypoplasia, TSEN54 gene, hyperekplexia, startle disease
Pontocerebellar hypoplasias (PCH) are inherited autosomal recessive neurodegenerative disorders with fetal onset1. Up to now eight subtypes (PCH1-8) have been defined on the basis of clinical, radiologic and pathologic findings2. All subtypes share common features including hypoplasia/atrophy of the cerebellum and pons, progressive microcephaly and variable cerebral involvement. PCH2 is the most frequent type. Children with PCH2 have generalized clonus; incoordination of sucking and swallowing; extrapyramidal features including chorea, athetosis, dystonia and spasticity; progressive microcephaly; severe impairment of cognitive and motor development; central visual impairment; and an increased risk for malignant hyperthermia. Epilepsy is present in approximately half of these cases. Neuroradiological findings in PCH2 show brainstem and cerebellar hypoplasia, with the cerebellar hemispheres more affected than the vermis. The cerebral cortex shows mild or severe atrophy3.
Startle reflex is a physiologic reflex, present from 6 weeks of age and persists throughout life. It is a basic alerting reaction consisting of facial grimacing with blinking, followed by involuntary movements of head flexion, adduction of the arms and flexion of the trunk and the knees, causing falling without a protective reaction4. Hyperekplexia (startle disease) is a rare, nonepileptic disorder characterized by an exaggerated, persistent startle reaction to unexpected auditory, somatosensory and visual stimuli. It is caused by genetic defects in different parts of the inhibitory glycinergic receptor. Glycine receptors are highly concentrated in the brainstem and spinal cord. Brainstem pathologies, either congenital or acquired, may cause secondary exaggerated startle response. The differential diagnosis of secondary startle syndromes also includes PCHs4,5. Here we present two cases with PCH2, in one family, who presented with exaggerated startle response in the neonatal period.
Case Report
The index case, patient 1, was born at 37 6/7 weeks of gestation with Apgar scores of 8 and 9 at the first and fifth minute respectively. He was admitted to a tertiary-level neonatal unit because of mild respiratory distress and marked jitteriness. He was the second child of parents who were first-degree cousins; his older sister was healthy. The pregnancy was uncomplicated, except that the mother felt the fetal movements more than was the case in her first pregnancy. His birth weight was 3890 g (>90p), length 53 cm (>90p) and head circumference 35 cm (75–90p). He had no dysmorphic features. Neurologic examination revealed marked hypertonia with a constant flexion posture in both the upper and lower extremities (persisting during sleep), increased deep tendon reflexes, clonus bilaterally and dramatic startle response to touch, noise and light. The startle response was characterized by small- to medium-amplitude oscillatory movements of all four limbs lasting from seconds to several minutes. These movements would stop either spontaneously or with gentle tactile pressure. He was noted to have inspiratory stridor, possibly due to laryngomalacia. Other systemic examinations were normal. The complete blood count, electrolytes, liver and renal function tests, blood gases and ammonia and lactate levels were normal. Cranial ultrasonography revealed cerebellar hypoplasia. Cranial magnetic resonance imaging (MRI) showed pontocerebellar hypoplasia, with the vermis relatively protected (Fig. 1). His respiratory distress was transient, requiring free-flow oxygen for a couple of days. He was fed through a nasogastric tube due to feeding problems. Clonazepam was started for the exaggerated startle reaction. After treatment, his hypertonia and startle response decreased but nonetheless persisted. During the course of his care, he underwent a gastrostomy because of severe feeding problems. He had frequent lower respiratory tract infections and died at the age of one year.
Fig. 1.

Cranial MRI images of patient 1 (a,b) during the neonatal period, and patient 2 (c,d) at the age of 8 years. Mid-sagittal T2-weighted (a,c) and coronal FLAIR (b,d) images show a flat ventral pons, cerebellar hypoplasia with the hemispheres being more affected and the vermis relatively spared (“dragonfly-like” pattern). In patient 2, cerebral atrophy is also present and cerebellar atrophy is more prominent, giving rise to an enlarged fourth ventricle.
The family history (Fig. 2) of the index patient revealed that his 8-year-old female paternal cousin, patient 2, had a similar history in the neonatal period. She was born at term by caesarean section; her birth weight was 3600 g, and she had respiratory problems after birth. During the neonatal period, she had marked jitteriness and seizures. Phenobarbital was started and continued for one year. She had poor swallowing ability and was fed with fluids. At the age of 8 years, she was experiencing two or three seizures every month. Her weight was 7.5 kg (<3p), length 84 cm (<3p) and head circumference 40.8 cm (<3p). Pectus excavatum deformity was present. She had muscle wasting, increased tone in the extremities, brisk reflexes and dyskinesia, with choreic movements. She also had profound developmental delay with poor head control and absence of eye contact, social smiling, voluntary movements and vocal communication. Although her startle response was not dramatic as that of case 1, she had eye blinking and jerky movements upon touch, noise and light. Cranial MRI of patient 2 showed similar findings to those of the index patient, along with cerebral atrophy. Myelination was complete (Fig. 1). She died at the age of 9 years, during a febrile infection.
Fig. 2.
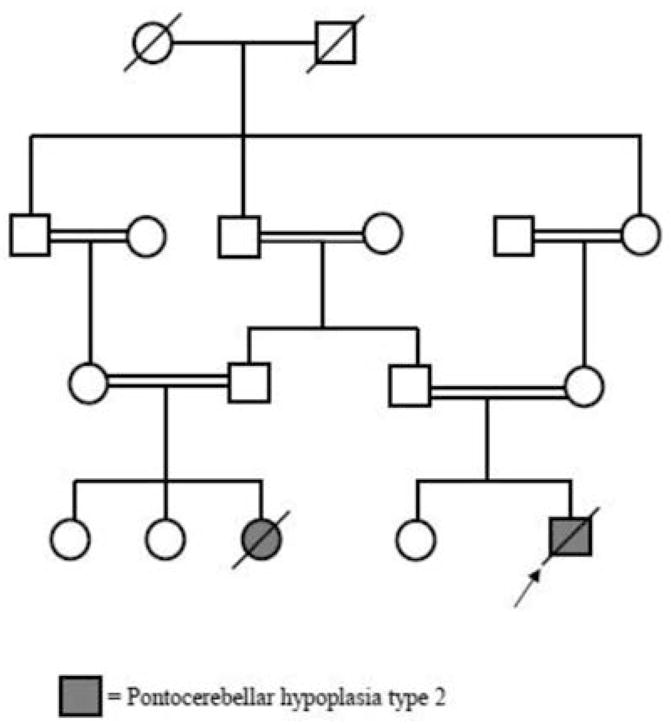
Pedigree chart of the family.
Based on the clinical and neuroradiological findings, the provisional diagnosis was PCH2. After genetic counseling and receipt of informed consent from the parents, molecular analysis was performed on a research basis. Case 2 was found to be homozygous mutant for an amino acid change of an alanine into a serine at position 307 (p.A307S, a common mutation) in the transferRNA (tRNA) splicing endonuclease subunit 54 gene (TSEN54). The blood sample from patient 1 was inadequate and could not be repeated after his death. Further analysis of the family and carrier testing were planned.
Discussion
We reported two cases with PCH2 in a family. Both cases presented with exaggerated startle response in the neonatal period and severe psychomotor retardation with no gain of developmental steps. The index case was more severe, with impaired swallowing and respiratory problems. Cranial MRI of both patients showed a flat ventral pons and hemisphere-dominant cerebellar atrophy. Cerebellar hypoplasia was more prominent in Case 2, giving rise to an enlarged fourth ventricle. On the basis of the neuroradiological and clinical findings, pontocerebellar hypoplasia, specifically PCH2, was given as the provisional diagnosis; patient 2 was found to be homozygous for the common TSEN54 mutation, p.A307S.
In a study of 169 patients (141 families) clinically diagnosed with PCH, Namavar et al.6 identified disease-causing mutations in 106 cases, representing a mutation frequency of 62.7%. TSEN54 mutations was found in 100 patients (59.2%); 88 (52.1%) of these patients had a homozygous p.A307S mutation in TSEN54. The researchers also found that all cases with the common mutation (p.A307S) had a “dragonfly-like” phenotype on coronal sections of the MRI, in which the cerebellar hemispheres were flat and severely reduced in size, and the vermis was to a relative degree spared. They suggested that MRI findings guide molecular diagnosis, since the PCH2 phenotype is distinct. Mild or severe atrophy of the cerebral cortex was observed in 40% of PCH2 cases6. The differential diagnoses as based on neuroradiological findings include other PCH types, congenital disorders of glycosylation, mitochondrial disorders, dystroglycanopathies, lissencephalies without known gene defects with microcephaly, cerebellar and pontine hypoplasia, and heterozygous CASK1 mutations with an X-linked dominant pattern affecting females and presenting with cortical dysplasia and brainstem hypoplasia2,3. Aside from these disorders, extreme prematurity also may cause cerebellar and brainstem hypoplasia. Messerschmidt et al.7 reported that extreme prematurity with birth weight <1500 g can cause severe reduction in cerebellar volume with symmetric involvement of both hemispheres and variable changes of the vermis.
PCH1 and PCH2 have similar neuroradiological findings but can be differentiated on the basis of the clinical findings. Anterior horn cell degeneration is seen in PCH1; patients with PCH1 also have findings similar to those displayed in spinal muscular atrophy type 1. PCH3 is rare, having been reported in only 2 families. Hypotonia and craniofacial dysmorphism, which are not seen in PCH2, were present in these cases. PCH2, PCH4 and PCH5 represent a spectrum of disorders caused by different mutations in the TSEN genes. TSEN encodes subunits of the enzyme tRNA endonuclease, which has a role in RNA processing. The mechanisms by which TSEN mutations result in PCH are still unknown8. PCH4 is a more severe form of PCH2; patients usually die in the neonatal period. PCH5 has been described in only one family; in it, cerebellar hypoplasia is more prominent in the vermis than in the hemispheres, in contrast to what is seen in the other PCHs. Other types (PCH6-8) are also very rare: PCH6 is a nuclear-coded mitochondrial disease, PCH7 was seen in a patient with vanishing testes, and PCH8 is a separate entity caused by the CHMP1A gene, in which patients have joint contractures and growth retardation2,3.
In our patients, an exaggerated startle response was present, but this was not identical with familial startle disease or hereditary hyperekplexia. There are three clinical symptoms in familial startle disease: generalized stiffness at birth, excessive startle reflex to unexpected stimuli and generalized stiffness following the startle reflex, which may mimic epileptic tonic spasms. Hypertonia is a common finding during the neonatal period in both PCH and familial startle disease. But in familial startle disease hypertonia is apparent only when the baby is awake; it disappears with sleep, increases with handling and normalizes during the first years of life5,8. Furthermore, in our patients, the startle reflex resembled generalized clonus or jitteriness, with oscillatory movements; there was no generalized stiffness after the startle reflex.
To conclude; PCH2, specifically the common TSEN54 mutation p.A307S, is a distinct entity with a characteristic “dragonfly-like” cerebellar pattern on MRI, which may guide molecular diagnosis among the various differentials. The symptom of exaggerated startle response may be seen as a result of brainstem pathology. In newborns with exaggerated startle response, brain imaging to exclude brainstem pathologies is important.
Acknowledgments
The molecular genetic analysis was performed on a research basis at the Gleeson Laboratory, Howard Hughes Medical Institute, University of California San Diego.
References
- 1.Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–422. doi: 10.1016/0387-7604(93)90080-r. [DOI] [PubMed] [Google Scholar]
- 2.Rudnik-Schöneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet. 2014;166:173–183. doi: 10.1002/ajmg.c.31403. [DOI] [PubMed] [Google Scholar]
- 3.Namavar Y, Barth PG, Poll-The BT, Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis. 2011;6:50. doi: 10.1186/1750-1172-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Praveen V, Patole SK, Whitehall JS. Hyperekplexia in neonates. Postgrad Med J. 2001;77:570–572. doi: 10.1136/pmj.77.911.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakker MJ, van Dijk JG, van den Maagdenberg AM, Tijssen MA. Startle syndromes. Lancet Neurol. 2006;5:513–524. doi: 10.1016/S1474-4422(06)70470-7. [DOI] [PubMed] [Google Scholar]
- 6.Namavar Y, Barth PG, Kasher PR, et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134:143–156. doi: 10.1093/brain/awq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Messerschmidt A, Brugger PC, Boltshauser E, et al. Disruption of cerebellar development: potential complication of extreme prematurity. AJNR Am J Neuroradiol. 2005;26:1659–1667. [PMC free article] [PubMed] [Google Scholar]
- 8.Namavar Y, Eggens VRC, Barth PG, Baas F. TSEN54-Related Pontocerebellar Hypoplasia. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle, WA: University of Washington, Seattle; 2009. Sep 8, [updated 2013 Oct 24] 1993–2014. Retrieved from: http://www.ncbi.nlm.nih.gov/books/NBK9673/ [Google Scholar]