

Abstract
Background
Restriction site associated DNA sequencing (RAD-seq), a next-generation sequencing technology, has greatly facilitated genetic linkage mapping studies in outbred species. RAD-seq is capable of discovering thousands of genetic markers for linkage mapping across many individuals, and can be applied in species with or without a reference genome. Although several analytical tools are available for RAD-seq data, alternative strategies are necessary for improving the marker quality and hence the genetic mapping accuracy.
Results
We demonstrate a strategy for constructing dense genetic linkage maps in hybrid forest trees by combining RAD-seq and whole-genome sequencing technologies. We performed RAD-seq of 150 progeny and whole-genome sequencing of the two parents in an F1 hybrid population of Populus deltoides × P. simonii. Two rough references were assembled from the whole-genome sequencing reads of the two parents separately. Based on the parental reference sequences, 3442 high-quality single nucleotide polymorphisms (SNPs) were identified that segregate in the ratio of 1:1. The maternal linkage map of P. deltoides was constructed with 2012 SNPs, containing 19 linkage groups and spanning 4067.16 cM of the genome with an average distance of 2.04 cM between adjacent markers, while the male map of P. simonii consisted of 1430 SNPs and the same number of linkage groups with a total length of 4356.04 cM and an average interval distance of 3.09 cM. Collinearity between the parental linkage maps and the reference genome of P. trichocarpa was also investigated. Compared with the result on the basis of the existing reference genome, our strategy identified more high-quality SNPs and generated parental linkage groups that nicely match the karyotype of Populus.
Conclusions
The strategy of simultaneously using RAD and whole-genome sequencing technologies can be applied to constructing high-density genetic maps in forest trees regardless of whether a reference genome exists. The two parental linkage maps constructed here provide more accurate genetic resources for unraveling quantitative trait loci and accelerating molecular breeding programs, as well as for comparative genomics in Populus.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3003-9) contains supplementary material, which is available to authorized users.
Keywords: Restriction site associated DNA, Whole-genome sequencing, Single nucleotide polymorphism, Genetic linkage map, Populus
Background
Forests cover about 30 % of Earth’s land area and are of significant economic and ecological importance [1]. Although most forest trees are characterized by their large and complex genomes, recent advances in DNA sequencing technology with assistances of available physical maps [2–4], have led to several tree genome sequence assemblies, which include two poplar species (Populus trichocarpa [5] and P. euphratica [6]), three conifer species (Picea abies [7], Picea glauca [8] and Pinus taeda [9]), and Eucalyptus grandis [10]. The availability of genome sequence information is essential for studying genomic architecture and evolution as well as for comparative genomics. However, for those tree species without a reference genome sequence, investigations on genome structure have to resort to genetic linkage maps that show the order and distance of a set of genome-wide genetic markers. Indeed, genetic linkage maps play an important role in genome comparisons with other species and assembling genome scaffold sequences or validating the integrity of an existing genome assembly [11–13]. More importantly, genetic linkage maps are prerequisites for identifying quantitative trait loci (QTLs) that control growth, wood quality, and other economically important traits, and thereby facilitating the genetic improvement of cultivated trees through marker-assisted selection and breeding.
Over the past two decades, more and more linkage maps have been constructed for a large number of forest trees [1, 14]. One of the major steps for constructing a linkage map is to obtain a set of genetic marker genotype data across many individuals in a mapping population [1]. Many previous tree linkage maps were established using molecular markers such as randomly amplified polymorphic DNA (RAPD), restriction fragment length polymorphisms (RFLP), amplified fragment length polymorphisms (AFLP), and simple sequence repeats (SSR). These traditional genetic markers were only developed to a small or moderate number due to instability or labor-intensive experiments, thus usually leading to sparse or unsaturated linkage maps, especially in outbred forest trees. Although recent single nucleotide polymorphism (SNP) array technologies have been applied to produce higher throughput marker data for constructing linkage maps in forest trees such as Eucalyptus [15] and Populus [16], they also have some limitations, including unreliable or useless genotype calls and only a small fraction of the studied loci being polymorphic [17].
Next-generation DNA sequencing (NGS) technologies can produce tremendous amounts of DNA sequence data at a consistently low cost, allowing us to obtain thousands of SNPs for genetic mapping. However, it is infeasible to apply whole-genome sequencing directly to hundreds of individuals in a mapping population because the total expense would be so high that most research projects cannot afford it. Therefore, several DNA library preparation methods for NGS have been developed to solve this problem by reducing genome complexity and adding DNA barcodes to samples [17]. These methods include restriction site associated DNA sequencing (RAD-seq) [18], genotyping-by-sequencing (GBS) [19], and specific locus amplified fragment sequencing (SLAF-seq) [20], and we focus on RAD-seq in the present study. RAD-seq uses NGS platforms for targeted sequencing of regions near restriction enzyme cut sites across genomes of many samples [18, 21]. RAD-seq has been extensively applied in constructing linkage maps in many organisms such as barley [22], ryegrass [23], moth [24], grape [25], gudgeon [26] and cotton [27]. Generally, there are two ways of discovering SNPs or other RAD markers, either with or without reference genome sequences. When a reference genome is available, RAD-seq reads can first be mapped to the reference sequences with tools such as BWA [28] and Bowtie2 [29], and then SNP calling or genotyping can be performed with tools such as SAMtools [30] and GATK [31]. For species without available reference genomes, de novo methods have to be employed to generate RAD markers, which include Stacks [32], RADtools [24], RaPiD [33], Rainbow [34] and PyRAD [21]. Although many tools are available for RAD-seq data analyses, there is still much room to improve the analytical strategies for obtaining more accurate and reliable SNP genotypes, particularly in highly heterozygous forest trees [35].
In this study, we performed RAD-seq data analysis for genetic mapping by combining the use of RAD-seq data from the progeny with the whole-genome sequencing data of their parents in an F1 hybrid population of Populus deltoides × P. simonii. The female parent P. deltoides has the characteristics of fast growth and resistance to disease but a poor rooting ability, while the male parent P. simonii has strong hardiness in cold, heat, drought and other bad conditions, and an excellent regeneration ability. The hybrids of the two parents display significant difference in morphological and physiological traits, providing a permanent material for mapping QTLs. Short paired-end (PE) reads of whole-genome sequencing data with high coverage from each parent were de novo assembled into contigs, forming a rough reference sequence of the parent. Based on each of the two parental reference sequences, two SNP datasets were identified and validated with each other to generate a high-quality SNP dataset for linkage mapping. Consequently, two high-quality parental linkage maps were constructed, each with a number of linkage groups that matched the karyotype of Populus perfectly. Collinearity between the parental linkage maps and the reference genome of P. trichocarpa was also investigated. Compared with the result of linkage mapping based solely on the reference genome of Populus [36], our strategy generated more accurate genetic linkage maps of the two parents. This strategy could be applied to construct high-density and high-quality genetic linkage maps, especially in outbred forest trees with or without a reference genome.
Methods
Plant materials and Illumina sequencing
The mapping material was a population of interspecific F1 hybrids between P. deltoides and P. simonii, which was generated in 2011 in Xiashu Forest Farm of Nanjing Forestry University, Jurong, Jiangsu Province, China [36]. We selected 150 individuals for genetic linkage mapping in this study. In the spring of 2013, young leaf tissue was collected and DNA was extracted from the two parents and 150 progeny using the CTAB protocol [37].
We performed RAD sequencing of the 150 progeny and whole-genome sequencing of the two parents. The RAD library for the progeny was constructed following the protocol described by Baird et al. [18] with a few modifications, details of which can be found in our previous study [36]. RAD sequencing was performed in seven lanes (PE, 90 bp) on an Illumina HiSeq 2000 at Beijing Genomics Institute (BGI), Shenzhen, China. For the whole-genome sequencing of the two parents, the DNA was randomly sheared by sonication and ligated with adapters. Fragments of 300–500 bp were selected using agarose gel electrophoresis. Two sequencing libraries for the two parents were constructed according to the Illumina protocol. The whole-genome sequencing was conducted on a HiSeq 2500 platform at Biomarker Technologies Co, Ltd. (BMK), Beijing, China.
The raw sequencing data were processed to clean the data with the same standard quality control pipelines in the two companies (i.e. BGI and BMK), and then to obtain high quality (HQ) data using NGS QC toolkit [38]. First, reads from each individual were segregated according to its unique molecular identifier. Second, paired reads containing primer/adapter sequence or having more than 10 % uncalled bases (N) were discarded. Third, paired reads were also discarded if more than 50 % of the bases in either of the reads have Phred quality score less than 5. And finally, we further filtered the clean data with NGS QC toolkit [38] to obtain HQ reads such that more than 70 % of the bases for each read have quality scores greater than or equal to 20.
De novo assembly, SNP discovery and genotyping
To improve the quality of genome assembly, we used the Perl program ErrorCorrectReads.pl in ALLPATHS-LG [39] to correct base calling errors in the HQ reads from the two parents. Each parental genome was then assembled from its corrected short reads using SOAPdenovo [40], which builds contigs using a de Bruijn graph algorithm. Different k-mer lengths were used and the optimal assembly was selected according to several parameters such as N50 and average contig length.
The two sets of contigs assembled above were considered to be the rough genome sequences of the female P. deltoides ‘I-69’ and the male P. simonii ‘L-3’. We performed SNP calling and genotyping across the hybrid F1 population based on the two parental rough genome sequences and the reference genome sequence of P. trichocarpa separately [5], using the software BWA [28], SAMtools and BCFtools (v1.2, [30]), and several in-house Perl scripts with the following steps:
mapping the filtered HQ reads from each individual to a reference genome sequence to generate a sequence alignment/map (SAM) format file using the BWA mem command with default parameters;
filtering out those records having an edit distance greater than 9 or best alignment score less than 60 or second-best alignment score greater than the best alignment score in the SAM file of each individual;
converting the filtered SAM file to BAM format and then sorting and indexing with SAMtools;
producing BCF files with the command samtools mpileup –g –I for all individuals;
generating VCF files with the command bcftools call –m –v for each parent;
filtering SNPs from the parental VCF files such that each SNP has a mapping quality score of at least 20 and a read coverage depth (DP) of at least 5, and merging the two parental SNP datasets into a list site file;
for all individuals, including the two parents, creating VCF files with the command bcftools call –m –f GQ –T using the list site file generated in step (6);
calling genotypes at all the list sites for each individual and filtering using stringent conditions with DP ≥ 10 and genotyping quality (GQ) > 50;
generating a SNP genotype dataset for the common SNP sites across the two parents and all 150 progeny.
Finally, three SNP genotype datasets were generated on the basis of the genome sequences of P. deltoides, P. simonii and P. trichocarpa, denoted by PD, PS and PT, respectively. The SNPs in those datasets were further filtered for linkage mapping according to Mendel’s law of segregation.
Linkage map construction
We performed chi-squared tests on all the SNPs in the PD, PS and PT datasets generated above to check whether they follow Mendel’s law of segregation. If a SNP deviated seriously from the Mendelian segregation ratio (p < .01) or had more than 10 % missing genotypes in the population, it was removed from linkage analysis. To use the SNPs called from the two parental rough genome sequences, the SNP loci that were identical between the filtered PD and PS datasets, at which each individual has the same genotype in the two datasets (i.e. the Hamming distance between PD and PS equal to 0 for a SNP locus), were chosen for linkage mapping. Because the overwhelming majority of SNPs were found to segregate in the ratio of 1:1 in the mapping population, we had to construct two parental linkage maps using the traditional pseudo-testcross mapping strategy [41] with the software packages JoinMap 4.1 [42] and FsLinkageMap 2.1 [43]. The maternal linkage map was constructed with the identical SNPs of segregation type ab×aa, and the paternal linkage map with the identical SNPs of segregation type aa×ab. For each linkage map construction, two-point linkage analysis was first performed for all pairs of SNP loci and then SNP markers were grouped under a logarithm of odds (LOD score) threshold using the software FsLinkageMap. Next, SNP markers in each linkage group were ordered three times using JoinMap with the maximum likelihood mapping method and once using FsLinkageMap. The optimal order was chosen as the mapping order of the linkage group according to the four ordering results with the two software packages and the ordering criterion of the minimum sum of adjacent recombination fractions [44]. Finally, map distances were calculated with the Kosambi mapping function, and linkage maps were first drawn in WMF format using FsLinkageMap and then modified in PDF or EPS format using the software Mayura Draw (http://www.mayura.com).
Results
Illumina sequencing and de novo assembly
We obtained 615,038,434 clean 101-bp reads from BMK, including 288,115,744 and 326,922,690 from whole genomes of the maternal P. deltoides ‘I-69’ and the paternal P. simonii ‘L-3’, respectively. RAD sequencing was performed at BGI, and 2,010,564,342 clean reads each 82–90 bp in length were generated with an average of 13,403,762 reads from each of the 150 progeny. We filtered out low-quality reads in which more than 30 % of bases had Phred quality score ≤ 20 using NGS QC toolkit for all individuals, and performed error correction only for each parental dataset with the standalone Perl script ErrorCorrectReads.pl in ALLPATHS-LG. This resulted in 231,587,056 (80 %) and 258,870,468 (79 %) high-quality reads for the parents ‘I-69’ and ‘L-3’, respectively, and an average of 13,001,952 (97 %) high-quality reads for the progeny. The final high-quality dataset of each parent was used for de novo assembly and each individual dataset for SNP genotype calling (Table 1).
Table 1.
Summary of whole-genome sequencing and RAD-seq data from BMK and BGI with averages in brackets
| Experiment | Sample | Number of sample | Number of clean reads | Clean reads data (Gb) | Number of high-quality reads | High-quality reads data (Gb) |
|---|---|---|---|---|---|---|
| BMK | Female parent | 1 | 288,115,744 | 29.10 | 231,587,056 | 23.39 |
| Male parent | 1 | 326,922,690 | 33.02 | 258,870,468 | 26.15 | |
| BGI | Progeny | 150 | 2,010,564,342 (13,403,762) |
137.37 (0.92) |
1,950,292,874 (13,001,952) |
123.23 (0.82) |
| Total | Total | 152 | 2,625,602,776 | 199.49 | 2,440,750,398 | 172.77 |
RAD-seq restriction site associated DNA sequencing
The high-quality sequence reads of each parent were assembled using SOAPdenovo at different k-mer lengths of 21, 27, 31, 37, 41, 47, 51, 57, 61 and 67 with contig cut-off length of 150 bp. We considered the contig dataset of each assembly result and compared statistics such as total number of contigs, N50 length, average contig length and longest contig length. The k-mer length was generally proportional to the number of contigs, total length and longest contig length for the assemblies of each parent (Additional file 1). We chose the optimal assembly that had the highest N50 contig length and largest average contig length among the different k-mers. We found that the optimal assemblies of the two parents both corresponded to a k-mer length of 37, which resulted in N50 length of 586 bp and average contig length of 441 bp for the female ‘I-69’, and N50 length of 873 bp and average contig length of 532 bp for the male ‘L-3’. These optimal assemblies contained 767,393 and 664,721 contigs spanning the genome sizes of 338.39 and 353.79 Mb for the female ‘I-69’ and male ‘L-3’, respectively (Additional file 1). We used the two assemblies as rough reference genome sequences for SNP discovery and genotype calling across the whole hybrid family of P. deltoides × P. simonii.
SNP discovery and genotype calling
We first mapped the high-quality reads of the two parents on the two rough reference genomes of P. deltoides and P. simonii and the reference genome of P. trichocarpa using the mapping tool BWA. As a result, more than 70 % of the high-quality reads from each parent were best mapped to their own rough reference sequence, while only 56–59 % were mapped to the other two reference sequences (Table 2). Each of these alignments had edit distance less than 9 and best alignment score at least 60 higher than that of the second-best alignment. With these mapping results in SAM format, we performed SNP calling using SAMtools and BCFtools. Based on the rough genome sequence of P. deltoides, 423,680 and 4,721,160 SNPs with coverage depth of at least 5 were discovered in the female parent ‘I-69’ and the male parent ‘L-3’, respectively. The number of SNPs found in both parents was 168,530, leading to 4,976,310 SNPs discovered in one or both parents. For convenience, we denote this total SNP dataset by PD. Similarly, we also found that the total numbers of SNPs in the two parents were 5,227,450 and 11,694,085 according to the reference sequences of P. simonii and P. trichocarpa, respectively (Table 2). These two SNP datasets are denoted by PS and PT.
Table 2.
Percentage of high-quality reads best mapped to the three reference sequences and number of SNPs identified accordingly for the female parent ‘I-69’ and the male parent ‘L-3’
| Parent | P. deltoides | P. simonii | P. trichocarpa | |||
|---|---|---|---|---|---|---|
| Mapped reads (%) | SNP | Mapped reads (%) | SNP | Mapped reads (%) | SNP | |
| I-69 | 71.34 | 423,680 | 56.77 | 4,955,722 | 56.23 | 6,597,917 |
| L-3 | 56.55 | 4,721,160 | 76.77 | 414,174 | 58.57 | 7,018,418 |
| Both | 168,530 | 142,446 | 1,933,395 | |||
| Total | 4,976,310 | 5,227,450 | 11,682,940 | |||
Next, the high-quality reads from each progeny were also mapped to the three reference sequences separately and the best alignments were retained for SNP genotype calling. We performed SNP genotype calling across the whole population (two parents and 150 progeny) based on the three SNP datasets PD, PS and PT separately. After a series of filtering procedures (detailed in Materials and Methods), 6513 SNPs with segregation type of ab×aa were genotyped on the basis of SNP dataset PD, which followed the Mendelian segregation ratio of 1:1 with p ≥ .01 and at which at least 90 % of the progeny were genotyped. This new SNP dataset containing 6513 SNPs is denoted by PD1 for convenience. On the basis of SNP datasets PS and PT, the numbers of SNPs were 23,221 and 26,865, respectively, and these two new SNP datasets were accordingly denoted by PS1 and PT1. Further analyses revealed that 2973 SNPs in PD1 each corresponded to one or more SNPs in PS1, at which each progeny shared the same genotype if the genotype was denoted by aa or ab. We called these SNPs the identical SNP loci between the two SNP datasets PD1 and PS1 and denoted the set by PDS1. In the same way, the identical SNP datasets between PD1 and PT1, between PS1 and PT1, and among the three datasets PD1, PS1 and PT1 were denoted by PDT1, PST1, and PDST1, and their numbers of SNPs were 3159, 13,769 and 2479, respectively (Fig. 1(a)). If the genotype was denoted using the base symbols A, C, G and T, such as AA, AC, and GT, the numbers of identical SNPs between any pair or among all three datasets were abruptly reduced, as shown in brackets in Fig. 1(a).
Fig. 1.
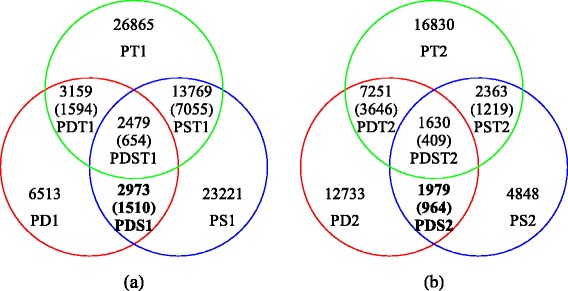
Venn diagram showing the numbers of identical SNPs between any two of or among three SNP data sets if the genotypes in the progeny are denoted as aa or ab. If the SNP genotypes are expressed using the nucleotide notations of A, C, G and T, the numbers are listed in brackets accordingly. (a) PD1, PS1, and PT1 are the SNP data sets calling from the two poplar parents based on the reference sequences of P. deltoides, P. simonii and P. trichocarpa, respectively, in which each SNP segregated in the Mendelian ratio of 1:1 with p ≤ 0.01 and the segregation type of ab×aa and was genotyped in at least 90 % of the progeny. (b) PD2, PS2, and PT2 are the similar SNP data sets defined as in (a), but each SNP has the segregation type of aa×ab
For those SNPs having the segregation type aa×ab, following the Mendelian segregation ratio of 1:1 with p ≥ .01 and genotyped in at least 90 % of the progeny, the datasets generated from PD, PS and PT were denoted by PD2, PS2 and PT2, respectively. Also, the identical SNP datasets between PD2 and PS2, between PD2 and PT2, between PS2 and PT2, and among the three datasets PD2, PS2 and PT2 were denoted by PDS2, PDT2, PST2, and PDST2, respectively. The numbers of SNPs contained in these datasets are shown in Fig. 1(b).
Genetic linkage maps
We used the two SNP datasets PDS1 and PDS2 generated above to construct genetic linkage maps of the maternal P. deltoides ‘I-69’ and the paternal P. simonii ‘L-3’, respectively. To improve mapping efficiency, when two or more SNPs were identical (i.e. complete linkage) within PDS1 or PDS2, and adjacent SNPs were within 1 kb on a contig, a single SNP was chosen to represent the group for linkage analysis. This reduced the number of SNPs in PDS1 from 2973 to 2012 and in PDS2 from 1979 to 1430. The final 2012 SNPs with segregation type ab×aa were assigned to 19 linkage groups (denoted as DLG1-19, Fig. 2), nicely matching the karyotype of Populus, at high LOD thresholds ranging from 6 to 18. After SNPs within each linkage group were ordered, a genetic linkage map of the female parent ‘I-69’ was constructed, spanning 4067.16 cM in total length with the individual groups ranging from 106.87 to 471.20 cM (Table 3). The distance between adjacent SNP markers on this genetic map ranged from 0.67 to 21.68 cM with an average of 2.04 cM (±1.69 SD). In the same way, the final 1430 SNPs with segregation type aa×ab were grouped into 19 linkage groups (denoted as SLG1-19, Fig. 3) at LOD thresholds ranging from 6 to 14, and they constituted a genetic linkage map of the male parent ‘L-3’ when SNPs within each linkage group were ordered. The total length of this paternal linkage map was 4356.04 cM, with the linkage group lengths varying from 118.28 to 512.67 cM, and the adjacent SNP intervals ranged from 0.67 to 19.05 cM with an average length of 3.09 cM (±2.41 SD) (Table 3). More detailed information on the two linkage maps is presented in Additional files 2 and 3, including SNP interval distance, cumulative distance, predicted linkage phase between adjacent SNPs, and the corresponding identical SNPs identified on the basis of the P. trichocarpa reference genome.
Fig. 2.

The genetic map of linkage groups DLG1-DLG19 for the maternal P. deltoides ‘I-69’. The length of each linkage group is presented under the linkage group name. Each SNP is named by the contig number of the rough reference sequence of P. deltoides and its position on it, prefixed with letter D
Table 3.
SNP number and length of linkage groups in the two parental genetic maps of P. deltoides ‘I-69’ and P. simonii ‘L-3’
| P. deltoides ‘I-69’ | P. simonii ‘L-3’ | Chromosome size (Mb)a | ||||
|---|---|---|---|---|---|---|
| Group | SNP number | Length (cM) | Group | SNP number | Length (cM) | |
| DLG1 | 240 (196)b | 471.20 | SLG1 | 155 (121) | 512.67 | 50.50 |
| DLG2 | 162 (133) | 328.55 | SLG2 | 119 (98) | 327.69 | 25.26 |
| DLG3 | 134 (113) | 279.22 | SLG3 | 94 (74) | 290.22 | 21.82 |
| DLG4 | 120 (87) | 254.00 | SLG4 | 80 (60) | 203.88 | 24.27 |
| DLG5 | 112 (92) | 225.29 | SLG5 | 86 (71) | 261.80 | 25.89 |
| DLG6 | 130 (109) | 303.32 | SLG6 | 106 (88) | 339.89 | 27.91 |
| DLG7 | 81 (69) | 186.87 | SLG7 | 70 (57) | 176.52 | 15.61 |
| DLG8 | 141 (125) | 242.47 | SLG8 | 107 (88) | 268.86 | 19.47 |
| DLG9 | 90 (77) | 165.32 | SLG9 | 82 (68) | 189.55 | 12.95 |
| DLG10 | 145 (119) | 249.87 | SLG10 | 89 (75) | 265.25 | 22.58 |
| DLG11 | 85 (70) | 172.13 | SLG11 | 45 (32) | 155.20 | 18.50 |
| DLG12 | 75 (61) | 150.29 | SLG12 | 46 (37) | 163.38 | 15.76 |
| DLG13 | 85 (71) | 167.25 | SLG13 | 64 (60) | 184.52 | 16.32 |
| DLG14 | 102 (83) | 176.57 | SLG14 | 74 (60) | 219.07 | 18.92 |
| DLG15 | 73 (62) | 151.34 | SLG15 | 48 (44) | 162.67 | 15.28 |
| DLG16 | 72 (61) | 155.82 | SLG16 | 30 (24) | 118.28 | 14.49 |
| DLG17 | 59 (47) | 135.89 | SLG17 | 64 (49) | 197.88 | 16.08 |
| DLG18 | 61 (47) | 144.89 | SLG18 | 36 (29) | 158.74 | 16.96 |
| DLG19 | 45 (32) | 106.87 | SLG19 | 35 (22) | 159.97 | 15.94 |
| Total | 2012 (1654) | 4067.16 | 1430 (1157) | 4356.04 | 394.51 | |
aThe genome size of P. trichocarpa (Tuskan et al. 2006)
bThe number (in brackets) of SNPs identical to the SNPs calling based on the reference genome of P. trichocarpa
Fig. 3.
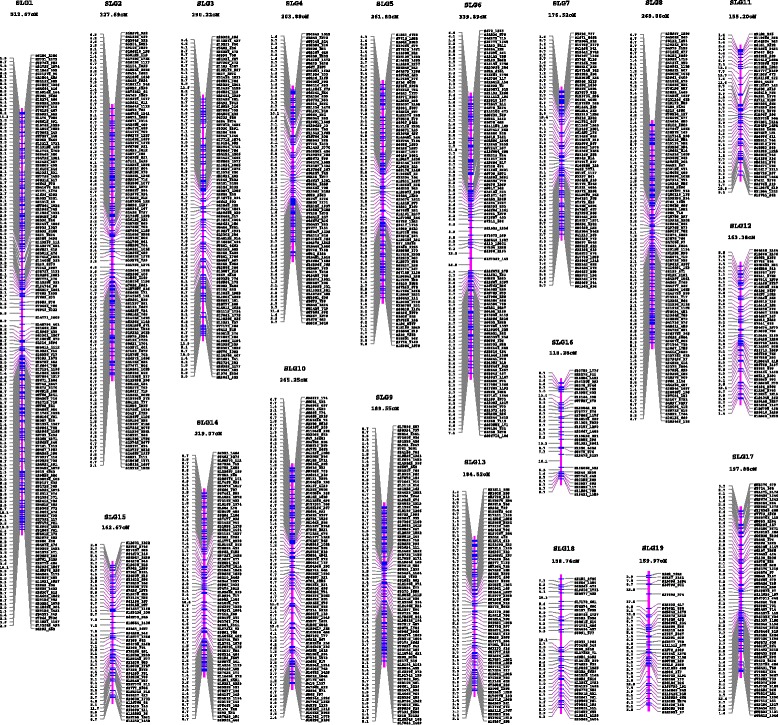
The genetic map of linkage groups SLG1-SLG19 for the paternal P. simonii ‘L-3’. The length of each linkage group is presented under the linkage group name. Each SNP is named by the contig number of the rough reference sequence of P. simonii and its position on it, prefixed with letter S
Further analyses revealed that there remained strong positive correlations between SNP number within a linkage group, linkage group length, and the physical size on the P. trichocarpa reference genome (Table 4). SNP number was highly correlated with linkage group length, with a correlation coefficient of 0.9731 for the maternal linkage map and 0.9406 for the paternal linkage map. The SNP number and length of the maternal linkage groups were also highly correlated with those of the paternal map with correlation coefficients more than 0.90. Moreover, the correlations between the linkage group lengths for the two parental maps and the physical size on the reference genome were also high with the coefficients of about 0.92. However, the correlations between the SNP numbers of linkage groups for the two parental maps and the physical size were relatively lower, but still having high coefficients over 0.80.
Table 4.
Correlations among the SNP number, genetic length and chromosome size for the linkage groups of the two parental maps
| SNP number in DLG | DLG Length | SNP number in SLG | SLG length | |
|---|---|---|---|---|
| DLG Length | 0.9731 | |||
| SNP number in SLG | 0.9334 | 0.9199 | ||
| SLG length | 0.9338 | 0.9495 | 0.9406 | |
| Chromosome size | 0.8852 | 0.9202 | 0.8018 | 0.9231 |
In spite of the high positive correlations described above, we observed that there existed some unusual patterns between the two parental linkage groups either in length or in the number of SNPs. When we compared the length of each male linkage group with the corresponding female linkage group, we found that the difference of >3 cM per 1 Mb occurred in two linkage groups, i.e. LG17 (3.86 cM) and LG19 (3.33 cM). Interestingly, it was noted that DLG19 has more number of SNPs (45) than SLG19 (35) while its length (106.87 cM) is significantly shorter than that of the later (159.97 cM). This could be explained by the recombination suppression phenomenon possibly occurred on chromosome 19 in the female parent due to the sex determination through a ZW system in Populus [45]. For the same reason, we inferred that recombination could be also suppressed on chromosome 17 in the maternal parent P. deltoides ‘I-69’.
Collinearity between genetic and physical maps
We found that 1654 (82.2 %) SNPs on the maternal linkage map and 1157 (80.9 %) SNPs on the paternal linkage map segregated identically to at least one SNP in the PT1 or PT2 dataset, in which each SNP has its position information on the reference genome of P. trichocarpa (Additional files 2 and 3). These identical SNPs connected the linkage groups of the two parents to the chromosomes of the reference genome, allowing direct comparisons between the genomes of P. deltoides, P. simonii and P. trichocarpa. Figure 4 presents scatter plots of the genetic map positions of the identical SNPs against their physical positions on the reference genome of P. trichocarpa for the 19 linkage groups of the two parents. On the whole, there is apparently a high level of collinearity between the parental linkage groups and the chromosomes of the reference genome. However, almost all of the linkage groups showed one or more local regions where SNP orders were inconsistent with the reference genome positions.
Fig. 4.
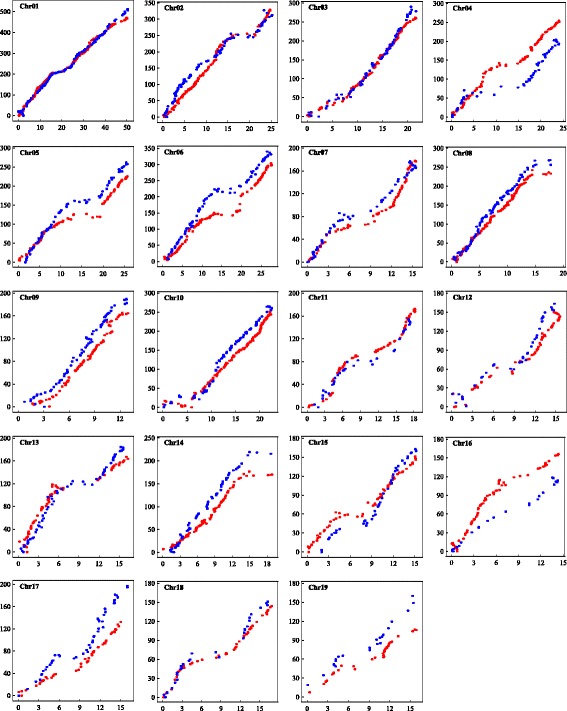
Collinear comparison between the parental genetic maps and the reference genome of P. trichocarpa. The x-axis indicates the reference sequence position with the unit of Mb; the y-axis indicates the genetic map position with the unit of cM. The red and blue points, respectively, indicate the SNP positions on the maternal and paternal genetic maps against the identical SNP positions on the reference genome
Discussion
Here we demonstrated a novel strategy for constructing high-quality genetic linkage maps in forest trees by combining the use of RAD-seq with whole-genome resequencing technologies. This genetic mapping strategy may be applicable to most outbred forest species in which no reference genome is available. With the plummeting cost of NGS, it is feasible for most laboratories to perform RAD-seq across tens to hundreds of individuals and to sequence the whole genomes of their two parents in an F1 hybrid population. Assemblies from the parental whole-genome sequencing reads can be used as rough references, and RAD-seq reads of each progeny as well as the reads from each parent can be aligned to them separately. Thus, hundreds of thousands of SNPs could be discovered and genotyped across the population with available software packages designed for NGS data, and then thousands of high-quality SNP markers may be selected for genetic linkage mapping, in terms of the Mendelian segregation ratio, the fraction of missing genotypes and other features such as mapping quality and read coverage depth. Two SNP genotype datasets are generally derived from the two parental rough reference genomes and can validate each other using the Hamming distance across the individuals to improve mapping data quality substantially. After obtaining such a large number of high-quality SNP genotypes across the F1 mapping population, current linkage mapping tools are applied with the strategy of choosing the best marker orders (as described in Materials and Methods) to construct parent-specific dense linkage maps.
RAD-seq has been extensively applied to SNP and RAD marker discovery across populations in species with or without a reference sequence [18, 35]. When a reference genome is unavailable, de novo methods can be used with several tools such as Stacks [32] and RADtools [24]. However, few comparison studies have been carried out to evaluate the performance of different RAD-seq analytical strategies, including assembly and SNP calling software packages. Here we have presented an alternative method for calling SNPs across the F1 population using RAD-seq data by incorporating the whole-genome sequencing data of the two parents. Although a reference sequence is available for poplar ([5], https://phytozome.jgi.doe.gov/pz/portal.html), the assembled rough reference sequences of the two parents, P. deltoides and P. simonii, may be more appropriate for SNP and genotype calling in our mapping population, because there are divergences between the P. trichocarpa reference genome and the two parental genomes. Furthermore, the reference genome assembly is not perfect with, to date, more than one thousand scaffolds still unassigned to any chromosomes. In a previous study [36], we mapped RAD-seq data from the same mapping population directly to the reference sequence of P. trichocarpa for SNP marker discovery and obtained 20 linkage groups for each parental linkage map, each with one linkage group ambiguously assigned to any chromosome. In contrast, in this study, we used the parental rough reference sequences and RAD-seq data from only half of the progeny to generate 19 linkage groups in each parental linkage map, which perfectly matches the karyotype of Populus (2n = 38).
Like most genetic mapping studies in forest trees [11, 46, 47], we obtained two parent-specific linkage maps using two SNP datasets generated from RAD-seq technology. The results of two sex-specific linkage maps from RAD-seq data can be found in other organisms such as ryegrass [23] and grape [25]. The main reason is that, for two diverged parents, at the overwhelming majority of SNP sites one parent has a heterozygous genotype and the other a homozygous genotype. Because a pair of SNPs, one with segregation type of aa×ab and the other ab×aa, cannot provide any recombination information [48], the two high-quality SNP datasets PDS1 and PDS2 generated in this study have to be analyzed separately, leading to two parental linkage maps. To construct an integrated genetic linkage map for the F1 hybrid population, a sufficient number of fully informative SNP markers with segregation type of ab×ab or ab×cd should be identified as bridges to link the two types of SNPs segregating in 1:1 ratio. With the decreasing cost of NGS, this could be resolved by resequencing the whole genomes instead of RAD-seq across many individuals in the mapping population to identify more fully informative SNPs.
We used 150 progeny for calling genotypes from RAD-seq data and for the subsequent construction of the genetic linkage maps. Such a moderate sample size can provide enough information to estimate the recombination fraction accurately between any two genetic markers that follow a Mendelian segregation ratio of 1:1. Because there are only four combined genotypes for a pair of markers each with a segregation ratio of 1:1, the average count of the combined genotypes was about 38 in our mapping population, which led to an expected LOD score of 5.36 for two moderately linked loci with a recombination fraction of 0.30 [49, 50]. This indicates that such a sample size could allow a large number of the moderately and tightly linked markers contained in a genetic linkage map, with a maximal genetic distance of 34.66 cM between two adjacent markers under the Kosambi mapping function. In summary, a moderate sample size of about 150 or more individuals is recommended for constructing parent-specific genetic linkage maps in an F1 hybrid population of forest trees with RAD-seq and whole-genome sequencing technologies.
Conclusions
Assembled contigs of whole-genome PE reads from each parent in an F1 hybrid population can be used as a rough reference for performing SNP calling and genotyping with RAD-seq data across the whole population. The two SNP genotype datasets each based on one parental reference can confirm each other to generate a high-quality genotype dataset for linkage mapping. This strategy could be applied to highly heterozygous undomesticated forest trees with or without a reference genome to construct high-density genetic linkage maps, which is difficult with traditional molecular markers. Both of the parental genetic linkage maps of P. deltoides and P. simonii constructed here with high density and quality perfectly match the karyotype of Populus, and provide important genetic resources for identifying QTLs, accelerating molecular breeding programs and performing comparative genomics in Populus.
Acknowledgements
We thank Huzhi Xu (Luoning Forest Bureau of Henan Provicne, China), Jiangtao Zhang (Forest Science Research Institute of Henan Province, China), Xiangjin Yan (Siyang Agroforestry Center of Jiangsu Province, China) and Jinhai Yang (Nanjing Qiaolin Forestry Science and Technology Co. Ltd., Nanjing, China) for their great help in our crossing experiments and seedling cultivation.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31270706) and the Priority Academic Program Development of the Jiangsu Higher Education Institutions (PAPD).
Availability of data and materials
The whole-genome and RAD sequencing raw data are available under accession numbers SRP071167 and SRP052929, respectively, at the NCBI Sequence Read Archive database (http://www.ncbi.nlm.nih.gov/Traces/sra). Additional results are available as Additional files 1, 2 and 3 in the end of the main manuscript.
Authors’ contributions
CT, HL and JS conceived and designed the experiments. MM, FL, ST and JW carried out the experiments. MM, CT, ST and JW performed the data analyses. CT and MM wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- BGI
Beijing Genomics Institute
- BMK
Beijing’s Biomarker Technologies Co, Ltd
- DP
coverage depth
- GQ
genotyping quality
- HQ
high quality
- NGS
next-generation DNA sequencing
- PE
paired-end
- QTL
quantitative trait loci
- RAD-seq
restriction site associated DNA sequencing
- SAM
sequence alignment/map
- SNP
single nucleotide polymorphism
Additional files
De novo assembly statistics of P. deltoides ‘I-69’ and P. simonii ‘L-3’ with different k-mers. (DOCX 20 kb)
Detailed information on genetic distance and linkage phase between adjacent SNP markers on the genetic linkage map of the female P. deltoides ‘I-69’. The corresponding identical SNPs identified based on the P. trichocarpa reference genome are also included. (XLS 452 kb)
Detailed information on genetic distance and linkage phase between adjacent SNP markers on the genetic linkage map of the male P. simonii ‘L-3’. The corresponding identical SNPs identified on the basis of the P. trichocarpa reference genome are also included. (XLS 349 kb)
Contributor Information
Mohaddeseh Mousavi, Email: mm.m2132@gmail.com.
Chunfa Tong, Phone: 86-025-85427760, Email: tongchf@njfu.edu.cn.
Fenxiang Liu, Email: fenxiangliu@hotmail.com.
Shentong Tao, Email: taoshentong@hotmail.com.
Jiyan Wu, Email: WuJiYan@hotmail.com.
Huogen Li, Email: hgli@njfu.edu.cn.
Jisen Shi, Email: jshi@njfu.edu.cn.
References
- 1.White TL, Adams WT, Neale DB. Forest genetics. 1. Cambridge, MA: CABI Publishing; 2007. [Google Scholar]
- 2.Kelleher CT, Chiu R, Shin H, Bosdet IE, Krzywinski MI, Fjell CD, et al. A physical map of the highly heterozygous Populus genome: integration with the genome sequence and genetic map and analysis of haplotype variation. Plant J. 2007;50(6):1063–78. doi: 10.1111/j.1365-313X.2007.03112.x. [DOI] [PubMed] [Google Scholar]
- 3.Mozo T, Dewar K, Dunn P, Ecker JR, Fischer S, Kloska S, et al. A complete BAC-based physical map of the Arabidopsis thaliana genome. Nat Gen. 1999;22(3):271–5. doi: 10.1038/10334. [DOI] [PubMed] [Google Scholar]
- 4.Schein J, Kucaba T, Sekhon M, Smailus D, Waterston R, Marra M. High-throughput BAC fingerprinting. In: Zhao S, Stodolsky M, editors. Bacterial artificial chromosomes. Vol. 1: Library Construction, Physical Mapping, and Sequencing. Totawa, NJ: Humana Press; 2004. pp. 143–56. [DOI] [PubMed] [Google Scholar]
- 5.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 6.Ma T, Wang J, Zhou G, Yue Z, Hu Q, Chen Y, et al. Genomic insights into salt adaptation in a desert poplar. Nat Commun. 2014; doi:10.1038/ncomms3797. [DOI] [PubMed]
- 7.Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497(7451):579–84. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- 8.Birol I, Raymond A, Jackman SD, Pleasance S, Coope R, Taylor GA, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics. 2013;29(12):1492–7. doi: 10.1093/bioinformatics/btt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimin A, Stevens KA, Crepeau MW, Holtz-Morris A, Koriabine M, Marcais G, et al. Sequencing and assembly of the 22-gb loblolly pine genome. Genetics. 2014;196(3):875–90. doi: 10.1534/genetics.113.159715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, et al. The genome of Eucalyptus grandis. Nature. 2014;510(7505):356–62. doi: 10.1038/nature13308. [DOI] [PubMed] [Google Scholar]
- 11.Bartholome J, Mandrou E, Mabiala A, Jenkins J, Nabihoudine I, Klopp C, et al. High-resolution genetic maps of Eucalyptus improve Eucalyptus grandis genome assembly. New Phytol. 2015;206(4):1283–96. doi: 10.1111/nph.13150. [DOI] [PubMed] [Google Scholar]
- 12.Fierst JL. Using linkage maps to correct and scaffold de novo genome assemblies: methods, challenges, and computational tools. Front Genet. 2015; doi:10.3389/fgene.2015.00220. [DOI] [PMC free article] [PubMed]
- 13.Tang H, Zhang X, Miao C, Zhang J, Ming R, Schnable JC, et al. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 2015; doi:10.1186/s13059-014-0573-1. [DOI] [PMC free article] [PubMed]
- 14.Neale DB, Kremer A. Forest tree genomics: growing resources and applications. Nat Rev Genet. 2011;12(2):111–22. doi: 10.1038/nrg2931. [DOI] [PubMed] [Google Scholar]
- 15.Hudson CJ, Freeman JS, Kullan AR, Petroli CD, Sansaloni CP, Kilian A, et al. A reference linkage map for Eucalyptus. BMC Genomics. 2012; doi:10.1186/1471-2164-13-240. [DOI] [PMC free article] [PubMed]
- 16.Muchero W, Guo J, DiFazio SP, Chen JG, Ranjan P, Slavov GT, et al. High-resolution genetic mapping of allelic variants associated with cell wall chemistry in Populus. BMC Genomics. 2015; doi:10.1186/s12864-015-1215-z. [DOI] [PMC free article] [PubMed]
- 17.Gardner KM, Brown P, Cooke TF, Cann S, Costa F, Bustamante C, et al. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3: Genes Genomes. Genetics. 2014;4(9):1681–7. doi: 10.1534/g3.114.011023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS One. 2008; doi:10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed]
- 19.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One. 2011; doi:10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed]
- 20.Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong WG, et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PloS One. 2013; doi:10.1371/journal.pone.0058700. [DOI] [PMC free article] [PubMed]
- 21.Eaton DA. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics. 2014;30(13):1844–9. doi: 10.1093/bioinformatics/btu121. [DOI] [PubMed] [Google Scholar]
- 22.Chutimanitsakun Y, Nipper RW, Guesta-Marcos A, Cistue L, Corey A, Filichkina T, et al. Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genomics. 2011;12:4. doi: 10.1186/1471-2164-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfender WF, Saha MC, Johnson EA, Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122:1467–80. doi: 10.1007/s00122-011-1546-3. [DOI] [PubMed] [Google Scholar]
- 24.Baxter SW, Davey JW, Johnston JS, Shelton AM, Heckel DG, Jiggins CD, et al. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PloS One. 2011; doi:10.1371/journal.pone.0019315. [DOI] [PMC free article] [PubMed]
- 25.Wang N, Fang L, Xin H, Wang L, Li S. Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Biol. 2012;12:148. doi: 10.1186/1471-2229-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kakioka R, Kokita T, Kumada H, Watanabe K, Okuda N. A RAD-based linkage map and comparative genomics in the gudgeons (genus Gnathopogon, Cyprinidae) BMC Genomics. 2013;14:32. doi: 10.1186/1471-2164-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Ning Z, Hu Y, Chen J, Zhao R, Chen H, et al. Molecular mapping of restriction-site associated DNA markers in allotetraploid upland cotton. PloS One. 2015; doi:10.1371/journal.pone.0124781. [DOI] [PMC free article] [PubMed]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genetics. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Catchen J, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes Genomes. Genetics. 2011;1:171–82. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willing EM, Hoffmann M, Klein JD, Weigel D, Dreyer C. Paired-end RAD-seq for de novo assembly and marker design without available reference. Bioinformatics. 2011;27(16):2187–93. doi: 10.1093/bioinformatics/btr346. [DOI] [PubMed] [Google Scholar]
- 34.Chong Z, Ruan J, Wu CI. Rainbow: an integrated tool for efficient clustering and assembling RAD-seq reads. Bioinformatics. 2012;28(21):2732–7. doi: 10.1093/bioinformatics/bts482. [DOI] [PubMed] [Google Scholar]
- 35.Davey JW, Cezard T, Fuentes-Utrilla P, Eland C, Gharbi K, Blaxter ML. Special features of RAD Sequencing data: implications for genotyping. Mol Ecol. 2013;22(11):3151–64. doi: 10.1111/mec.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong CF, Li HG, Wang Y, Li XR, Ou JJ, Wang DY, et al. Construction of high-density linkage maps of Populus deltoides × P. simonii using restriction-site associated DNA sequencing. PloS One. 2016; doi:10.1371/journal.pone.0150692. [DOI] [PMC free article] [PubMed]
- 37.Doyle JJ, Doyle JL. A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytologist Bulletin. 1987;19:11–5. [Google Scholar]
- 38.Patel RK, Jain M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PloS One. 2012; doi:10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed]
- 39.Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, et al. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18(5):810–20. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grattapaglia D, Sederoff R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics. 1994;137:1121–37. doi: 10.1093/genetics/137.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Ooijen JW. JoinMap 4, Software for the calculation of genetic linkage maps in experimental populations. Wageningen: Kyazma B. V; 2006. [Google Scholar]
- 43.Tong CF, Zhang B, Shi JS. A hidden Markov model approach to multilocus linkage analysis in a full-sib family. Tree Genet Genomes. 2010;6(5):651–62. doi: 10.1007/s11295-010-0281-2. [DOI] [Google Scholar]
- 44.Falk CT. A simple scheme for preliminary ordering of multiple loci: application to 45 CF families. In: Elston RC, Spence MA, Hodge SE, MacCluer JW, editors. Multipoint mapping and linkage based upon affected pedigree members, Genetic Workshop 6. New York: Liss; 1989. pp. 17–22. [Google Scholar]
- 45.Yin T, DiFazio S, Gunter LE, Zhang X, Sewell MM, Woolbright SA, et al. Genome structure and emerging evidence of an incipient sex chromosome in Populus. Genome Res. 2008;18:422–30. doi: 10.1101/gr.7076308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Tong CF, Yin T, Zhang X, Zhuge Q, Huang M, et al. Detection of quantitative trait loci influencing growth trajectories of adventitious roots in Populus using functional mapping. Tree Genet Genomes. 2009;5:539–52. doi: 10.1007/s11295-009-0207-z. [DOI] [Google Scholar]
- 47.Berlin S, Lagercrantz U, von Arnold S, Ost T, Ronnberg-Wastljung AC. High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus. BMC Genomics. 2010;11:129. doi: 10.1186/1471-2164-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maliepaard C, Jansen J, Van Ooijen JW. Linkage analysis in a full-sib family of an outbreeding plant species: overview and consequences for applications. Gen Res. 1997;70:237–50. doi: 10.1017/S0016672397003005. [DOI] [Google Scholar]
- 49.Weir BS. Genetic data analysis II: methods for discrete population genetic data. 2. Sunderland, MA, USA: Sinauer Associates Inc; 1996. [Google Scholar]
- 50.Wu RL, Ma CX, Casella G. Statistical genetics of quantitative traits: linkage, maps and QTL. New York: Springer; 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The whole-genome and RAD sequencing raw data are available under accession numbers SRP071167 and SRP052929, respectively, at the NCBI Sequence Read Archive database (http://www.ncbi.nlm.nih.gov/Traces/sra). Additional results are available as Additional files 1, 2 and 3 in the end of the main manuscript.