

Abstract
Background
Results from genome-wide association studies (GWAS) identified many loci and biological pathways that influence adult body mass index (BMI). We aimed to identify if biological pathways related to adult BMI also affect infant growth and childhood adiposity measures.
Methods
We used data from a population-based prospective cohort study among 3,975 children with a mean age of 6 years. Genetic risk scores were constructed based on the 97 SNPs associated with adult BMI previously identified with GWAS and on 28 BMI related biological pathways based on subsets of these 97 SNPs. Outcomes were infant peak weight velocity, BMI at adiposity peak and age at adiposity peak, and childhood BMI, total fat mass percentage, android/gynoid fat ratio, and preperitoneal fat area. Analyses were performed using linear regression models.
Results
A higher overall adult BMI risk score was associated with infant BMI at adiposity peak and childhood BMI, total fat mass, android/gynoid fat ratio, and preperitoneal fat area (all p-values < 0.05). Analyses focused on specific biological pathways showed that the membrane proteins genetic risk score was associated with infant peak weight velocity, and the genetic risk scores related to neuronal developmental processes, hypothalamic processes, cyclicAMP, WNT-signaling, membrane proteins, monogenic obesity and/or energy homeostasis, glucose homeostasis, cell cycle, and muscle biology pathways were associated with childhood adiposity measures (all p-values <0.05). None of the pathways were associated with childhood preperitoneal fat area.
Conclusions
A genetic risk score based on 97 SNPs related to adult BMI was associated with peak weight velocity during infancy and general and abdominal fat measurements at the age of 6 years. Risk scores based on genetic variants linked to specific biological pathways, including central nervous system and hypothalamic processes, influence body fat development from early life onwards.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0425-y) contains supplementary material, which is available to authorized users.
Keywords: Genome-wide association study; Body mass index; Polymorphism, single nucleotide; Genetics; Pediatrics
Background
Childhood overweight and obesity are associated with various adverse short- and long-term consequences, including cardiovascular disease and type 2 diabetes [1–4]. Besides the well-known lifestyle-related risk factors, overweight and obesity have a strong genetic component with heritability estimates from twin studies reported to be up to 80 % [5, 6]. Large genome-wide association studies (GWAS) have identified many single nucleotide polymorphisms (SNPs) associated with body mass index (BMI) in adults [7, 8]. Less is known about the genetic background of BMI in childhood. Three recent studies revealed a total of 15 genetic loci associated with childhood BMI, most of which are also associated with adult BMI [9–11]. We previously reported that a genetic risk score based on 29 SNPs related to adult BMI was associated with infant growth and childhood adiposity measures [12]. A recent GWAS increased the number of adult BMI associated SNPs to 97 [8]. These SNPs are located in or close to genes linked to several biological pathways. In adults especially central nervous system processes seem to play a role [8]. The role of these pathways in body fat development during early life is not known yet. Thus far, GWAS in children did not report any specific biological pathways [11]. Knowledge on the biological pathways influencing BMI from early life onwards may help to better understand the development of overweight and obesity in children.
In this study, we used data from 3,975 children participating in a population-based cohort study to examine the associations of genetic risk scores for adult BMI, both overall and based on specific biological pathways, with infant weight growth patterns and childhood adiposity measures. For comparison, we also examined the associations of genetic risk scores based on the 49 SNPs related with adult waist-hip-ratio (WHR) and on the 15 SNPs associated with childhood BMI with the same infant and childhood outcomes [11, 13].
Methods
Study design and population
This study was embedded in the Generation R Study, a population-based, prospective cohort study from fetal life onwards in Rotterdam, the Netherlands [14]. All pregnant women with an expected delivery date between April 2002 and January 2006 and living in Rotterdam were asked to participate. The study was approved by the local Medical Ethical Committee and written consent was obtained for each participating child. GWA scans were available for 59 % of all children (N = 5,732) [15]. The Generation R Study is a multi-ethnic cohort. Participants of European origin constitute the largest ethnic group (56 %), and the largest other groups are Surinamese (9 %), Turkish (7 %) and Moroccan (6 %) [14]. Our present study included all singleton live births with GWA data and information on at least one of the outcomes of interest (N = 4,151). A participant flowchart is shown in Additional file 1: Figure S1.
Genetic variants and risk scores
DNA was isolated from cord blood or, in a small minority of children with missing cord blood samples, at 6 years of age. For genome-wide association analysis the Illumina 610 and 660 W Quad platforms were used [16]. Stringent quality checks were performed in which individuals with low sample call rates (<97.5 %) or sex mismatches were excluded. Imputation of genotypes to the cosmopolitan panel of HapMap ii (release 22) was done using MACH software [17, 18]. Prior to imputation, we excluded SNPs with a high level of missing data (SNP call rate <98 %), significant deviations from Hardy-Weinberg equilibrium (P < 1*10−6), or low minor allele frequencies (<0.1 %). Information about the SNPs of interest for the current study was extracted from the GWAS dataset. The average imputation quality for all SNPs included in this study was 0.96, ranging from 0.55 to 1.00, demonstrating overall good imputation. For 93 out of the 97 known BMI SNPs information was available in our GWA dataset. We used proxies (R2 > 0.96, D’ = 1) for the remaining four BMI SNPs: rs13012571 was used as a proxy for rs13021737, rs1978487 for rs9925964, rs6445197 for rs2365389, and rs9636202 for rs17724992. Thus, the total number of SNPs used in the analysis was 97 (Additional file 2: Table S1). These SNPs were combined into weighted BMI genetic risk scores (see below). The same procedure was used for the 49 WHR and 15 child BMI SNPs [11, 13]. For 46 of the 49 WHR SNPs information was available in the GWA dataset. Rs4607103 was used as a proxy for rs2371767 (R2 = 0.90, D’ = 1). For the WHR SNPs rs8042543 and rs6556301 no perfect proxy was available leading to a total number of SNPs of 47 for WHR. For all but one SNPs identified for childhood BMI, information was available in our dataset. We used rs3751812 as a proxy for rs1421085 (R2 = 0.93, D’ = 0.97) (Additional file 2: Table S1).
In the paper on adult BMI, the 97 adult BMI SNPs were categorized into pathway categories. The authors performed a literature search, which brought about 405 genes within 500 kb on either side and with r2 > 0.2 of the 97 SNPs [8]. Based on their biological function, these genes were then catagorized into 28 pathways. We used this same categorization, but we excluded categories consisting of one SNP only. For each pathway category, we combined SNPs into a weighted genetic risk score. Some SNPs were included in more than one category based on their biological function (Additional file 3: Table S2). The number of overlapping SNPs between the biological categories is shown in Additional file 4: Table S3. As a comparison we ran a pathway analysis using QIAGEN’s Ingenuity® Pathway Analysis software (IPA) (IPA®, QIAGEN Redwood City,www.qiagen.com/ingenuity).
Infant weight growth and childhood general and abdominal adiposity
We used repeated growth measurements to derive infant peak weight velocity (PWV), BMI at adiposity peak (BMIAP) and age at adiposity peak (AGEAP), as described previously [19–23]. Briefly, the Reed1 model was used for boys and girls separately, to obtain PWV during infancy. BMIAP and AGEAP were obtained by fitting a cubic mixed effects model on log (BMI) from 2 weeks to 1.5 years of age while adjusting for sex.
At the median age of 6.0 years (95 % range, 5.7, 7.4) we measured general and abdominal adiposity measures as described in detail previously [24]. Briefly, BMI (kg/m2) was calculated from height and weight measured without shoes and heavy clothing. Total, android, and gynoid fat mass were measured by Dual-energy X-ray absorptiometry (DXA) (iDXA, GE-Lunar, 2008, Madison, WI, USA) [24]. Total fat mass (kg) was calculated as a percentage of total body weight (kg). Android/gynoid fat ratio provides the ratio of central body fat distribution in the abdomen (android fat) and hip (gynoid fat) regions [25]. Preperitoneal fat area, which is a measure of visceral abdominal fat, was measured by abdominal ultrasound [24, 26, 27].
Statistical analysis
We constructed a weighted genetic risk score combining the 97 adult BMI SNPs summing the number of outcome increasing risk alleles from the GWA dosage data, weighted using effect estimates of risk increasing alleles in adults. The risk score was rescaled to standard deviation scores (SDS, (observed value-mean)/standard deviation (SD)). Similarly, we constructed genetic risk scores based on SNPs involved in 28 different biological categories, and based on 47 adult WHR SNPs and 15 childhood BMI SNPs. For the biological categories and the WHR SNPs, we used the effect estimates from the original papers as weights [8, 11, 13]. For the 15 childhood SNPs, weights were obtained from the GWAS meta-analysis without the Generation R data [11]. We used linear regression analyses to examine the associations of the risk scores with PWV, BMIAP, and AGEAP in infancy, and BMI, total fat mass percentage, android/gynoid fat ratio, and preperitoneal fat area in childhood. The variance explained by the risk scores was considered to be the increase in the unadjusted R2 between the model containing all covariates and the risk score or separate SNPs, and the same model without the risk score. For all analyses, we natural logarithm transformed total fat mass, android/gynoid fat ratio, and preperitoneal fat area to obtain a normal distribution. Standard deviation scores were created for all outcome measures to allow comparison of effect estimates. For BMI, age-adjusted SD scores were created using the Dutch reference growth curves (Growth Analyzer 3.0, Dutch Growth Research Foundation, Rotterdam, the Netherlands) [12]. To enable comparison with our current risk scores, we rescaled the previously published 29 adult BMI SNPs risk score to SD scores. All models were adjusted for sex plus the first four principal components from the genetic data to adjust for ethnic background [28]. Models for general and abdominal adiposity measures were additionally adjusted for age except for BMI which was already age adjusted. Models for total fat mass, android/gynoid fat ratio, and preperitoneal fat area were additionally adjusted for height. [24] We also tested whether the associations of the child and adult BMI risk scores with the childhood adiposity outcomes were explained by infant growth by adding PWV and BMIAP separately to the regression models. For the analyses of the 28 biological pathways, we applied Bonferroni correction and considered a p-value of <0.0018 (0.05/28) as significant. All analyses were performed using the Statistical Package for the Social Sciences version 21.0 for Windows (SPSS; IBM, Chicago, IL, USA).
Results
Characteristics of the study population
Characteristics of all children are listed in Table 1. The children had a median age of 6.0 years (95 % range 5.7, 7.4). The median BMI at that age was 15.8 (95 % range 13.7, 21.2).
Table 1.
Characteristics of the study population
| Characteristics | Full group (N = 3,975) | European (N = 2,566) | Turkish (N = 300) | Surinamese (N = 287) | Moroccan (N = 234) | Other (N = 588) |
|---|---|---|---|---|---|---|
| Birth | ||||||
| Boys | 50.2 % | 49.6 % | 53.7 % | 53.3 % | 50.4 % | 49.2 % |
| Gestational age at birth (weeks)a | 40.1 (36.4; 42.3) | 40.3 (33.3; 42.0) | 40.0 (36.2; 42.3) | 39.7 (35.7; 42.0) | 40.6 (36.4; 42.2) | 40.0 (36.4; 42.1) |
| Weight at birth (grams) | 3458 (514) | 3506 (514) | 3402 (480) | 3238 (536) | 3496 (426) | 3379 (506) |
| Infant | ||||||
| Peak weight velocity (kg/year) | 12.2 (2.1) | 12.0 (2.0) | 13.1 (2.4) | 12.5 (2.2) | 12.6 (2.1) | 12.4 (2.2) |
| Body mass index at adiposity peak (kg/m2) | 17.6 (0.8) | 17.5 (0.8) | 17.9 (0.9) | 17.5 (0.9) | 17.8 (0.8) | 17.7 (0.8) |
| Age at adiposity peak (years) | 0.7 (0.04) | 0.7 (0.04) | 0.7 (0.04) | 0.7 (0.04) | 0.7 (0.04) | 0.7 (0.04) |
| Childhood | ||||||
| Age at visit (years)a | 6.0 (5.7; 7.8) | 6.0 (5.7; 7.5) | 6.1 (5.7; 7.7) | 6.1 (5.5; 8.2) | 6.1 (5.7; 8.3) | 6.1 (5.7; 8.2) |
| Height (cm) | 119.6 (6.0) | 119.5 (5.6) | 119.0 (5.7) | 119.9 (7.0) | 119.1 (5.9) | 120.1 (4.9) |
| Weight (kg) | 23.3 (4.2) | 22.9 (3.6) | 24.5 (5.3) | 23.5 (5.3) | 23.9 (4.1) | 24.0 (4.9) |
| Body mass index (kg/m2)a | 15.8 (13.7; 21.3) | 15.7 (13.7; 19.8) | 16.6 (13.6; 24.2) | 15.7 (13.2; 23.3) | 16.4 (14.0; 22.0) | 16.2 (13.6; 22.0) |
| Total fat mass percentagea | 24.0 (16.3; 38.6) | 23.5 (16.4; 36.4) | 26.6 (18.3; 43.5) | 24.1 (14.8; 41.4) | 25.9 (17.8; 39.9) | 24.3 (15.9; 39.4) |
| Android-gynoid fat ratioa | 0.2 (0.2; 0.4) | 0.2 (0.2; 0.4) | 0.3 (0.2; 0.5) | 0.2 (0.2; 0.5) | 0.2 (0.2; 0.4) | 0.2 (0.1; 0.4) |
| Preperitoneal fat area (cm2)a | 0.4 (0.2; 1.2) | 0.4 (0.2; 1.0) | 0.5 (0.2; 1.9) | 0.4 (0.2; 1.7) | 0.4 (0.2; 1.6) | 0.4 (0.2; 1.3) |
| Overweight (%)b | 12.9 | 10.5 | 23.7 | 11.8 | 19.7 | 17.8 |
| Obese (%)b | 4.1 | 2.1 | 11.0 | 8.0 | 7.7 | 6.1 |
N = 3,975
Values are means (standard deviations) unless otherwise specified
aMedian (95 % range)
bThe IOTF-classification was used to define overweight and obesity [41]
Infant weight growth patterns
The overall adult BMI genetic risk score was associated with BMIAP (Table 2; Fig. 1a-c), but not with other infant weight growth measures. BMIAP increased by 0.048 SDS (95 % confidence interval (CI) 0.015, 0.081) per SD increase in the genetic risk score. Of the 28 adult BMI genetic risk scores based on biological pathways, only the membrane proteins pathway genetic risk score was associated with PWV (p-value <0.002). Effect estimates for the unweighted and weighted 97 adult BMI SNPs risk scores were similar (Additional file 5: Table S4). As a comparison, the overall adult WHR genetic risk score was not associated with any infant growth measure (Table 2; Additional file 6: Figure S2a-c), whereas the childhood BMI genetic risk score was associated with PWV and BMIAP (0.048 SDS (95 % CI 0.016, 0.079) and 0.051 SDS (0.017, 0.084), respectively, per SD increase in the genetic risk score) (Table 2; Additional file 7: Figure S3a-c). The genetic risk score based on 29 adult BMI SNPs showed lower effect estimates per SD increase than our 97 SNPs adult BMI risk score for PWV and BMIAP, and a higher effect estimate for AGEAP, although none of the associations were significant for the 29 SNP genetic risk score (Additional file 8: Table S5). The largest variance explained by the adult BMI and pathway risk scores was obtained for the membrane proteins pathway with PWV (0.33 %) (Additional file 9: Table S6).
Table 2.
Associations of BMI, WHR, and childhood BMI genetic risk scores with infant growth (N = 2,955)a
| Risk score (number of SNPs in risk score) | Peak weight velocityb | BMI at adiposity peakb | Age at adiposity peakb | |||
|---|---|---|---|---|---|---|
| Beta (CI 95 %) | P-value | Beta (CI 95 %) | P-value | Beta (CI 95 %) | P-value | |
| Main risk scores* | ||||||
| Adult BMI (N = 97) | 0.027 (−0.004; 0.058) | 0.093 | 0.048 (0.015; 0.081) | 0.005 | 0.015 (−0.021; 0.051) | 0.418 |
| Secondary risk scores | ||||||
| Adult WHR (N = 47) | −0.022 (−0.054; 0.010) | 0.180 | −0.010 (−0.044; 0.025) | 0.587 | −0.016 (−0.053; 0.022) | 0.411 |
| Child BMI (N = 15) | 0.038 (0.007; 0.070) | 0.018 | 0.039 (0.006; 0.073) | 0.023 | 0.027 (−0.010; 0.063) | 0.153 |
| Adult BMI pathway genetic risk scores** | ||||||
| Neuronal | ||||||
| Neuronal developmental processes (N = 29) | 0.036 (0.003; 0.070) | 0.031 | 0.049 (0.013; 0.084) | 0.007 | −0.020 (−0.058; 0.019) | 0.311 |
| Neurotransmission (N = 10) | −0.009 (−0.040; 0.022) | 0.558 | −0.001 (−0.034; 0.032) | 0.948 | 0.002 (−0.034; 0.038) | 0.901 |
| Hypothalamic expression and regulation (N = 13) | 0.001 (−0.030; 0.033) | 0.932 | 0.008 (−0.025; 0.042) | 0.637 | 0.023 (−0.013; 0.059) | 0.203 |
| Neuronal expression (N = 12) | −0.034 (−0.065; −0.003) | 0.034 | −0.010 (−0.044; 0.024) | 0.559 | 0.026 (−0.010; 0.062) | 0.159 |
| Lipid biosynthesis and metabolism (N = 10) | 0.002 (−0.030; 0.033) | 0.918 | 0.006 (−0.028; 0.040) | 0.358 | 0.020 (−0.017; 0.056) | 0.291 |
| Bone development (N = 9) | 0.017 (−0.014; 0.048) | 0.290 | 0.017 (−0.017; 0.050) | 0.336 | 0.001 (−0.035; 0.037) | 0.957 |
| Signaling | ||||||
| MAPK1/extracellular signal-regulated kinases (N = 9) | 0.009 (−0.022; 0.040) | 0.579 | 0.008 (−0.025; 0.042) | 0.625 | 0.011 (−0.025; 0.047) | 0.534 |
| JAK (N = 2) | −0.007 (−0.038; 0.025) | 0.679 | −0.005 (−0.038; 0.029) | 0.779 | −0.006 (−0.042; 0.030) | 0.750 |
| CyclicAMP (N = 5) | −0.020 (−0.052; 0.013) | 0.233 | 0.019 (−0.015; 0.054) | 0.368 | −0.016 (−0.053; 0.021) | 0.391 |
| WNTSignaling (N = 6) | 0.033 (0.001; 0.064) | 0.041 | 0.017 (−0.016; 0.051) | 0.311 | 0.019 (−0.017; 0.055) | 0.293 |
| G-protein coupled receptor | ||||||
| Notch signaling (N = 2) | 0.010 (−0.021; 0.041) | 0.531 | 0.009 (−0.024; 0.043) | 0.581 | 0.012 (−0.024; 0.048) | 0.508 |
| Mitochondrial (N = 8) | 0.010 (−0.023; 0.043) | 0.559 | 0.004 (−0.032; 0.039) | 0.840 | 0.039 (0.001; 0.077) | 0.046 |
| Retinoic acid receptors (N = 6) | 0.019 (−0.013; 0.050) | 0.245 | 0.025 (−0.009; 0.058) | 0.144 | 0.019 (−0.017; 0.055) | 0.308 |
| Endocytosis/exocytosis (N = 14) | 0.004 (−0.027; 0.036) | 0.778 | 0.005 (−0.028; 0.038) | 0.776 | 0.007 (−0.029; 0.043) | 0.699 |
| Eye-related (N = 5) | 0.010 (−0.022; 0.042) | 0.548 | 0.010 (−0.025; 0.045) | 0.567 | −0.030 (−0.067; 0.007) | 0.116 |
| Tumorigenesis (N = 11) | 0.018 (−0.015; 0.050) | 0.285 | 0.018 (−0.017; 0.052) | 0.320 | −0.001 (−0.038; 0.036) | 0.954 |
| Apoptosis (N = 13) | 0.027 (−0.004; 0.059) | 0.087 | 0.018 (−0.016; 0.052) | 0.294 | 0.033 (−0.004; 0.069) | 0.077 |
| Membrane proteins (N = 12) | 0.057 (0.025; 0.088) | 3.88*10 −4 | 0.048 (0.015; 0.082) | 0.005 | 0.028 (−0.008; 0.065) | 0.124 |
| Hormone metabolism/regulation (N = 4) | −0.009 (−0.041; 0.022) | 0.564 | −0.009 (−0.042; 0.025) | 0.610 | 0.010 (−0.027; 0.046) | 0.604 |
| Purine/pyrimidine cycle (N = 4) | 0.009 (−0.022; 0.041) | 0.557 | 0.039 (0.006; 0.073) | 0.023 | −0.025 (−0.061; 0.011) | 0.178 |
| Monogenic obesity/energy homeostasis (N = 9) | −0.013 (−0.045; 0.018) | 0.406 | −0.014 (−0.048; 0.020) | 0.413 | 0.026 (−0.011; 0.062) | 0.168 |
| Immune system (N = 15) | 0.045 (0.014; 0.076) | 0.005 | 0.049 (0.015; 0.082) | 0.004 | −0.003 (−0.039; 0.033) | 0.868 |
| Limb development (N = 3) | 0.018 (−0.014; 0.049) | 0.267 | 0.022 (−0.011; 0.056) | 0.195 | 0.001 (−0.035; 0.037) | 0.945 |
| Ubiquitin pathways (N = 6) | −0.006 (−0.038; 0.025) | 0.684 | 0.007 (−0.027; 0.040) | 0.693 | −0.025 (−0.061; 0.011) | 0.168 |
| Glucose homeostasis/diabetes (N = 11) | 0.023 (−0.009; 0.054) | 0.160 | 0.021 (−0.013; 0.055) | 0.219 | 0.026 (−0.010; 0.063) | 0.156 |
| Cell cycle (N = 23) | 0.008 (−0.023; 0.039) | 0.611 | 0.011 (−0.023; 0.044) | 0.538 | −0.001 (−0.037; 0.035) | 0.959 |
| DNARepair | ||||||
| Nuclear trafficking (N = 4) | −0.015 (−0.047; 0.017) | 0.362 | −0.023 (−0.057; 0.011) | 0.187 | −0.032 (−0.068; 0.005) | 0.092 |
| Muscle biology (N = 6) | −0.011 (−0.043; 0.020) | 0.479 | −0.0003 (−0.034; 0.033) | 0.985 | 0.014 (−0.022; 0.050) | 0.446 |
* Bold font indicates P-value < 0.05. ** Bold font indicates significant after Bonferroni correction for the 28 pathways (p-value < 0.0018)
aAnalyses were performed in children with complete data on genetic variants, at least one outcome under study, and covariates
bValues are linear regression coefficients for models adjusted for sex and the first four genetic principal components and represent the difference in standard deviation scores of the outcome measures for each additional average risk allele in the risk scores
Fig. 1.
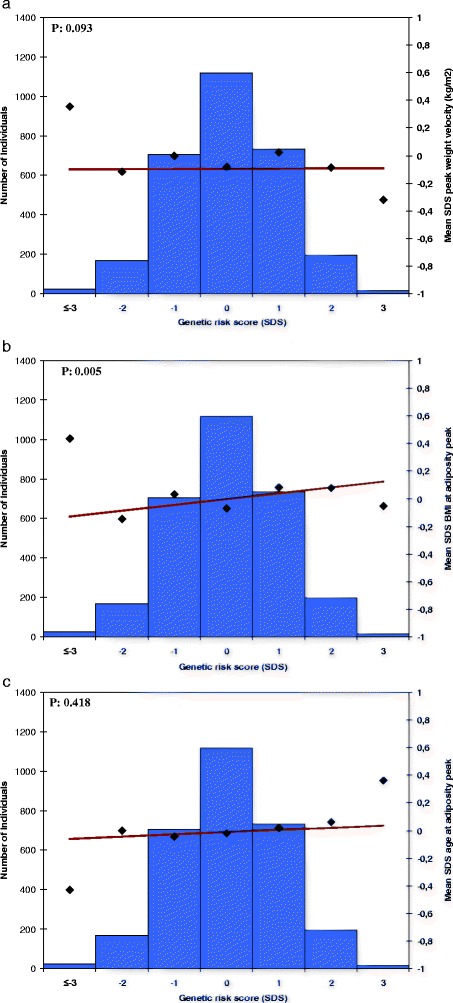
Association of adult body mass index genetic risk score with infant growth measures (N = 2,955). The x axis represents the categories of the risk score (overall sum of risk alleles, weighted by previously reported effect estimates, rescaled to SDS. The risk score ranged from −4 to 3 SDS and was rounded to the nearest integer for clarity of presentation. The right y axis shows mean SDS and corresponds to the dots and the line representing the regression of the mean SDS values for each category of the risk score. The y axis on the left corresponds to the histogram representing the number of individuals in each risk-score category. P-value is based on the continuous risk score, as presented in Table 2. Graphs represent; a peak weight velocity, b BMI at adiposity peak, and c age at adiposity peak
General and abdominal adiposity at school-age
The overall adult BMI genetic risk score was associated with all childhood general and abdominal adiposity measures. For each SD increase in the genetic risk score, childhood BMI increased by 0.112 SDS (95 % CI 0.084, 0.141), total fat mass increased by 0.092 SDS (95 % CI 0.065, 0.119), android/gynoid fat ratio increased by 0.077 SDS (95 % CI 0.045, 0.108), and increased preperitoneal fat area by 0.034 SDS (95 % CI 0.001, 0.066) (Table 3; Fig. 2a-d). Effect estimates for the unweighted and weighted 97 adult BMI SNPs risk scores were similar (Additional file 5: Table S4). Addition of PWV to the regression models did not materially change the effect estimates for the association of the BMI risk scores with BMI, total fat mass percentage, and android/gynoid fat ratio. However, the effect estimate for the association of the adult BMI risk score with childhood preperitoneal fat area was no longer significant. We observed similar findings when we added BMIAP instead of PWV to these regression models. However, the effects on the associations of the BMI risk scores with BMI and total fat mass were somewhat larger. Effect estimates for the associations of the child BMI risk score with BMI and total fat mass were 10–15 % lower after additional adjustment for PWV. Effect estimates for android/gynoid fat ratio and preperitoneal fat area did not materially change. We observed similar findings after additional adjustment for BMIAP (Additional file 10: Table S7 and Additional file 11: Table S8).
Table 3.
Associations of BMI, WHR, and childhood BMI genetic risk scores with childhood adiposity (N = 3,975)a, b
| Risk score (number of SNPs in risk score) | Body mass indexc | Total fat massc,d,e | Android/gynoid ratioc,d,e | Preperitoneal fat areac,d,e | ||||
|---|---|---|---|---|---|---|---|---|
| Beta (CI 95 %) | P-value | Beta (CI 95 %) | P-value | Beta (CI 95 %) | P-value | Beta (CI 95 %) | P-value | |
| Main risk scores* | ||||||||
| Adult BMI (N = 97) | 0.112 (0.084; 0.141) | 1.01*10 −14 | 0.092 (0.065; 0.119) | 3.89*10 −11 | 0.077 (0.045; 0.108) | 2.00*10 −6 | 0.034 (0.001; 0.066) | 0.042 |
| Secondary risk scores | ||||||||
| Adult WHR (N = 47) | −0.012 (−0.042; 0.017) | 0.405 | −0.012 (−0.040 0.016) | 0.402 | 0.073 (0.041; 0.105) | 8.00*10 −6 | 0.029 (−0.004; 0.061) | 0.088 |
| Child BMI (N = 15) | 0.091 (0.063; 0.119) | 3.43*10 −10 | 0.073 (0.046; 0.100) | 1.40*10 −7 | 0.081 (0.050; 0.112) | 3.75*10 −7 | 0.038 (0.006; 0.070) | 0.020 |
| Adult BMI pathway genetic risk scores** | ||||||||
| Neuronal | ||||||||
| Neuronal developmental processes (N = 29) | 0.018 (0.014; 0.023) | 2.25*10-5 | 0.032 (0.003; 0.061) | 0.031 | 0.038 (0.004; 0.071) | 0.029 | 0.008 (−0.026; 0.042) | 0.654 |
| Neurotransmission (N = 10) | 0.013 (−0.015; 0.042) | 0.370 | −0.003 (−0.030; 0.024) | 0.827 | 0.002 (−0.029; 0.034) | 0.876 | −0.009 (−0.040; 0.023) | 0.595 |
| Hypothalamic expression and regulation (N = 13) | 0.099 (0.071; 0.128) | 5.81*10 −12 | 0.089 (0.062; 0.115) | 1.29*10 −10 | 0.080 (0.049; 0.111) | 5.30*10 −7 | 0.041 (0.009; 0.073) | 0.013 |
| Neuronal expression (N = 12) | 0.017 (−0.012; 0.046) | 0.240 | 0.020 (−0.008; 0.047) | 0.165 | 0.036 (0.004; 0.068) | 0.027 | 0.009 (−0.023; 0.041) | 0.583 |
| 0.023 (−0.005; 0.052) | 0.112 | 0.013 (−0.014; 0.041) | 0.341 | 0.016 (−0.016; 0.048) | 0.320 | −0.001 (−0.033; 0.032) | 0.972 | |
| Bone development (N = 9) | 0.018 (−0.011; 0.064) | 0.226 | 0.006 (−0.021; 0.033) | 0.656 | 0.015 (−0.016; 0.047) | 0.340 | 0.004 (−0.028; 0.036) | 0.811 |
| Signaling | ||||||||
| MAPK1/extracellular signal-regulated kinases (N = 9) | 0.034 (0.006; 0.062) | 0.018 | 0.037 (0.010; 0.064) | 0.008 | 0.023 (−0.008; 0.054) | 0.149 | 0.014 (−0.017; 0.046) | 0.378 |
| JAK (N = 2) | 0.033 (0.005; 0.062) | 0.023 | 0.020 (−0.007; 0.047) | 0.150 | 0.012 (−0.020; 0.043) | 0.457 | 0.007 (−0.025; 0.039) | 0.676 |
| CyclicAMP (N = 5) | 0.046 (0.017; 0.075) | 0.002 | 0.052 (0.024; 0.079) | 2.75*10 −4 | 0.039 (0.006; 0.071) | 0.019 | 0.026 (−0.007; 0.058) | 0.123 |
| WNTSignaling (N = 6) | 0.058 (0.030; 0.087) | 6.10*10 −5 | 0.029 (0.002; 0.057) | 0.034 | 0.039 (0.007; 0.070) | 0.016 | 0.032 (0.000; 0.064) | 0.047 |
| G-protein coupled receptor | ||||||||
| Notch signaling (N = 2) | −0.027 (−0.056; 0.001) | 0.059 | −0.028 (−0.055; 0.000) | 0.046 | −0.028 (−0.059; 0.003) | 0.075 | −0.028 (−0.060; 0.003) | 0.080 |
| Mitochondrial (N = 8) | 0.040 (0.010; 0.070) | 0.009 | 0.041 (0.012; 0.069) | 0.005 | −0.002 (−0.035; 0.031) | 0.905 | −0.003 (−0.037; 0.031) | 0.877 |
| Retinoic acid receptors (N = 6) | 0.045 (0.017; 0.074) | 0.002 | 0.037 (0.010; 0.065) | 0.007 | 0.016 (−0.015; 0.047) | 0.313 | 0.017 (−0.015; 0.049) | 0.293 |
| Endocytosis/exocytosis (N = 14) | −0.012 (−0.041; 0.016) | 0.400 | −0.003 (−0.030; 0.024) | 0.840 | −0.021 (−0.053; 0.010) | 0.178 | −0.020 (−0.051; 0.012) | 0.218 |
| Eye-related (N = 5) | 0.012 (−0.016; 0.041) | 0.398 | 0.015 (−0.012; 0.043) | 0.276 | −0.012 (−0.044; 0.020) | 0.456 | 0.003 (−0.029; 0.035) | 0.845 |
| Tumorigenesis (N = 11) | 0.041 (0.012; 0.070) | 0.006 | 0.020 (−0.008; 0.048) | 0.161 | 0.017 (−0.016; 0.049) | 0.312 | 0.013 (−0.020; 0.046) | 0.431 |
| Apoptosis (N = 13) | 0.025 (−0.003; 0.054) | 0.084 | 0.020 (−0.007; 0.047) | 0.151 | −0.008 (−0.039; 0.024) | 0.621 | −0.036 (−0.068; −0.004) | 0.028 |
| Membrane proteins (N = 12) | 0.075 (0.046; 0.103) | 2.44*10 −7 | 0.044 (0.017; 0.071) | 0.002 | 0.059 (0.028; 0.090) | 1.93*10 −4 | 0.011 (−0.021; 0.044) | 0.495 |
| Hormone metabolism/regulation (N = 4) | 0.021 (−0.008; 0.049) | 0.161 | 0.043 (0.015; 0.070) | 0.002 | 0.026 (−0.005; 0.057) | 0.103 | 0.004 (−0.028; 0.036) | 0.812 |
| Purine/pyrimidine cycle (N = 4) | 0.013 (−0.016; 0.041) | 0.379 | 0.004 (−0.023; 0.031) | 0.762 | −0.017 (−0.048; 0.014) | 0.285 | −0.007 (−0.038; 0.024) | 0.661 |
| Monogenic obesity/energy homeostasis (N = 9) | 0.074 (0.045; 0.102) | 4.74*10 −7 | 0.068 (0.041; 0.095) | 1.00*10 −6 | 0.065 (0.034; 0.096) | 5.00*10 −5 | 0.030 (−0.003; 0.062) | 0.072 |
| Immune system (N = 15) | 0.045 (0.017; 0.074) | 0.002 | 0.037 (0.010; 0.065) | 0.008 | 0.021 (−0.011; 0.052) | 0.193 | −0.008 (−0.040; 0.024) | 0.620 |
| Limb development (N = 3) | 0.035 (0.007; 0.064) | 0.015 | 0.024 (−0.006; 0.049) | 0.125 | 0.028 (−0.003; 0.060) | 0.076 | 0.004 (−0.028; 0.036) | 0.794 |
| Ubiquitin pathways (N = 6) | −0.007 (−0.036; 0.021) | 0.617 | 0.006 (−0.021; 0.034) | 0.656 | −0.011 (−0.043; 0.020) | 0.483 | −0.015 (−0.047; 0.017) | 0.359 |
| Glucose homeostasis/diabetes (N = 11) | 0.050 (0.021; 0.079) | 0.001 | 0.023 (−0.004; 0.051) | 0.096 | 0.042 (0.011; 0.074) | 0.008 | 0.009 (−0.023; 0.042) | 0.575 |
| Cell cycle (N = 23) | 0.044 (0.016; 0.073) | 0.002 | 0.047 (0.019; 0.074) | 0.001 | 0.024 (−0.007; 0.055) | 0.135 | 0.007 (−0.025; 0.039) | 0.684 |
| DNARepair | ||||||||
| Nuclear trafficking (N = 4) | −0.005 (−0.034; 0.023) | 0.716 | −0.009 (−0.036; 0.018) | 0.518 | −0.004 (−0.036; 0.028) | 0.804 | −0.008 (−0.041; 0.024) | 0.608 |
| Muscle biology (N = 6) | 0.048 (0.020; 0.077) | 0.001 | 0.029 (0.002; 0.057) | 0.038 | 0.025 (−0.007; 0.056) | 0.127 | 0.022 (−0.010; 0.054) | 0.181 |
* Bold font indicates P-value < 0.05. ** Bold font indicates significant after Bonferroni correction for the 28 pathways (p-value < 0.0018)
aAnalyses were performed in children with complete data on genetic variants, at least one outcome under study, and covariates
bValues are linear regression coefficients for models adjusted for sex and the first four genetic principal components and represent the difference in standard deviation scores of the outcome measures for each additional average risk allele in the risk scores
cValues are additionally adjusted for age
dValues are additionally adjusted for height
eRegression coefficients are based on standard deviation scores of ln-transformed outcome measures
Fig. 2.
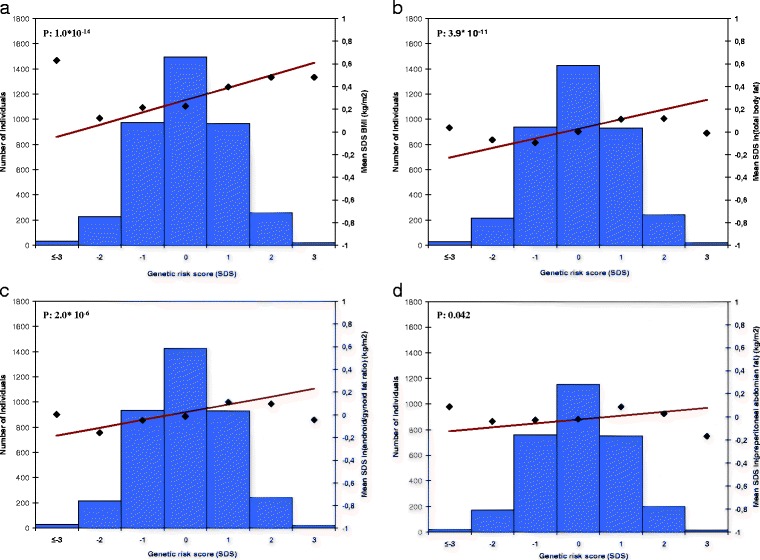
Association of adult body mass index genetic risk score with childhood adiposity measures (N = 3975). The x axis represents the categories of the risk score (overall sum of risk alleles, weighted by previous reported effect estimates, rescaled to SDS. The risk score ranged from −4 to 3 SDS and was rounded to the nearest integer for clarity of presentation). The right y axis shows the mean SDS and corresponds to the dots and a line representing the regression line of the mean SDS values for each category of the risk score. The y axis on the left corresponds to the histogram representing the number of individuals in each risk-score category. P-value is based on the continuous risk score, as presented in Table 3. Graph a-d represent; a BMI in kg/m2, b ln (fat mass percentage), c ln (android/gynoid fat ratio), and d ln (preperitoneal fat area)
Of the 28 adult BMI genetic risk scores based on the biological pathways, those based on neuronal developmental processes, hypothalamic expression and regulation, WNT-signaling, membrane proteins, monogenic obesity/energy homeostasis, glucose homeostasis/diabetes, and muscle biology were associated with childhood BMI (all p-values <0.0018). Genetic risk scores based on hypothalamic expression and regulation, cyclicAMP, monogenic obesity/energy homeostasis, and cell cycle were associated with total fat mass, whereas for android/gynoid fat ratio only the genetic risk scores based on hypothalamic expression and regulation, membrane proteins, and monogenic obesity/energy homeostasis show significant associations (all p-values <0.0018). None of the pathways were associated with preperitoneal fat area (Table 3). We based our pathway risk scores on these biological categories to keep our analysis as close as possible to the analysis of the original paper as possible [8]. As a comparison, we also ran a pathway analysis using IPA. Results were comparable regarding the major categories (eg. neurological development and function, cell cycle, lipid metabolism, apoptosis). However, the IPA software showed a larger subdivision with 74 different pathways instead of 28 as suggested by the GIANT consortium (Additional file 12, Table S9). The overall adult WHR genetic risk score was only associated with android/gynoid fat ratio (Table 3; Additional file 13: Figure S4a-d). The childhood BMI genetic risk score was associated with all childhood adiposity measures (Table 3; Additional file 14: Figure S5a-d). The genetic risk score based on 29 SNPs showed higher effect estimates per SD increase than our 97 SNPs adult BMI risk score for the childhood adiposity outcomes, especially for preperitoneal fat area (Additional file 8, Table S5). The 97 adult BMI SNPs explained 4.9 % of childhood BMI when added into our model as individual SNPs. When the 97 SNPs were combined into the weighted risk score and added to our model, the risk score explained 1.4 % of childhood BMI (Additional file 15: Table S10).
Discussion
We observed that a higher overall adult BMI genetic risk score based on 97 SNPs was associated with BMIAP during infancy, and with BMI, total fat mass, android/gynoid fat ratio, and preperitoneal fat area during childhood. A genetic risk score based on SNPs in or close to genes in the membrane proteins pathway was associated with infant PWV, whereas genetic risk scores based on pathways for neuronal developmental processes, hypothalamic processes, cyclicAMP, WNT-signaling, membrane proteins, monogenic obesity/energy homeostasis, glucose homeostasis, cell cycle, and muscle biology were associated with childhood adiposity measures. None of the pathway risk scores were associated with preperitoneal fat area.
Interpretation of main findings
Previous studies revealed a total of 97 loci related to adult BMI [8]. In a previous study, we reported on the association of a genetic risk score based on 29 adult BMI SNPs known at that time with infant growth and childhood adiposity measures [12]. This risk score was associated with a higher AGEAP and with a higher BMI, total fat mass, android/gynoid fat ratio, and preperitoneal fat area. In the current study, we aimed to identify the effects of updated and more detailed risk scores based on the 97 currently known loci and on subgroups of loci representing specific biological pathways on the same infant growth and childhood adiposity measures. Infant weight growth patterns are known to be strongly associated with BMI in childhood and adulthood, and childhood BMI is associated with obesity and cardiovascular disease in adulthood [1–4, 20, 22, 24]. Thus, it is important to understand the molecular pathways underlying childhood adiposity.
Our results suggest a modest effect of the adult BMI risk score on infant weight growth measures. We observed an association of the overall adult BMI genetic risk score with BMIAP only. In our previous study, based on 29 adult BMI SNPs, the genetic risk score was associated with AGEAP only [12]. A recent study among 9,328 children reported an association of a genetic risk score of 32 adult BMI-associated SNPs, including the 29 included in our previous risk score, with BMIAP, which is in line with our current finding. Additionally, a weak inverse association was found of this risk score with AGEAP [29]. The difference in associations between the previously published 29 SNP adult BMI risk score and our current 97 SNP adult BMI risk score may imply that the increased number of SNPs in the current genetic risk score adds noise to the association of the 97 SNP adult BMI risk score with childhood adiposity outcomes. Also, the analyses were run in a slightly different population, as siblings were excluded for the current study. The added SNPs may be more representative of BMIAP. The childhood BMI genetic risk score was associated with infant PWV and BMIAP, which are both strongly associated with increased risk of overweight in childhood [23]. The overall adult BMI genetic risk score was also associated with all childhood adiposity measures, which is in line with previous studies [12, 29, 30]. Some of these associations are partly explained by infant growth. The WHR risk score was associated with childhood android/gynoid fat ratio only, which is not surprising given the close relation of android/gynoid fat ratio to WHR. Results for the childhood BMI risk score were similar to the associations found with the adult BMI risk score, except that effect estimates were much larger for the child BMI risk score. Larger effect estimates may reflect stronger effects of the childhood-specific SNPs in children. Our results suggest that genetic risk scores based on adult BMI, WHR and childhood BMI influence childhood adiposity outcomes, and also BMI growth patterns from infancy onwards.
The 97 SNPs in our risk score explained 2.7 % of the adult BMI variance in the original paper [8]. In the current study we found that the same SNPs, when added simultaneously to our regression model, account for 4.9 % of childhood BMI suggesting a larger effect of these SNPs in childhood than in adulthood. This may be due to a relative increase in the effects of environmental factors over time. It should be noted that this estimate represents the upper bound of the phenotypic variation accounted for by the 97 SNPs, due to the method of entering all SNPs simultaneously to the model rather than combined into a risk score. When combined into a weighted risk score the 97 SNPs explained only 1.5 % of childhood BMI. We previously reported on a genetic risk score combining only 29 adult BMI SNPs, which explained 2.4 % of the variance in BMI in children of the Generation R Study [12]. Increasing the number of adult SNPs from 29 to 97 thus seemed to add noise to our risk score. It may be that some genetic loci show age-dependent associations with BMI, with different effects in children as compared to adults [31, 32]. Previous work has described an inverse association of the fat mass and obesity related locus (FTO) with BMI before the age of 2.5 years, no association between 2.5 and 5 years, and a positive association from around the age of 5 years onwards. The association then strengthens with age, reaching its peak at the age of 20 years and subsequently weakens again [31, 32]. A similar age dependent pattern has been observed for the melanocortin 4 receptor (MC4R) locus [32].
Our results showed that during infancy only the membrane proteins pathway affects PWV. This pathway involves membrane proteins that play a role regulating different cell processes involved in e.g., apetite, cholesterol synthesis, and gene expression [8]. Our findings suggest that these processes are also important for weight growth during early life. None of the other pathways were associated with infant growth measures. In line with previous adult studies, a strong role was observed for central nervous system related processes in pathways associated with childhood adiposity measures [8, 33]. Especially the hypothalamic expression and regulation pathway is suggested to be important, which is confirmed in our analyses [8, 31]. Mutations in some of the genes in the monogenic obesity/energy homeostasis related pathways are also suggested to act via central nervous system related processes [33]. The hypothalamic expression and regulation and the monogenic obesity/energy homeostasis pathways were associated with childhood BMI, total fat mass, and android/gynoid fat ratio. The other pathways that were associated with BMI suggest a role for fasting/feeding related processes, glucose homeostasis, signaling, and diabetes related pathways. Our findings suggest a stronger role for the predefined categories during childhood than during infancy, which may be because the childhood measures are more closely related to adult BMI.
Reported total heritability estimates for childhood BMI from twin studies are as high as 80 %. In the current study we found that a risk score based on the known SNPs only explained 1.5 % of the variation in child BMI, emphasizing that a large part of the heritability remains to be discovered [5, 6]. In addition to SNPs, other sources of (epi-) genetic variation, such as copy number variants (CNV) and differences in methylation, may also contribute [34, 35]. A large part of the common CNVs have been efficiently tagged by SNPs in GWA studies [36]. However, associations of rarer CNVs with BMI and obesity showed mixed results [35, 37, 38]. Recently, methylation at specific sites in the DNA has been associated with BMI in adults and children [34, 39, 40]. Additional research in larger study populations is needed to further disentanble the (epi-) genetic background of BMI in children and adults.
Methodological considerations
The large number of participants and available detailed phenotypes is a major strength of the study. Of all children with genetic data, information on infant growth measures was available for 54 %. Measures of childhood general and abdominal adiposity were available in 72 % of all children. Children without information on infant growth measures had a higher BMI, total fat mass, android/gynoid fat ratio, and preperitoneal fat area (all p-values < 0.001) compared with the participants included in our analyses. This may have resulted in an underestimation of the association for the risk scores with infant growth and childhood adiposity measures. Detailed measurements of childhood abdominal adiposity were performed. Both DXA and abdominal ultrasound are considered valid methods for such measurements [24, 26]. Not all SNPs were available in our GWAS dataset. We used a limited number of proxies in very high linkage disequilibrium to complete the sets of SNPs for adult BMI, WHR, and child BMI. No good proxies were available for two WHR SNPs. Given the high number of SNPs available, all risk scores are considered a good representative of the original set of adult SNPs. Although our population is relatively large, we still may have had limited power for these analyses, leaving a possibility of underestimating the number of associated pathways.
Conclusions
A genetic risk score based on 97 loci associated with adult BMI was associated with PWV during infancy and with general and abdominal fat measurements in childhood. Our results suggest that the genetic background and the pathways involved in adult and childhood adiposity at least partly overlap. Adult BMI related biological pathways involved in neuronal developmental processes, hypothalamic expression and regulation, cyclicAMP, WNT-signaling, membrane proteins, monogenic obesity/energy homeostasis, glucose homeostasis, and cell cycle likely influence adiposity from early life onwards. Further studies are needed to identify more (rare) loci and unravel the underlying mechanisms of childhood adiposity.
Acknowledgements
The Generation R Study is conducted by the Erasmus Medical Center in close collaboration with the School of Law and Faculty of Social Sciences of the Erasmus University Rotterdam, the Municipal Health Service Rotterdam area, Rotterdam, the Rotterdam Homecare Foundation, Rotterdam and the Stichting Trombosedienst and Artsenlaboratorium Rijnmond (STAR), Rotterdam. We gratefully acknowledge the contribution of participating mothers, general practitioners, hospitals, midwives and pharmacies in Rotterdam.
Availability of data and material
All data generated or analysed during this study are included in this published article and its supplementary information files.
Funding
The general design of Generation R Study is made possible by financial support from the Erasmus Medical Center, Rotterdam, the Erasmus University Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Netherlands Organisation for Scientific Research (NWO), the Ministry of Health, Welfare and Sport and the Ministry of Youth and Families. This research also received funding from the European Union’s Seventh Framework Programme (FP7/2007–2013), project EarlyNutrition under grant agreement no. 289346. VWJ received an additional grant from the Netherlands Organization for Health Research and Development (VIDI 016.136.361) and a Consolidator Grant from the European Research Council (ERC-2014-CoG-64916). JFF has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 633595 (DynaHEALTH).
Authors’ contributions
CM contributed to all steps, designed the study with VWVJ and JFF, analyzed the data, and drafted the manuscript. SV contributed to data analysis and critical review of the manuscript. CJK contributed to data collection and cleaning as well as critical review of the manuscript. VWVJ contributed to study design, critical appraisal of the data analysis of the data, and writing and critical revision of the manuscript. JFF contributed to study design, analysis of the data, and writing and critical revision of the manuscript. All authors read and approved the final manuscript.
Authors’ information
For correspondence please contact JFF.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by the Medical Ethical Committee of the Erasmus Medical Center, Rotterdam. A written consent was obtained for each participating child.
Abbreviations
- AGEAP
Age at adiposity peak
- BMI
Body mass index
- BMIAP
Body mass index at adiposity peak
- CI
Confidence interval
- CNV
Copy number variant
- GWAS
Genome-wide association studies
- IPA
Ingenuity pathway analysis
- PWV
Peak weight velocity
- SD
Standard deviation
- SDS
Standard deviation scores
- SNP
Single nucleotide polymorphism
- WHR
Waist-hip ratio
Additional files
Flow chart of participants. (DOCX 26 kb)
List of SNPs included in the main BMI and WHR risk scores. (XLS 23 kb)
List of SNPs included per risk score for each biological category. (XLS 34 kb)
Matrix of the number of overlapping SNPs per biological category. (XLS 13 kb)
Associations of unweighted genetic risk score for BMI with infant growth and childhood adiposity. (DOC 32 kb)
Association of WHR risk score with average peak weight velocity (a), body mass index at adiposity peak (b), and age at adiposity peak (c) (N = 2,955). (DOCX 50 kb)
Association of child BMI risk score with average peak weight velocity (a), body mass index at adiposity peak (b), and age at adiposity peak (c) (N = 2,955). (DOC 42 kb)
Associations of a 29 adult BMI SNPs genetic risk score with infant growth and childhood adiposity. (DOC 31 kb)
Phenotypic variance in measures of infant growth explained by genetic risk scores based on adult BMI and WHR SNPs (N = 3,975). (DOC 54 kb)
Associations of adult BMI genetic risk score with childhood adiposity, additionally adjusted for infant peak weight velocity (N = 3,975). (DOC 31 kb)
Associations of adult BMI genetic risk score with childhood adiposity, additionally adjusted for BMI at adiposity peak (N = 3,975). (DOC 30 kb)
Pathway analysis using Ingenuity Pathway Analysis software. (XLS 171 kb)
Association of WHR risk score with average BMI (a), total fat mass (b), android/gynoid fat ratio (c), and preperitoneal fat area (d) (N = 3,975). (DOC 47 kb)
Association of child BMI risk score with average BMI (a), total fat mass (b), android/gynoid fat ratio (c), and preperitoneal fat area (d) (N = 3,975). (DOC 48 kb)
Phenotypic variance in measures of childhood body composition explained by genetic risk scores based on adult BMI and WHR SNPs (N = 3,975). (DOC 61 kb)
Contributor Information
Claire Monnereau, Email: c.monnereau@erasmusmc.nl.
Suzanne Vogelezang, Email: s.vogelezang@erasmusmc.nl.
Claudia J. Kruithof, Email: c.kruithof@erasmusmc.nl
Vincent W. V. Jaddoe, Email: v.jaddoe@erasmusmc.nl
Janine F. Felix, Phone: +31107043997, Email: j.felix@erasmusmc.nl
References
- 1.World Health Organisation. Obesity and Overweight. Fact sheet No311. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/, Accessed July 2015.
- 2.Reilly JJ, Methven E, McDowell ZC, Hacking B, Alexander D, Stewart L, et al. Health consequences of obesity. Arch Dis Child. 2003;88(9):748–52. doi: 10.1136/adc.88.9.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker JL, Olsen LW, Sorensen TI. Childhood body-mass index and the risk of coronary heart disease in adulthood. N Engl J Med. 2007;357(23):2329–37. doi: 10.1056/NEJMoa072515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis CE, McTigue KM, Burke LE, Poirier P, Eckel RH, Howard BV, et al. Mortality, health outcomes, and body mass index in the overweight range: a science advisory from the American Heart Association. Circulation. 2009;119(25):3263–71. doi: 10.1161/CIRCULATIONAHA.109.192574. [DOI] [PubMed] [Google Scholar]
- 5.Maes H, Neale M, Eaves L. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27(4):325–51. doi: 10.1023/A:1025635913927. [DOI] [PubMed] [Google Scholar]
- 6.Wardle J, Carnell S, Haworth C, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87(2):398–404. doi: 10.1093/ajcn/87.2.398. [DOI] [PubMed] [Google Scholar]
- 7.Speliotes E, Willer C, Berndt S. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–48. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Locke A, Kahali B, Berndt S, Justice A, Pers T, Day F, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warrington N, Howe L, Paternoster L, Kaakinen M, Herrala S, Huikari V, et al. A genome-wide association study of body mass index across early life and childhood. Int J Epidemiol. 2015;44(2):700–12. doi: 10.1093/ije/dyv077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stergiakouli E, Gaillard R, Tavaré J, Balthasar N, Loos R, Taal H, et al. Genome-wide association study of height-adjusted BMI in childhood identifies functional variant in ADCY3. Obesity. 2014;22(10):2252–9. doi: 10.1002/oby.20840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felix J, Bradfield J, Monnereau C, van der Valk R, Stergiakouli E, Chesi A, et al. Genome-wide association analysis identifies four new susceptibility loci for childhood body mass index. Hum Mol Genet. 2016;25(2):389–403. doi: 10.1093/hmg/ddv472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogelezang S, Monnereau C, Gaillard R, Renders C, Hofman A, Jaddoe V, et al. Adult adiposity susceptibility loci, early growth and general and abdominal fatness in childhood: the Generation R Study. Int J Obes. 2015;39(6):1001–9. doi: 10.1038/ijo.2015.12. [DOI] [PubMed] [Google Scholar]
- 13.Shungin D, Winkler T, Croteau-Chonka D, Ferreira T, Locke A, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518(7538):187–96. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaddoe V, van Duijn C, Franco O, van der Heijden A, van Ijzendoorn M, de Jongste J, et al. The generation R study: design and cohort update 2012. Eur J Epidemiol. 2012;27(9):739–56. doi: 10.1007/s10654-012-9735-1. [DOI] [PubMed] [Google Scholar]
- 15.Kruithof C, Kooijman M, van Duijn C, Franco O, de Jongste J, Klaver C, et al. The generation R study: biobank update 2015. Eur J Epidemiol. 2014;29(12):911–27. doi: 10.1007/s10654-014-9980-6. [DOI] [PubMed] [Google Scholar]
- 16.Jaddoe V, van Duijn C, van der Heijden A, Mackenbach J, Moll H, Steegers E, et al. The generation R study: design and cohort update 2010. Eur J Epidemiol. 2010;25(11):823–41. doi: 10.1007/s10654-010-9516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Willer C, Ding J, Scheet P, Abecasis G. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berkey C, Reed R. A model for describing normal and abnormal growth in early childhood. Hum Biol. 1987;59(6):973–87. [PubMed] [Google Scholar]
- 20.Mook-Kanamori D, Durmuş B, Sovio U, Hofman A, Raat H, Steegers E, et al. Fetal and infant growth and the risk of obesity during early childhood: the Generation R Study. Eur J Endocrinol. 2011;165(4):623–30. doi: 10.1530/EJE-11-0067. [DOI] [PubMed] [Google Scholar]
- 21.Sovio U, Bennett A, Millwood I, Molitor J, O’Reilly P, Timpson N, et al. Genetic determinants of height growth assessed longitudinally from infancy to adulthood in the northern Finland birth cohort 1966. PLoS Genet. 2009;5(3) doi: 10.1371/journal.pgen.1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzoulaki I, Sovio U, Pillas D, Hartikainen A, Pouta A, Laitinen J, et al. Relation of immediate postnatal growth with obesity and related metabolic risk factors in adulthood: the northern Finland birth cohort 1966 study. Am J Epidemiol. 2010;171(9):989–98. doi: 10.1093/aje/kwq027. [DOI] [PubMed] [Google Scholar]
- 23.Kruithof C, Gishti O, Hofman A, Gaillard R, Jaddoe V. Infant weight growth velocity patterns and general and abdominal adiposity in school-age children. The generation R study. Eur J Clin Nutr. 2016. [DOI] [PMC free article] [PubMed]
- 24.Gishti O, Gaillard R, Manniesing R, Abrahamse-Berkeveld M, van der Beek E, Heppe D, et al. Fetal and infant growth patterns associated with total and abdominal fat distribution in school-age children. J Clin Endocrinol Metab. 2014;99(7):2557–66. doi: 10.1210/jc.2013-4345. [DOI] [PubMed] [Google Scholar]
- 25.Helba M, Binkovitz L. Pediatric body composition analysis with dual-energy X-ray absorptiometry. Pediatr Radiol. 2009;39(7):647–56. doi: 10.1007/s00247-009-1247-0. [DOI] [PubMed] [Google Scholar]
- 26.Mook-Kanamori D, Holzhauer S, Hollestein L, Durmus B, Manniesing R, Koek M, et al. Abdominal fat in children measured by ultrasound and computed tomography. Ultrasound Med Biol. 2009;35(12):1938–46. doi: 10.1016/j.ultrasmedbio.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki R, Watanabe S, Hirai Y, Akiyama K, Nishide T, Matsushima Y, et al. Abdominal wall fat index, estimated by ultrasonography, for assessment of the ratio of visceral fat to subcutaneous fat in the abdomen. Am J Med. 1993;95(3):309–14. doi: 10.1016/0002-9343(93)90284-V. [DOI] [PubMed] [Google Scholar]
- 28.Medina-Gomez C, Felix J, Estrada K, Peters M, Herrera L, Kruithof C, et al. Challenges in conducting genome-wide association studies in highly admixed multi-ethnic populations: the Generation R Study. Eur J Epidemiol. 2015;30(4):317–30. doi: 10.1007/s10654-015-9998-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warrington N, Howe L, Wu Y, Timpson N, Tilling K, Pennell C, et al. Association of a body mass index genetic risk score with growth throughout childhood and adolescence. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0079547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belsky D, Moffitt T, Houts R, Bennett G, Biddle A, Blumenthal J, et al. Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Arch Pediatr Adolesc Med. 2012;166(6):515–21. doi: 10.1001/archpediatrics.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sovio U, Mook-Kanamori D, Warrington N, Lawrence R, Briollais L, Palmer C, et al. Association between common variation at the FTO locus and changes in body mass index from infancy to late childhood: the complex nature of genetic association through growth and development. PLoS Genet. 2011;7(2) doi: 10.1371/journal.pgen.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardy R, Wills A, Wong A, Elks C, Wareham N, Loos R, et al. Life course variations in the associations between FTO and MC4R gene variants and body size. Hum Mol Genet. 2010;19(3):545–52. doi: 10.1093/hmg/ddp504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willer C, Speliotes E, Loos R, Li S, Lindgren C, Heid I, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41(1):25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dick K, Nelson C, Tsaprouni L, Sandling J, Aïssi D, Wahl S, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383(9933):1990–8. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 35.Bochukova E, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463(7281):666–70. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conrad D, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–12. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henrichsen C, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18(R1):R1–8. doi: 10.1093/hmg/ddp011. [DOI] [PubMed] [Google Scholar]
- 38.Peterson R, Maes H, Lin P, Kramer J, Hesselbrock V, Bauer L, et al. On the association of common and rare genetic variation influencing body mass index: a combined SNP and CNV analysis. BMC Genomics. 2014;15:368. doi: 10.1186/1471-2164-15-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demerath E, Guan W, Grove M, Aslibekyan S, Mendelson M, Zhou YH, Hedman ÅK, et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24(15):4464–79. doi: 10.1093/hmg/ddv161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang R, Garratt E, Pan H, Wu Y, Davis E, Barton S, et al. Genome-wide methylation analysis identifies differentially methylated CpG loci associated with severe obesity in childhood. Epigenetics. 2015;10(11):995–1005. doi: 10.1080/15592294.2015.1080411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole T, Bellizzi M, Flegal K, Dietz W. Establishing a standard definition for child overweight and obesity worldwide: international survey. BMJ. 2000;320(7244):1240–3. doi: 10.1136/bmj.320.7244.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]