

Abstract
Background:
The purpose of this study was to develop ibuprofen (IB)-polyethylene glycol (PEG) 8000 solid dispersions (SDs) and investigate them for in vitro dissolution and in vivo anti-inflammatory activity.
Materials and Methods:
IB-PEG 8000 SDs were prepared by fusion method using varying combination ratios of IB and PEG 8000. Characterization based on surface morphology, particle size, absolute drug content, and Fourier transform infrared (FT-IR) spectroscopy was carried out on the SDs. The in vitro release of IB from the SDs was performed in simulated gastric fluid (SGF, pH 1.2) and simulated intestinal fluid (SIF, pH 7.4) without enzymes, whereas the anti-inflammatory activity was evaluated using egg albumin-induced rat paw edema model.
Results:
Greenish brown, discrete, and irregularly shaped SDs of mean particle size range 113.5 ± 2.5-252.5 ± 1.9 μm, which were stable over 3 months, were obtained. The drug content of the SDs ranged from 73.4 ± 2.9 % to 83.5 ± 2.7%. Although the drug content increased with increased concentration of PEG 8000 in the SDs, the mean particle size decreased with increased concentration of PEG 8000 in the SDs. The FT-IR results indicate no strong chemical interaction of IB and PEG 8000 in the SDs. There was marked increase in the dissolution rate of IB from the SDs (P < 0.05) as compared to pure IB and physical mixture. The dissolution was better in SIF than in SGF. The increased dissolution rate of IB may be due to the formation of microcrystals, increased wettability and dispersibility in PEG 8000. The SDs showed good anti-inflammatory properties achieving up to 90% edema inhibition at 6 h while the pure sample of IB had 77% edema inhibition at 6 h.
Conclusion:
SDs based on IB-PEG 8000 is a good approach to enhance the dissolution rate and anti-inflammatory activity of IB, thus, encouraging further development of the SDs.
Key words: Ibuprofen, in vitro dissolution, edema inhibition, polyethylene glycol 8000, solid dispersion
INTRODUCTION
The oral route of administration is the most common and preferred route of drug delivery, but for many drugs it can be a problematic and inefficient mode of delivery for a number of reasons including limited drug absorption resulting in poor bioavailability.[1,2,3] Drug absorption from the gastrointestinal (GI) tract can be limited by a variety of factors with the most significant contributors being poor aqueous solubility and/or poor membrane permeability of the drug molecule. Thus, a drug with poor aqueous solubility will typically exhibit dissolution rate limited absorption, and a drug with poor membrane permeability will typically exhibit permeation rate limited absorption. Hence, the two areas of pharmaceutical research that focus on improving the oral bioavailability of active agents include enhancing solubility and dissolution rate of poorly water-soluble drugs and enhancing the permeability of poorly permeable drugs.[4,5,6,7,8,9,10]
Solid dispersion (SD) technology has attracted considerable interest as an efficient means of improving the dissolution rate, and hence, the bioavailability of a range of hydrophobic drugs. SD is the dispersion of one or more active ingredients in inert carriers at solid state prepared by various methods.[1,2,3] SD can help overcome the delivery problems of new classes of active molecules and may also extend the therapeutic potential of established drugs.[11,12,13,14,15,16,17,18,19,20]
Ibuprofen (IB) ([2RS]-1[4-[2-methyl propyl] phenyl] propionic acid) [Figure 1] is a nonsteroidal anti-inflammatory drug (NSAID) that has been widely used in the treatment of mild to moderate pain and fever. It is a nonselective inhibitor of cyclooxygenase-1 (COX-1) and COX-2. Although its anti-inflammatory properties may be weaker than those of some other NSAIDs, it has a prominent analgesic and antipyretic effect. Its effects are due to the inhibitory actions on COXs, which are involved in the synthesis of prostaglandins, agents that play an important role in the production of pain, inflammation, and fever.[21,22] Because of high membrane permeability, the extent IB absorption approaches up to 100%. Dissolution thus becomes the rate limiting step for absorption.[23] Various formulations, such as prodrugs, inclusion complexes, and microcapsules of IB, were developed.[24,25,26] However, the dissolution rate and the oral bioavailability of IB from these formulations differed widely. The formulation methods were time-consuming and costly, and some formulations were bulky with poor flow characteristics and handling difficulties. SDs of poorly water soluble drugs in hydrophilic carrier matrices have been reported to improve their solubility and dissolution rate.[27,28,29] Moreover, they are also proven to enhance their bioavailability by increasing their saturation solubility in GI fluids. However, IB SDs using solvent or solvent-melting method could be problematic because of the difficulty of finding a common solvent. Large volumes of solvents and long duration of heating might be necessary for complete dissolution of both components. The common methods such as vacuum drying, spray drying, spraying on sugar beads using a fluidized bed coating system, or lyophilization used for the removal of organic solvents from SDs could make the process relatively more complicated, tedious, and costly. In addition, they might also associate with the solvent related environmental problems.[30] Although SDs by melting could be problematic (for drugs with high melting temperature) because of the possible thermal instability of the components, and the hardening of melts resulting into difficulties in the pulverization for subsequent formulation, the low melting temperature of IB may permit SD with low melting point. The traditional melting methods have been reported to be associated with many processing difficulties such as the temperature and shear rate control, reproducibility, and scalability. For many drugs including IB, SDs by melt agglomerations in high shear mixers using a hot solution of meltable hydrophilic carriers as a binding solution have been claimed to be advantageous industrially.[31,32]
Figure 1.
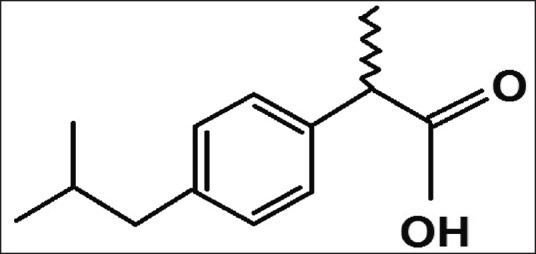
Structural formula of ibuprofen
Polyethylene glycols (PEGs) are semi-crystalline polymers that have been used extensively in the SDs preparation for their wetting, solubilizing, and surface active properties.[7] They have been reported to enhance the solubility, dissolution, and bioavailability of many poorly water soluble drugs using various techniques including melting agglomeration and melting.[31,32,33,34,35] The extent of their absorption appears to be dependent on their molecular weights. Complete absorptions have been reported for PEGs with lower molecular weights. However, the absorption is much more limited in the case of PEGs with high molecular weights. A particular advantage of PEGs for the formation of SDs is that they also have good solubility in many organic solvents. These relatively low melting points of PEGs are advantageous in the manufacture of SDs by the melting method.[31,32] In this study, to improve its solubility, dissolution, bioavailability, and low melting temperature IB were utilized to make SDs with PEG 8000. The SDs were evaluated for their in vitro dissolution and in vivo anti-inflammatory properties. PEG 8000 was empirically selected as a meltable polymer for its low melting point, surfactant properties, and oral safety.[2,7]
Consequently, the aim of this study was to evaluate, in vitro dissolution and in vivo anti-inflammatory properties of IB-loaded PEG 8000 SDs prepared by the fusion method for the controlled delivery of IB.
MATERIALS AND METHODS
Materials
IB (Spectrum, Houston, TX), PEG-8000 (Carl Roth, Karlsruhe, Germany), concentrated hydrochloric acid, sodium chloride, sodium hydroxide (Merck, Germany), and monobasic potassium phosphate (Sigma Chemical Co., USA) were used as procured from the manufacturers without further purification. All other reagents were analytical grade and used as such. Distilled water was obtained from an all glass still.
Preparation of ibuprofen physical mixture
IB to PEG 8000 ratio of 1:1 was prepared for comparative study. The samples were weighed and mixed thoroughly in a mortar and pestle to obtain a homogeneous physical mixture (PM). This was sieved through a 100 mesh screen and transferred into an amber colored bottle and stored in a desiccator for further use. It was coded PM.
Preparation of ibuprofen solid dispersions
The fusion (melt) method was adopted.[1,36] Briefly, an accurately weighed amount of PEG 8000 was placed in a crucible on a water bath and melted with constant stirring at a temperature of about 60°C. An accurately weighed amount of IB was incorporated into the melted carrier with stirring to ensure homogeneity. The mixture was heated until a clear homogeneous melt was obtained. The crucible was then removed from the water bath and allowed to cool at room temperature. The prepared SDs were placed in glass vials stored in a desiccator for further studies. The formulations were coded PB1, PB2, PB3, and PB4 based on IB:PEG 8000 ratios (1:1), (1:2), (1:3), and (1:4), respectively.
Characterization of the solid dispersions
Determination of the percentage yield
The percentage yield was calculated from the weight of dried SDs (W1) recovered from each of the batches and the sum of the initial dry weight of starting materials (W2) using the following equation:[27,28]
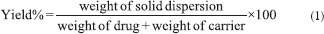
Estimation of drug content and encapsulation efficiency
Approximately, 100 mg of the SDs was weighed accurately and dissolved in 100 mL of phosphate buffered saline (PBS, pH 7.4). The solution was shaken vigorously and filtered, and the filtrate was spectrophotometrically (Unico 2102 PC UV/Vis Spectrophotometer, USA) analyzed at 275 nm for IB content. The amount of drug encapsulated in the SDs was calculated with reference to a standard Beer's plot for IB to obtain the percentage encapsulation efficiency using the formula below:[17,31,32]

The above procedure was repeated to obtain the encapsulation efficiency as the mean from the replicate determinations.
Particle size analysis and morphological characteristics
The particle size of the SDs was determined by computerized image analysis on a photomicroscope (Leica, Germany). Samples from each of the batches were dispersed in PBS and mounted on a slide and observed under a light microscope. With the aid of software in the microscope, the projected diameters of the particles corresponding to the particle sizes of the SDs were determined and the mean calculated. The particle morphologies were also observed and captured by the photomicroscope.
Fourier transform infrared spectroscopy
The FT-IR spectra of IB, PEG-8000, and SD in different ratios were recorded with FT-IR spectrophotometer. The SDs of the ratios 1:1, 1:2, 1:3, 1:4, and 1:5 of drug and polymer, respectively, were evaluated for their drug polymer interaction in the FT-IR spectrum after the translucent pellets were made using KBr. FT-IR spectroscopy was conducted using a Shimadzu FT-IR 8300 spectrophotometer (Shimadzu, Tokyo, Japan) and the spectrum was recorded in the wavelength region 4000-400/cm with threshold of 1.303, sensitivity of 50, and resolution of 2/cm range. The procedure consisted of dispersing a sample in KBr and compressing into discs by applying a pressure of 5 tons for 5 min in a hydraulic press. The pellet was placed in the light path and the spectrum obtained.
In vitro drug dissolution
In vitro dissolution profile for each SD as well as pure drug was performed using USP XXII rotating paddle apparatus (Erweka, Germany). Beer's plot for IB at different concentrations was made at a wavelength of 245 nm in simulated gastric fluid (SGF, pH 1.2) and at a wavelength of 272 nm in simulated intestinal fluid (SIF, pH 7.4). The dissolution medium consisted of 250 mL of freshly prepared SGF, without pepsin (pH 1.2) maintained at 37 ± 1°C. The polycarbonate dialysis membrane used was pretreated by soaking it in the dissolution medium for 24 h prior to the commencement of each release experiment. In each case, 100 mg of the formulated SDs was placed in the dialysis membrane containing 5 mL of the dissolution medium, securely tied with a thermo-resistant thread and then immersed in the dissolution medium under agitation provided by the paddle at 50 rpm. At predetermined time intervals, 2 mL portions of the dissolution medium were withdrawn, filtered, and analyzed spectrophotometrically (Unico 2102 PC UV/Vis Spectrophotometer, USA) at 245 nm. For each sample withdrawn, an equivalent volume (2 mL) of SGF maintained at the same temperature was added to the contents of the dissolution medium to maintain sink conditions throughout the release period. The amount of drug released at each time interval was determined with reference to the standard Beer's plot for IB in SGF. A positive control was set up for each batch by similarly weighing amounts of pure IB equivalent to that in the SDs. The release study was repeated using freshly prepared SIF without pancreatin (pH 7.4) as the release medium. Four replicate release studies were carried out in each case.
In vitro anti-inflammatory studies
This study was conducted in accordance with Ethical (Guidelines of Animal Care and Use) Committee (Research Ethics Committee) of University of Nigeria, Nsukka, following the Federation of European Laboratory Animal Science Association and European Community Council Directive of November 24, 1986 (86/609/ECC/). Male Wister rats (100-200 g) were housed in cages under controlled temperature and humidity in a photoperiod schedule of 12 h light/12 h duration. They were fed a standard laboratory animal diet and tap water ad libitum. The rats had free access to food and water prior to the commencement and throughout the duration of the experiment in order to mitigate the gastro-erosive side effects of the administered loaded drug. The animals were divided into six experimental groups of five rats per group. The IB SD equivalent to 20 mg/kg body weight was administered orally to the rats. The reference group received 20 mg/kg of a pure sample of IB, while the control group received 1% (w/v) polyethylene glycol 8000 (PEG 8000) suspension, serving as vehicle control. Thirty minutes posttreatment, edema was induced by injection of 0.2 mL fresh undiluted egg albumin into the sub-planter region of the right hind paw of the rats. The volumes of the distilled water displaced by treated right hind paw of the rats were measured using plethysmometer before and at 30 min, 1, 2, 3, 4, 5, and 6 h after egg albumin injection.
Average edema at every interval was assessed in terms of the difference in volume displacement (Vt-Vo). The percentage inhibition of edema was calculated using the relation:
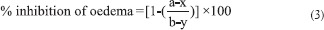
where “a” is the mean paw volume of treated rats after egg albumin injection, “x” is the mean paw volume of treated rats before egg albumin injection, “b” is the mean paw volume of control rats after egg albumin injection, and “y” is the mean paw volume of control rats before egg albumin injection.
Stability studies
Stability studies were carried out by keeping the samples in screw cap vials in a desiccator at 25 ± 2°C and 60 ± 5% relative humidity. Samples were drawn and analyzed for drug content and dissolution studies after 3 months.
Statistical analysis
The results obtained were analyzed using One-way analysis of variance. Descriptive and inferential statistics were employed in data analysis. P < 0.05 was considered to be statistically significant.
RESULTS AND DISCUSSION
The percentage yields of SD obtained for all batches of the SDs are shown in Table 1, and were between 63% (for batch PB1) and 81% (for batch PB4) while the percentage yield of the PM was 56%. The percentage yield increased with increase in the concentration of PEG 8000 in the SDs. High values of the SDs recovered from the formulations are a strong indication that the formulation technique adopted is reliable and amenable to validation, consistent with similar reports.[12,37,38]
Table 1.
Formulation compositions and some physicochemical properties of ibuprofen SDs
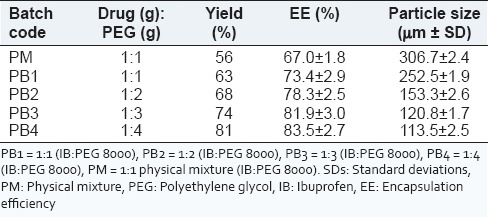
The encapsulation efficiencies of the SDs [Table 1] were good for all batches, ranging from 73.4 ± 2.9% to 83.5 ± 2.7% for batches PB1 and PB4, respectively. The encapsulation efficiency of the PM was lower than those of the PMs (67.0 ± 1.8), perhaps due to its crystalline nature. It is evident from Table 1 that the drug contents were dependent on the composition of the SDs, consistent with similar reports.[17,39] The encapsulation efficiency increased with increase in the concentration of PEG 8000 in the SDs. The higher values of the encapsulation efficiency observed in the SDs may be due to increase in the core material in the SDs. Relatively higher entrapment efficiency of the drug in batch PB4 may be due to the enhanced solubilizing effect of higher quantity of PEG 8000.
Figures 2a, 2b and 2c show the photomicrographs of IB, IB-PEG 8000 PM, and SD (optimized formulation – PB4), respectively while the particle sizes of the SDs are shown in Table 1. The SDs showed different surface characteristics that varied with the compositions of the SDs. The particle size ranges of the SDs were from 113.5 ± 2.5 µm to 252.5 ± 1.9 µm for batches PB1 and PB4, respectively, while that of the PM was 306.7 ± 2.4 µm. The higher the polymer (PEG 8000), the smaller the particle sizes of the SDs. However, the particle sizes of all the batches lie within the micrometer range. The drug and the formulated SDs had variable shapes and were greenish brown in color. The range of particle diameters for SDs would be useful in oral, intramuscular, and intravenous delivery of various classes of drugs since the size of SDs is known to play a critical role in determining the route of delivery of various drugs.[11,13,14] The surface morphology of SDs almost resembles that of pure PEG 8000, indicating that IB was absorbed into the PEG 8000 and homogeneously dispersed into the polymer. The photomicrographs suggest that the individual surface properties of PEG 8000 and IB were lost during melting and solidification indicating the formation of effective SD systems.
Figure 2.
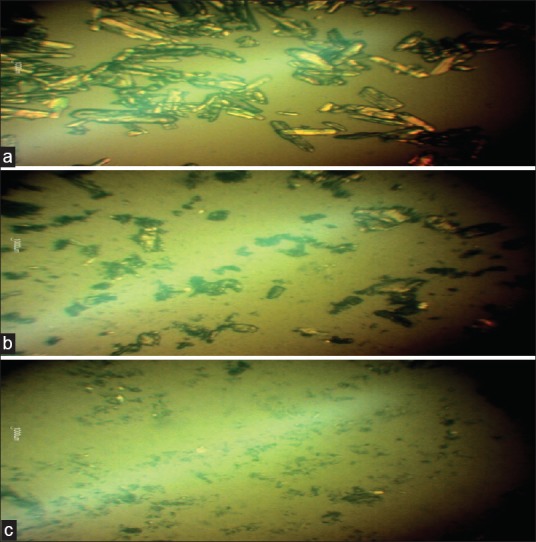
(a) Ibuprofen pure sample (IB) (b) ibuprofen-PEG 8000 physical mixture 1:1 (PM) and (c) ibuprofen-PEG 8000 solid dispersion 1:4 (optimised formulation, batch PB4)
Figures 3–9 show FT-IR spectra of IB, PEG 8000, IB-PEG 8000 PM, and SDs, respectively. The FT-IR was carried out as fingerprints to identify PM and SDs of IB relative to IB pure sample. The FT-IR spectrum of IB showed that principal peaks were observed at wave numbers of 511.15 – 683.7, 772.52, 938.40, 1074.39, 1241.23, 1329.00, 1435.09, 1715.74, and 2942.51/cm corresponding to aromatic C-H deformation, aromatic C-H deformation (two adjacent free H's), aromatic C-H deformation (one adjacent free H's), C-N vibration, C-O stretching, O-H bend, C=H deformation (CH3CH2), C=C stretching of α-β unsaturated ring, and C-H stretching, respectively. The spectrum of pure IB showed an intense, well-defined infrared band of carbonyl stretching of the isopropionic acid group. PEG 8000 showed a characteristic broad spectrum of O-H stretching vibration from 3,300 to 3,600/cm, C-H stretching of OC2H5 groups from 2,800 to 2,900/cm, and C-O stretching from 1,000 to 1,200/cm. SDs showed IR spectra almost similar to those of their corresponding PMs. The FT-IR spectrum of the PM of IB and PEG 8000 (batch PM) showed that principal peaks were observed at wave numbers of 523.69 – 668.36, 847.74, 946.12, 1116.82, 1254.74, 1346.36, 1447.62, 1719.6, 2884.64/cm corresponding to aromatic C-H deformation, aromatic C-H deformation (two adjacent free H's), aromatic C-H bending (one adjacent free H's), C-N vibration, C-O stretching (strong), O-H bend, C=H deformation, C=C stretching of α-β unsaturated ring, and O=C-H stretching, respectively. The FT-IR spectrum of IB-PEG 8000 SD (batch PB1) showed that principal peaks were observed at wave numbers of 520.8 – 666.43, 845.81, 946.12, 1117.79, 1257.63, 1347.32, 1473.66, 1720.56, 2883.68/cm corresponding to aromatic C-H deformation, aromatic C-H bending (two adjacent free H's), aromatic C-H bending (one adjacent free H's), C-O stretching (strong), C-O stretching, O-H bend, C=H deformation (CH3CH2), C=C stretching of α-β unsaturated ring, and O=C-H stretching, respectively. The FT-IR spectrum of IB-PEG 8000 solid dispersion (batch PB2) showed that principal peaks were observed at wave numbers of 524.66, 844.85, 954.8, 1111.03, 1236.41 – 1279.81, 1349.25, 1464.02, 1719.6, 2883.68/cm corresponding to aromatic C-H deformation, aromatic C-H bending (two adjacent), aromatic C-H bending (one adjacent free H's), C-O stretching (strong), C-O stretching, O-H bend, C=H deformation (CH3CH2), C=C stretching of α-β unsaturated ring, and O=C-H stretching, respectively. The FT-IR spectrum of IB-PEG 8000 SD (batch PB3) showed that principal peaks were observed at wave numbers of 842.92, 945.15, 1118.75, 1263.42, 1351.18, 1473.66, 1720.56, 2893.32/cm corresponding to aromatic C-H bending (two adjacent free H's), aromatic C-H bending (one adjacent free H's), C-O stretching (strong), C-O stretching, O-H bend, C=H deformation (CH3CH2), C=C stretching of α-β unsaturated ring, and O=C-H stretching, respectively. FT-IR spectrum of IB-PEG 8000 SD (batch PB4) showed that principal peaks were observed at wave numbers of 843.88, 950.94, 1118.75, 1264.38, 1352.14, 1464.02, 1721.53, 2884.64/cm corresponding to aromatic C-H bending (two adjacent free H's), aromatic C-H bending (one adjacent free H's), C-O stretching (strong), C-O stretching, O-H bend, C=H deformation (CH3CH2), C=C stretching of α-β unsaturated ring, and O=C-H stretching, respectively. FT-IR is a very powerful technique for detecting the presence of interaction in drug carrier SDs.[6,8,40] The appearance or disappearance of peaks and/or the shift of their positions are often an indication of interactions such as hydrogen bonding.[20,34,35] As principal peaks of FT-IR spectrum of the pure drug appeared in the IB-PEG 8000 PM and SDs, it can be concluded that there was no strong chemical interaction between the drug and the polymers employed in the formulations. Sharper and major peaks of IB were obtained more in the SDs than in the PMs. By implication, the drug (IB) was solubilized more in the former than in the latter. Some additional peaks were also observed in the spectra of IB SDs, which could be due to the presence of PEG 8000. The disappearance of some major peaks of IB in IB SDs is an indication of overlapping due to hydrogen bonding or overlapping of peak corresponding to the drug and the polymers.
Figure 3.
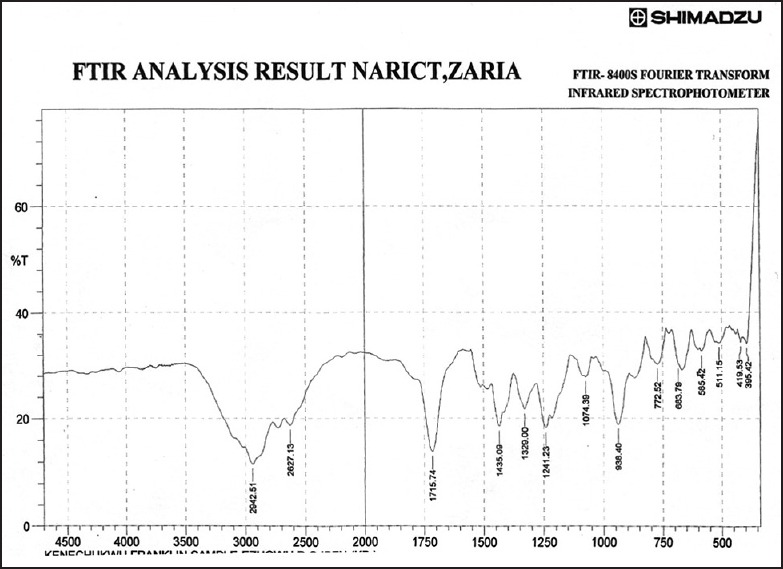
Fourier transform infrared spectrum of ibuprofen pure sample
Figure 9.
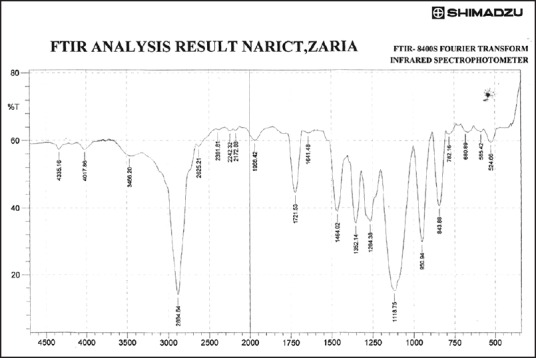
Fourier transform infrared spectrum of ibuprofen solid dispersion using ibuprofen and polyethylene glycol 8000 in the ratio of 1:4 (batch PB4)
Figure 4.
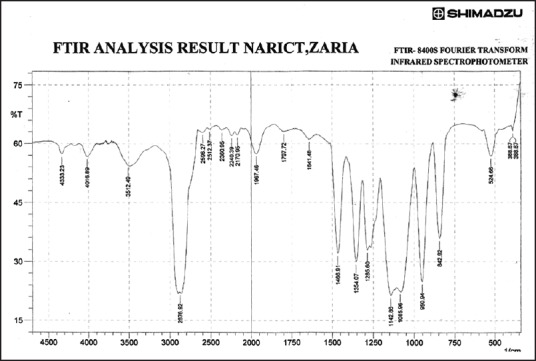
Fourier transform infrared spectrum of polyethylene glycol 8000
Figure 5.
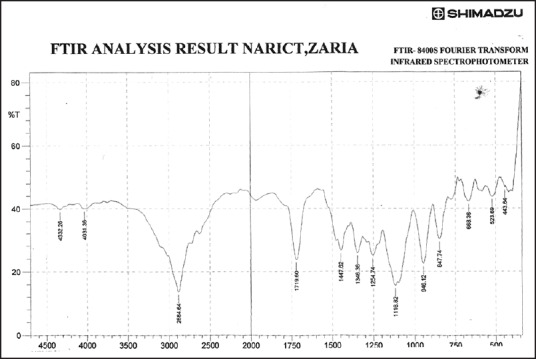
Fourier transform infrared spectrum of physical mixture using ibuprofen and polyethylene glycol 8000 in the ratio of 1:1 (batch physical mixture)
Figure 6.
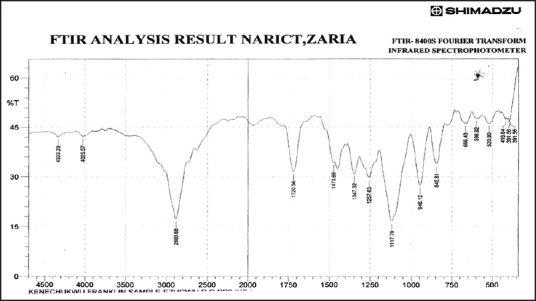
Fourier transform infrared spectrum of ibuprofen solid dispersion using ibuprofen and polyethylene glycol 8000 in the ratio of 1:1 (batch PB1)
Figure 7.
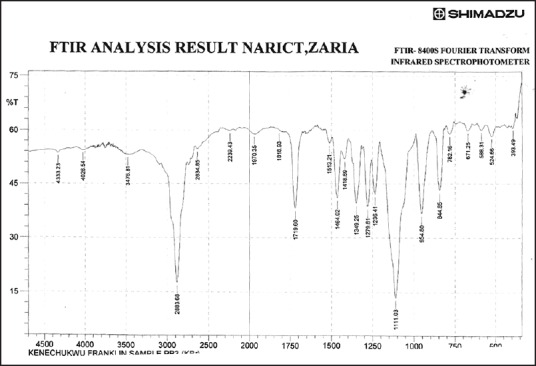
Fourier transform infrared spectrum of ibuprofen solid dispersion using ibuprofen and polyethylene glycol 8000 in the ratio of 1:2 (batch PB2)
Figure 8.
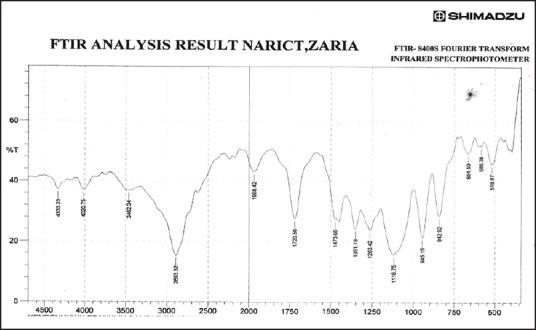
Fourier transform infrared spectrum of ibuprofen solid dispersion using ibuprofen and polyethylene glycol 8000 in the ratio of 1:3 (batch PB3)
The dissolution profiles of pure IB, the PM, and the SDs in SGF (pH 1.2) and in SIF (pH 7.4) are shown in Figures 10 and 11, respectively. The release profiles for IB from the SDs were investigated in SGF and SIF. These were used as the release media to simulate the GI environment to aid in the prediction of the release profile of the various formulations of the drugs in the GI tract.[1] The PM and SDs showed improved dissolution of IB over that of pure IB. The improved dissolution of IB is mainly attributed to increased wettability and improved solubility due to the higher level of hydrophilicity achieved by the use of polymeric carrier, PEG 8000, which sterically stabilized the surface of the hydrophobic drug (IB). The drug is then adsorbed on the surface of the carrier in an extremely fine state of subdivision. The resulting decrease in particle size and the concomitant increase in the surface area served to increase the thermodynamic activity of the drug, which in turn greatly enhanced the dissolution of the drug as compared to the pure drug alone.[8] In addition, all of the SD samples revealed more improved IB dissolution than the PM. This indicates that the increased dissolution of IB from IB SD was due to the presence of the drug in an amorphous state when compared with the PMs and pure drug, where the drug is present in the crystalline state.[40] The increased dissolution of IB from the SDs was due to the presence of the drug in the amorphous state as compared to the crystalline pure drug.[8,40] Other mechanisms such as reduction of particle size of incorporated drug, partial transformation of the crystalline drug to the amorphous state, formation of solid solution and complexes, reduction of aggregation and agglomeration, improved wetting of drug, and solubilization of the drug by the carrier of the diffusion layer have been reported to be responsible for improving the aqueous solubility or dissolution properties of SDs.[19,20,34,35]
Figure 10.
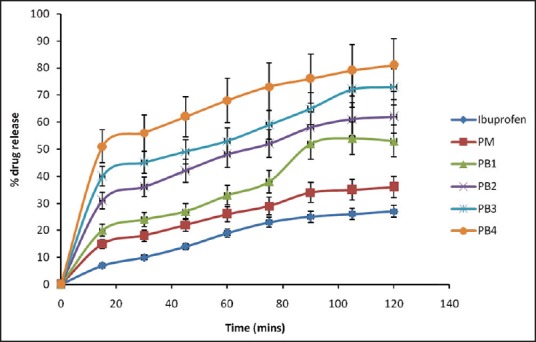
Dissolution profiles of solid dispersions in simulated gastric fluid (pH 1.2). Keys: 1B = ibuprofen, PM = physical mixture, PB1=1:1 (ibuprofen: polyethylene glycol 8000), PB2 = 1:2 (ibuprofen: Polyethylene glycol 8000), PB3 = 1:3 (ibuprofen: Polyethylene glycol 8000), PB4 = 1:4 (ibuprofen: Polyethylene glycol 8000). Data are expressed as mean ± standard deviation (n = 3)
Figure 11.
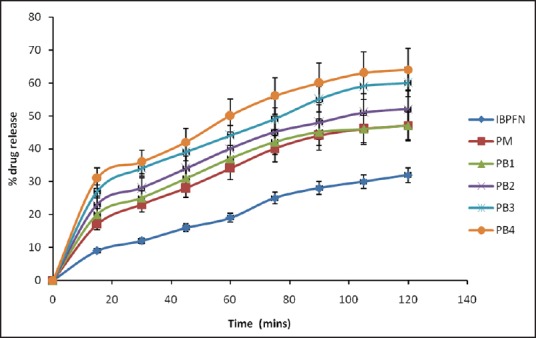
Dissolution profiles of solid dispersions in simulated intestinal fluid (pH 7.2). Keys: 1B = ibuprofen, PM= physical mixture, PB1=1:1 (ibuprofen: Polyethylene glycol 8000), PB2 = 1:2 (ibuprofen: Polyethylene glycol 8000), PB3 = 1:3 (ibuprofen: Polyethylene glycol 8000), PB4 = 1:4 (ibuprofen: Polyethylene glycol 8000). Data are expressed as mean ± standard deviation (n = 3)
There was burst release in SGF unlike in SIF, where the SDs exhibited sustained drug dissolution. The burst release was characterized by an initial drug release which occurred rapidly in <10 min into the receptor compartment in which more than 20% of the loaded drug was released from the various batches of the SDs. This initial “burst release” was followed by a more gradual and extended release over the next 2 h. In SGF, the amounts of drug released from the formulations [Figure 10] in 30 min were 10.12% of pure sample, 18.20% of PM, and 24.01%, 36.10%, 45.14%, and 56.24% of PB1, PB2, PB3, and PB4, respectively. In SIF, however, the amounts of drug released from the formulations [Figure 11] in 30 min were 12.05% of pure sample, 23.07% of PM, and 25.22%, 28.17%, 34.20%, and 36.13% of PB1, PB2, PB3, and PB4, respectively. It was also observed that more than 50% of drug was released after 60 min from batches PB3 and PB4 of SDs of IB when compared with the pure sample of IB, which achieved 19% drug release after 60 min in SGF. Overall, the SDs with 1:5 and 1:4 carrier ratio of IB:PEG 8000 exhibited higher dissolution rate than those with lower carrier content (1:1, 1:2, 1:3) in both SIF and SGF. The order of dissolution rate shown by the SDs was found to be 1:5 >1:4 >1:3 >1:2, 1> 1:1). The amounts of IB released as a result of burst effect may likely represent the amounts that adhered weakly to the surface of the formulated SDs. The remaining amounts which were released in a more gradual pattern most likely represented the amounts that were entrapped into the core (matrix) of the SDs. Burst release resulting in biphasic release pattern may be utilized in the therapeutic design of dosage forms, and this has severally been reported for SDs.[16,17] It may be an advantage because it would lead to the high initial blood concentration of the drug and a gradual release of the remaining drug. The bolus dose, when required for instance in the management of painful and inflammatory conditions, will be provided by the initial burst as seen in the SD formulations. The improved dissolution was more sustained in SIF than in SGF. This could be related to the fact that the carrier (PEG 8000) is more soluble at higher pH (SIF, pH 7.4) than lower pH (SGF, pH 1.2).
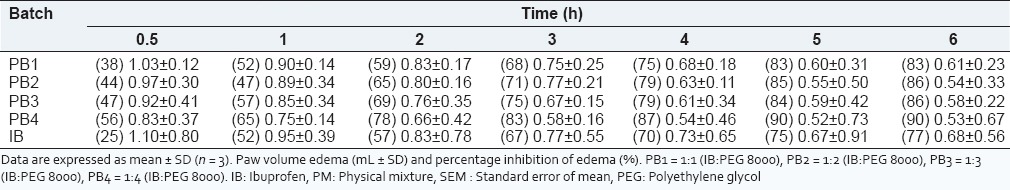
From Table 2, the SDs (PB1 – PB4) showed a significant increase in anti-inflammatory effect, in the egg albumin induced paw edema as compared to the pure sample of IB after egg albumin injection. IB inhibits inflammation by antagonizing the COX enzyme required for prostaglandins synthesis.[4] The formulations exhibited edema inhibition values significantly different from the control (P < 0.05), indicating a high degree of bioavailability of the administered SDs formulations, consistent with previous studies on IB SDs.[5,28,29,30,31,32] At 0.5 h, the SDs inhibited the size of the edema by 38-56% as compared to the pure sample of IB which had 25% edema inhibition after 0.5 h. It was also observed that the rate of inhibition increases as the ratio of IB to PEG 8000 increased. This shows that the polymer is effective in enhancing the solubility and release of the drug. The SD formulated inhibited more than 50% inhibition of edema at 2 h. All the batches of IB SDs showed good anti-inflammatory properties with achieving up to 90 % edema after 6 h comparable to the pure sample of IB, which had 77% edema inhibition at 6 h. This study clearly shows that the addition of hydrophilic carrier such as PEG 8000 to IB improves the dissolution rate of the drug. The SDs performed better than the PM or pure IB powder. Polyethylene glycol 8000 (PEG 8000) is a good meltable polymer for IB because of its low melting point, surfactant properties, and oral safety.[31,32,33] As the serum concentrations and anti-inflammatory effects of IB are correlated, rapid IB absorption could be a prerequisite for the quick onset of its anti-inflammatory action. Dissolution thus becomes the rate limiting step for the IB absorption.[30] Quick release of IB in the GI tract following oral administration is desirable. In this study, rapid release of IB was achieved in a relatively easy and simple manner. Quicker dissolution of SDs in rat intestine resulted into their rapid absorption and improved bioavailability as compared to that of pure drug. The ulcerogenic effect of IB SD should be carried out to check its effect in the intestine.
Table 2.
Anti-inflammatory properties of IB solid dispersion
In order to determine the change in the physicochemical parameter and in vitro dissolution profile on storage, stability study was carried out.[6] The physicochemical parameter of the optimized formulation was not significantly changed on storage. The in vitro dissolution profiles of trandolapril before and after storage indicate that the formulation was stable after storage for more than 3 months.
CONCLUSION
This study clearly shows that the dispersion of IB in a hydrophilic carrier such as PEG 8000 improved the dissolution rate and anti-inflammatory activity of the drug. PEG 8000 is a good meltable polymer for IB because of its low melting point, surfactant properties, and oral safety. The solubilization effect of this hydrophilic carrier resulted in the reduction of particle aggregation of the drug, elimination of crystallinity, increased wettability and dispersibility, and alteration of the surface properties of the drug particles. These were probably responsible for the enhanced dissolution rate and improved anti-inflammatory effect of IB in the SDs.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
This work is a product of research project for the award of M. Pharm. by the Department of Pharmaceutics, University of Nigeria, Nsukka, Nigeria. The authors wish to thank Pastor Ishaya Gando and Usuf Usman both of the National Research Institute for Chemical Technology, Zaria, Kaduna State, Nigeria, for assistance in FT-IR analyses, as well as Ph. Eur. Carl Roth GmbH + Co. KG Karlsruhhe Germany for the provision of PEG 8000 used in the study.
REFERENCES
- 1.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 2.Serajuddin AT. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–66. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 3.Dhirendra K, Lewis S, Udupa N, Atin K. Solid dispersions: A review. Pak J Pharm Sci. 2009;22:234–46. [PubMed] [Google Scholar]
- 4.Chowdari KP, Srinivas L. Physical stability and dissolution rate of ibuprofen suspension formulated employing solid dispersion. Indian J Pharm Sci. 2000;62:253–6. [Google Scholar]
- 5.Park YJ, Kwon R, Quan QZ, Oh DH, Kim JO, Hwang MR, et al. Development of novel ibuprofen-loaded solid dispersion with improved bioavailability using aqueous solution. Arch Pharm Res. 2009;32:767–72. doi: 10.1007/s12272-009-1516-3. [DOI] [PubMed] [Google Scholar]
- 6.Goracinova K, Klisarova L, Simov A, Fredro-Kumbaradzi E, Petrusevska-Tozi L. Preparation, physical characterisation, mechanisms of drug/polymer interactions and stability studies of controlled release of solid dispersion granules containing weak base as active substance. Drug Dev Ind Pharm. 1996;22:255–62. [Google Scholar]
- 7.Craig DQ. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int J Pharm. 2002;231:131–44. doi: 10.1016/s0378-5173(01)00891-2. [DOI] [PubMed] [Google Scholar]
- 8.Owusu-Ababio G, Ebube NK, Reams R, Habib M. Comparative dissolution studies for mefenamic acid-polyethylene glycol solid dispersion systems and tablets. Pharm Dev Technol. 1998;3:405–12. doi: 10.3109/10837459809009868. [DOI] [PubMed] [Google Scholar]
- 9.Ghebremeskel AN, Vemavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble API: Selection of polymer-surfactant combinations using solubility parameters and testing the processability. Int J Pharm. 2007;328:119–29. doi: 10.1016/j.ijpharm.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Dannenfelser RM, He H, Joshi Y, Bateman S, Serajuddin AT. Development of clinical dosage forms for a poorly water soluble drug I: Application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93:1165–75. doi: 10.1002/jps.20044. [DOI] [PubMed] [Google Scholar]
- 11.Van den Mooter G, Augustijns P, Blaton N, Kinget R. Physico-chemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm. 1998;164:67–80. [Google Scholar]
- 12.Lu JW, Wang MZ, Ding P, Pan ZS. Preparation and dissolution of nimodipine PEG solid dispersions. Chin Pharm J. 1995;30:23–5. [Google Scholar]
- 13.Patil MP, Gaikwad NJ. Preparation and characterization of gliclazide-polyethylene glycol 4000 solid dispersions. Acta Pharm. 2009;59:57–65. doi: 10.2478/v10007-009-0001-3. [DOI] [PubMed] [Google Scholar]
- 14.Weuts I, Kempen D, Verreck G, Decorte A, Heymans K, Peeters J, et al. Study of the physicochemical properties and stability of solid dispersions of loperamide and PEG6000 prepared by spray drying. Eur J Pharm Biopharm. 2005;59:119–26. doi: 10.1016/j.ejpb.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Dordunoo SK, Ford JL, Rubinstein MH. Preformulation studies on solid dispersions containing triamterne or tamezapam in polyethylene glycols or gelucire 44/14 for liquid filling of hard gelatin capsules. Drug Dev Ind Pharm. 1991;17:1685–713. [Google Scholar]
- 16.Li Z, Zhu H, De-Ping W, Jia-Bi Z. Enhancement of nitrendipine dissolution by PEG 6000 and polysorbate 80. Zhongguo Yaoke Daxue Xuebao. 2000;31:21–4. [Google Scholar]
- 17.El-Badry M, Fetih G, Fathy M. Improvement of solubility and dissolution rate of indomethacin by solid dispersions in Gelucire 50/13 and PEG4000. Saudi Pharm J. 2009;17:217–25. doi: 10.1016/j.jsps.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akiladevi D, Basak S. Dissolution enhancement of paracetamol by solid dispersion technique. J Pharm Res. 2010;3:2846–9. [Google Scholar]
- 19.Leonard D, Barrera MG, Lamas MC, Salomón1 CJ. Development of prednisone: Polyethylene glycol 6000 fast-release tablets from solid dispersions: Solid-state characterization, dissolution behavior, and formulation parameters. AAPS PharmSciTech. 2007;8:E1–8. doi: 10.1208/pt0804108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arias MJ, Ginés JM, Moyano JR, Rabasco AM. Dissolution properties and in vivo behaviour of triamterene in solid dispersions with polyethylene glycols. Pharm Acta Helv. 1996;71:229–35. doi: 10.1016/s0031-6865(96)00017-9. [DOI] [PubMed] [Google Scholar]
- 21.Tripathi KD. Essentials of Medical Pharmacology. 5th ed. New Delhi: Jaypee Brothers; 2003. Non-steroidal anti-inflammatory drugs and anti-pyretic analgesics; p. 176. [Google Scholar]
- 22.Wagner W, Khanna P, Furst DE. Non-steroidal anti-inflammatory drugs, disease modifying anti-rheumatic drugs, non-opioid analgesics and drugs used in gout. In: Katzung BG, editor. Basic and Clinical Pharmacology. 9th ed. Boston: McGraw Hill; 2004. p. 585. [Google Scholar]
- 23.Vale JA, Meredith TJ. Acute poisoning due to non-steroidal anti-inflammatory drugs. Clinical features and management. Med Toxicol. 1986;1:12–31. doi: 10.1007/BF03259825. [DOI] [PubMed] [Google Scholar]
- 24.Murtha JL, Ando HY. Synthesis of the cholesteryl ester prodrugs cholesteryl ibuprofen and cholesteryl flufenamate and their formulation into phospholipid microemulsions. J Pharm Sci. 1994;83:1222–8. doi: 10.1002/jps.2600830907. [DOI] [PubMed] [Google Scholar]
- 25.Ghorab MK, Adeyeye MC. Enhancement of ibuprofen dissolution via wet granulation with beta-cyclodextrin. Pharm Dev Technol. 2001;6:305–14. doi: 10.1081/pdt-100002611. [DOI] [PubMed] [Google Scholar]
- 26.Adeyeye CM, Price JC. Development and evaluation of sustained-release ibuprofen-wax microspheres. II. In vitro dissolution studies. Pharm Res. 1994;11:575–9. doi: 10.1023/a:1018931002991. [DOI] [PubMed] [Google Scholar]
- 27.Ali A, Sharma SN. Preparation and evaluation of solid dispersions of ibuprofen. Indian J Pharm Sci. 1991;53:233–6. [Google Scholar]
- 28.Newa M, Bhandari KH, Li DX, Kwon TH, Kim JA, Yoo BK, et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm. 2007;343:228–37. doi: 10.1016/j.ijpharm.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Li SM, Sunada H. Preparation and evaluation of Ibuprofen solid dispersion systems with kollidon particles using a pulse combustion dryer system. Chem Pharm Bull (Tokyo) 2007;55:1545–50. doi: 10.1248/cpb.55.1545. [DOI] [PubMed] [Google Scholar]
- 30.Uddin R, Saffoon N, Huda NH, Jhanker YM. Effect of water soluble polymers on dissolution enhancement of ibuprofen solid dispersion prepared by fusion method. Stamford J Pharm Sci. 2010;3:63–7. [Google Scholar]
- 31.Newa M, Bhandari KH, Lee DX, Sung JH, Kim JA, Yoo BK, et al. Enhanced dissolution of ibuprofen using solid dispersion with polyethylene glycol 20000. Drug Dev Ind Pharm. 2008;34:1013–21. doi: 10.1080/03639040701744095. [DOI] [PubMed] [Google Scholar]
- 32.Newa M, Bhandari KH, Kim JA, Yoo BK, Choi HG, Yong CS, et al. Preparation and evaluation of fast dissolving ibuprofen-polyethylene glycol 6000 solid dispersions. Drug Deliv. 2008;15:355–64. doi: 10.1080/10717540801952431. [DOI] [PubMed] [Google Scholar]
- 33.Franco M, Trapani G, Latrofa A, Tullio C, Provenzano MR, Serra M, et al. Dissolution properties and anticonvulsant activity of phenytoin-polyethylene glycol 6000 and -polyvinylpyrrolidone K-30 solid dispersions. Int J Pharm. 2001;225:63–73. doi: 10.1016/s0378-5173(01)00751-7. [DOI] [PubMed] [Google Scholar]
- 34.Law D, Krill SL, Schmitt EA, Fort JJ, Qiu Y, Wang W, et al. Physicochemical considerations in the preparation of amorphous ritonavir-poly(ethylene glycol) 8000 solid dispersions. J Pharm Sci. 2001;90:1015–25. doi: 10.1002/jps.1054. [DOI] [PubMed] [Google Scholar]
- 35.Veiga MD, Diaz P, Gines JM, Rabasco AM. Preparation and evaluation of mequitazine/PEG-6000 binary systems. Pharmazie. 1994;49:906–9. [Google Scholar]
- 36.Ofokansi KC, Kenechukwu FC, Isah AB, Ogbonna JD. Solid dispersion as an approach for dissolution enhancement and delivery of trandolapril, a poorly water-soluble ACE inhibitor. Indian J Novel Drug Deliv. 2012;4:284–94. [Google Scholar]
- 37.Palmieri GE, Antonini I, Martelli S. Characterization and dissolution studies of PEG-4000/ fenofibrate solid disperseions. STP Pharm Sci. 1996;6:178–84. [Google Scholar]
- 38.Jaymin C, Shah JR, Chen DC. Preformulation study of etoposide: II. Increased solubility and dissolution rate by solid-solid dispersions. Int J Pharm. 1995;113:103–11. [Google Scholar]
- 39.Ahmed SM, Abdel-Rahman AA, Saleh SI, Ahmed MO. Comparative dissolution characteristics of bropirimine-beta-cyclodextrin inclusion complex and its solid dispersions with PEG-6000. Int J Pharm. 1993;96:5–11. [Google Scholar]
- 40.Guyot M, Fawaz F, Bildet J, Bomono F, Laguni M. Physiochemical characterization and dissolution of norfloxacin cyclodextrin complex and PEG solid dispersion. Int J Pharm Sci. 1995;123:53–63. [Google Scholar]