

Abstract
Bone sarcomas are rare tumors, approximating 0.2% of all cancers, with osteosarcoma (OGS), chondrosarcoma, and Ewing sarcoma being the most common cancers in this subset. The formation of disease management groups/clinics focused on sarcomas has resulted in better understanding and management of these uncommon tumors. Multiple large-scale retrospective data from Tata Memorial Hospital (TMH) and All India Institute of Medical Sciences have reported outcomes comparable to Western data in the field of OGS and Ewing sarcoma, with interesting prognostic factors identified for further evaluation. Soft tissue sarcomas are a rare heterogeneous group of tumors, more than 50 different tumor entities. The common subtypes identified in India include Ewing sarcoma and synovial sarcoma. Valuable work regarding brachytherapy has been done by radiation oncologists from the TMH, especially in pediatric patients.
Keywords: Bone sarcomas, Indian data, soft tissue sarcomas
Bone Sarcomas
Background
Primary bone sarcomas are rare tumors, comprising approximately 0.2% of all cancers. Their true incidence is difficult to estimate because of their rarity.[1] Osteosarcoma (OGS), chondrosarcoma, and Ewing sarcoma/primitive neuroectodermal tumor (PNET) are the common bone sarcomas, as per Western data, with rare tumors such as fibrosarcomas, chordomas, and undifferentiated pleomorphic sarcoma (UPS) constituting as the remaining subtypes. Bone sarcomas constitute as the third most common cause of mortality in adolescents. Remarkable achievements with multimodality management of these tumors have led to an increase in their 5-year survival rates, from approximately 50% in the year 1970, to the range of 75–80%, presently, in the adolescent age-group.[2]
On-going studies in molecular diagnostics have also helped in differentiating this heterogeneous group of tumors. Attempts are being made to identify prognostic and predictive markers for these tumors. Over the last decade, certain centers across the country have formed groups focused toward sarcoma treatment, for example, bone and soft tissue disease management group at the Tata Memorial Hospital (TMH), Mumbai, Maharashtra, India and bone and soft tissue sarcoma (STS) joint clinic at the All India Institute of Medical Sciences (AIIMS), New Delhi, India which has resulted in better understanding and management of these uncommon, but diagnostically, as well as therapeutically challenging tumors, eventually leading to treatment benefit for such patients. Through this summary, we have attempted to capture data from various centers across India, on various aspects of bone sarcomas, with a major focus on OGS and Ewing sarcoma.
Demographics and distribution
Various hospital-based registries have attempted to gauge the incidence and distribution of sarcomas in the Indian population; however, there is no recent, population-based evaluation of the incidence of bone sarcomas in India. In as early as 1998, Yeole and Jussawalla published the incidence of bone malignancy in Mumbai, based on the data from the Bombay Cancer Registry. As per that registry, bone malignancies were found to represent 0.9% of all cancers with Ewing sarcoma as the most common bone malignancy.[3] As per hospital-based cancer registry, 2001, TMH, Mumbai, Maharashtra, India OGS was found to be the most common bone sarcoma, followed by Ewing sarcoma/PNET and chondrosarcoma.[4] Based on retrospectively collected data over 36 years in Karnataka, Rao et al. found that out of the 523 bone tumors, 39% were malignant, with OGS and Ewing sarcoma constituting 45.7% and 19.4% of malignant bone tumors, respectively.[5] More recent data come from the registry of JSS Medical College and Hospital, Mysore, Karnataka, India where a review of 117 cases over a period of 8 years was conducted, showed that the majority of malignant bone tumors were OGS (35.14%) with a median age of presentation being 26.87 years.[6]
Osteosarcoma
The outcomes in OGS experienced a major turnaround with the addition of multiagent chemotherapy to surgery, based on the pioneering work by Rosen et al. in the late 70’s and early 80’s.[7] After the initial progress, however, over the last two decades, outcomes have stagnated with 5-year overall survival (OS) of 65–70%, with no major improvement in the subset of cases of metastatic OGS that continue to have poor outcomes. Research in this field is on-going within major cancer centers in India.
Diagnostics, prognosis, and management
Routine diagnosis of an OGS is based on results from a well-exposed radiograph and a biopsy that unravels the histopathological subtype of OGS. Invariably neoadjuvant chemotherapy (NACT) is offered in cases of high-grade OGS, followed by surgical resection. In a study from TMH, including 100 cases of high-grade extremity-based OGS, a significant association between grade of tumor necrosis and clinical outcome was noted.[8]
Further, there has been considerable interest in the characterization of OGS cell lines and their response to various therapeutic modalities. Kumar et al. carried out in vitro cell studies in OGS cell lines MG-63 to evaluate the combined effect of anticancer drug camptothecin (CPT) and (177) Lu-EDTMP, which is a known therapeutic radionucleotide. The expression of proteins such as BCL-2, PARP, and mitogen-activated protein kinase that were related to apoptotic signaling pathways was assessed by Western blotting after incubation of the cell lines (MG-63) with both (177) Lu-EDTMP and CPT. Their results indicated that cellular toxicity and apoptosis were relatively higher in MG-63 cells that were treated with CPT before treating with (177) Lu-EDTMP, in comparison with the corresponding individual controls.[9]
In terms of prognosis, vascular endothelial growth factor (VEGF) expression in cases of OGS, at the time of biopsy has been shown to have a negative influence on survival. Whether it continues to be expressed and influences outcome in tumors after NACT, has not been determined. In a study, Bajpai et al. evaluated VEGF expression in 31 patients and compared VEGF expression at baseline and with grade of tumor. The authors compared pre- and post-NACT VEGF expression with histological necrosis. This study confirmed the preexisting data that positive VEGF expression was concordant with high histological grade (90% concordance). The authors also revealed that postneoadjuvant VEGF expression, as well as VEGF change following NACT significantly, correlated with histological necrosis.[10] The same study group also analyzed surrogates of tumor angiogenesis through radiological imaging. In another study, they evaluated tumor angiogenesis by qualitative analysis of dynamic contrast enhanced-magnetic resonance imaging (DCE-MRI) through the concept of time-intensity curves (TICs). TIC representing low microvascular permeability is the persistent type, while that of high permeability are plateau and washout types. The results in this 31 patients, cohort showed that TIC could correctly identify all 28 VEGF positive samples at baseline and 24/25 (96%) of VEGF positive samples and 5/6 (83%) of VEGF-negative samples after NACT correctly. While VEGF expression was found to correlate with CDE-MRI, the same was not found to correlate with diffusion-weighted-MRI and positron emission tomogram-computed tomography (PET-CT).[11]
In a recent study, using MRI and clinical findings as alternate parameters to evaluate response to NACT in patients (12 of 14 patients with OGS), Amit et al. observed that both dynamic MRI slope, as well clinical indicators such as change in pain, tumor girth, maximum tumor diameter, and surface temperature correlated with histological response (P < 0.01). In another study, the same group also reported on the comparison of DCE-MRI (via slope) and volumetric assessment via static MRI as predictors of histological necrosis postchemotherapy. In that study, more than 60% reduction in slope of curve on DCE-MRI proved to be an indicator of good histological response (positive predictive value = 80%), while change in tumor volume, as measured by static MRI failed to show a significant correlation (P = 0.071).[12,13]
Like DCE-MRI, PET-CT have also been evaluated by as a noninvasive predictor of response to chemotherapy. In a study of 31 patients, Bajpai et al. identified two independent variables in stepwise multivariable analysis as predictive of histological necrosis: in cases V1 (prechemotherapy volume) was ≤300 mL and SUV2:SUV1 ratio was ≤0.48, observed good histologic response proportion was 83%, whereas in cases V1 was more than300 mL and SUV2:SUV1 ratio exceeded 0.48, the same was 0%.[14] While metabolic burden seems to be a sensitive substitute for response evaluation, further validation is required.
A number of studies have been published over the last decade with regards to outcomes in OGS from India. The earliest work in this field came as early as 1993 when Pathak et al. published their data on 29 patients with high-grade OGS, who received sequential adriamycin and cisplatin as adjuvant therapy after definitive surgery. The relapse-free survival was 72%, and OS for the entire group was 69%, which was superior to outcomes in historical controls treated with surgery alone.[15]
A major controversy that persists in the management of OGS is the role of methotrexate in the treatment of OGS. Most of cancer centers in India offer nonmethotrexate-based chemotherapy regimens in cases of high-grade OGS. The study group from AIIMS recently published their data of 237 patients with an attempt to establish prognostic factors, as well as report outcomes in a cohort of localized OGS patients treated by a uniform protocol. They showed a 5-year event-free survival (EFS) and OS of 36.6% and 50.3% respectively, with baseline factors of symptom duration more than 4 months. They noted that good performance status predicted for better PFS, while good PS and normal serum alkaline phosphatase predicted for better OS, on multivariate analysis.[16] The group from AIIMS also evaluated whether a high tumor volume at presentation was a predictor for local recurrence (LR). They used MRI images to calculate baseline tumor volume. However, in this retrospective analysis, the mean tumor volume in the group without LR was 406.74 ± 771.67 cc, as compared to 195.77 ± 226.8 cc in the group with LR, which was not found to be statistically significant.[17]
Recently, at TMH, Mumbai, the study group conducted an analysis of 239 cases of nonmetastatic OGS (unpublished, accepted for and presented at SIOP, 2015) who received an in-house chemotherapy protocol, OGS 2012.[18] Essentially, this study included dose-dense chemotherapy with doxorubicin, ifosfamide, and cisplatin in patients enrolled between November 2011 and December 2014 (4 years). The investigators found high rates of nutritional depletion at baseline in patients (Vitamin B12-48%, iron deficiency - 59%, anemia - 17%), besides low albumin levels, as predictive of poor OS. They also observed albumin and age as predictive variables for development of Grades 3 and 4 thrombocytopenia in such cases. Their estimated disease-free survival (DFS) at 2 and 3.5 years was 77% and 70%.[18]
While standard chemotherapeutic regimens are fairly well established with the drugs of importance, such as ifosfamide, doxorubicin, cisplatin, methotrexate and to some extent, etoposide, the issue of compliance to such dose dense regimens, especially in Indian patients is a major issue that requires evaluation. In a premier study at TMH, Bajpai et al. evaluated 124 patients of OGS, who received NACT and found that there was a significant association between histological response and compliance (P = 0.031). However, after a median follow-up of 7.9 months, there was no difference in survival, which was likely because of the short duration of follow-up in their study.[19]
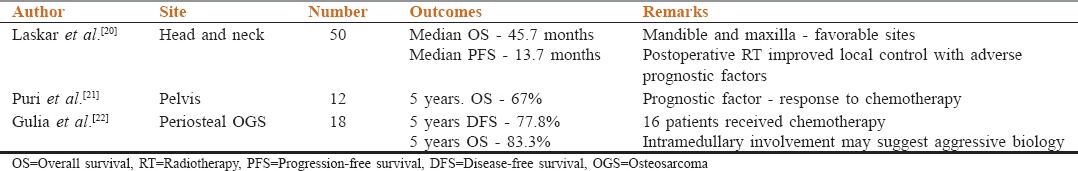
Retrospective studies have also been published on rare sites of OGS, including their treatment modalities and outcomes. These studies have been enlisted in Table 1.[20,21,22]
Table 1.
Site specific data in osteogenic sarcoma from India
Another significant study regarding follow-up criteria, conducted by the TMH group was the trial for optimal survival of sarcomas study that evaluated differences in survival in more versus less intensive follow-up, as well as the requirement of a CT scan versus simple chest radiograph, for radiological follow-up for detecting pulmonary metastases. In a set of 500 nonmetastatic patients, the authors observed 3-year OS of 64% and 69% in 6- and 3-monthly groups, respectively and 3-year OS of 67% and 66% in chest radiography and CT scan groups, respectively, and, the authors concluded that while less frequent visits detected metastases and LR adequately, it could be conclusively revealed that 6 monthly interval of follow-up was noninferior as against 3 monthly follow-up visits.[23]
Ewing sarcoma family of tumors
Diagnostics
Ewing sarcoma family of tumor (EWSFT) includes Ewing sarcoma of the bone, extraosseous Ewing sarcoma, also referred to as extraskeletal Ewing sarcoma (tumor growing outside of the bone), PNET, peripheral neuroepithelioma, Askin’s tumor (Ewing sarcoma of the chest wall), and atypical Ewing sarcoma.
Ewing sarcoma/PNET is an undifferentiated small blue round cell tumor of bone and soft tissues, predominantly seen in children and young adults characterized by recurrent balanced translocations, involving in almost all cases, EWSR1 gene on chromosome 22 that leads to formation of fusion oncogenes such as EWS-FLI and EWS-ERG that play key role in its pathogenesis. This tumor is invariably diagnosed by histopathology with demonstration of positive immunohistochemical staining of antibody markers, such as CD99/MIC2 (diffuse, cytoplasmic membranous pattern of staining) and FLI1 in most cases, along with negative expression of Tdt in all cases. This tumor has a wide clinicopathological spectrum and in cases with equivocal immunohistochemical results and in tumors occurring at unusual locations or in older age group patients, should be ideally confirmed with molecular testing. Rekhi et al. from TMH, Mumbai, Maharashtra, India have published the first Indian study on molecular diagnostics of Ewing sarcoma, including validation of EWSR1 gene rearrangement by fluorescent in situ hybridization (FISH) technique in Ewing sarcomas, comparing it with results from polymerase chain reaction (PCR) that is another molecular technique for identifying specific fusion genes. They observed that the FISH technique was more sensitive, as compared to PCR that detects specific chimeric transcripts. At the same time, they noted EWSR1 rearrangement was also noted in other tumors. Therefore, they suggested that molecular results should be interpreted with clinic pathological, including immunohistochemical profile in all cases of Ewing sarcoma/PNET.[24] Furthermore, Indian researchers have contributed to some more interesting insights in the field of molecular biology and diagnostics of EWSFT. In a prospective study, Tilak et al. evaluated peripheral blood circulating Tregs in PNET patients (in contrast to bone marrow Tregs) at baseline and during chemotherapy and observed that peripheral blood Tregs are higher in patients than healthy controls. They observed a significant reduction in Tregs with chemotherapy treated cases and their rise at progression. However, there was no significant difference observed in this study in the EFS or OS between the high and low Treg cell groups.[25] Jully et al. at the Cancer Institute (WIA), Adyar, Chennai further contributed to the characterization of the junction region of this fusion protein by analyzing an in silico model of EWS-FLI fusion protein for ligand binding sites. Their analysis indicated that junction region (AA 251-343) encompasses potential ligand biding sites in the EWS-FLI1 protein and when expressed in bacteria were present as soluble form. They also observed ectopically expressing this region in Ewing sarcoma cells inhibited tumorigenicity, and EWS-FLI1 target genes indicating a dominant negative biological effect.[26] Cathepsin L is a cysteine protease is involved in bone remodeling and expressed in activated macrophages. It is highly expressed in metastatic tumor tissue, especially with bone metastases. Biswas et al. evaluated the immunohistochemical expression of cathepsin L in tumor cells and tumor-associated macrophages (TAMs) in chemo-naive EWSFT in a set of 62 archived specimens. They found that low (13%) immunohistochemical expression of cathepsin L in their patient cohort and noted that this marker did not influence the outcome. In that study, while cathepsin L expression was found to be associated with poor response to NACT, Grade 3 TAMs with cathepsin L expression was associated with improved EFS.[27]
Radiological imaging plays an important role in the diagnosis and assessment of response in EWSFT. Researchers at TMH retrospectively attempted to correlate fluorodeoxyglucose (FDG) uptake in PET scans with tumor behavior as well as evaluated whether PET-CT scan could be used as a noninvasive method to predict response to NACT. They concluded that increased FDG uptake, as measured by mean SUVmax, was indicative of metastatic potential of the tumor, in view of the raised SUV in metastatic versus nonmetastatic patients. They also concluded that there was significant correlation between change in metabolic activity of the primary tumor and histopathological response after NACT suggesting that FDG-PET might be an ideal noninvasive method to assess tumor behavior and response to therapy in EWS.[28]
Prognosis and management
EWSFT are optimally treated by multimodality management group that requires an entire dedicated team consisting of a surgical-, medical-, radiation-oncologist, and pathologist, along with rehabilitative services, orthopedicians, and counselors. While trial data sets the benchmark for treatment modalities, over time, there is a growing sense that real world hospital practice in a logistically constrained setting might put forward different management strategies. There have been several large studies from India that have been published over the last few years, and this has contributed to our understanding of EWSFT.[29,30,31]
In a recent large retrospective analysis conducted at AIIMS, including 224 patients with localized EWSFT, the authors evaluated potential prognostic factors and suggested that patients with combination of ESFT of spine/abdominopelvic region and baseline white blood cell (WBC) count >11 × 109/L had a significantly inferior OS (P < 0.001) and patients with combination of ESFT with symptom duration >4 months, tumor diameter >8 cm, and baseline WBC count >11 × 109/L had significantly inferior EFS (P = 0.002).[29] These results are at par with worldwide data which have shown inferior clinical outcomes in such cases with high volume disease (>200 mL) and pelvic primaries; though baseline WBC has not been previously identified as a prognostic indicator. Another study from the same group, in cases of metastatic tumor settings, disclosed hypoalbuminemia as a unique prognostic factor associated with EFS (P = 0.003), though age ≤15 years and radical radiotherapy as local treatment also had a trend toward inferior local control (LC) rates, although the values were statistically not significant.[30]
The study group from AIIMS also published their data regarding prognostic factors and outcomes in extremity EWSFT. In their cohort of 158 patients, while metastases and radiotherapy (vs. surgery as primary local treatment) predicted for poorer outcomes in the entire cohort, radiotherapy was found to be noninferior to surgery in the subset of patients with localized tumors with regard to tumor diameter (P = 0.8) or post-NACT response (P = 0.1). The 5-year EFS, OS, and LC rates were 24.1%, 43.5%, and 55%, respectively, for the entire cohort and 36.4%, 57.6%, and 58.2% respectively, for patients without loco-regional disease.[31]
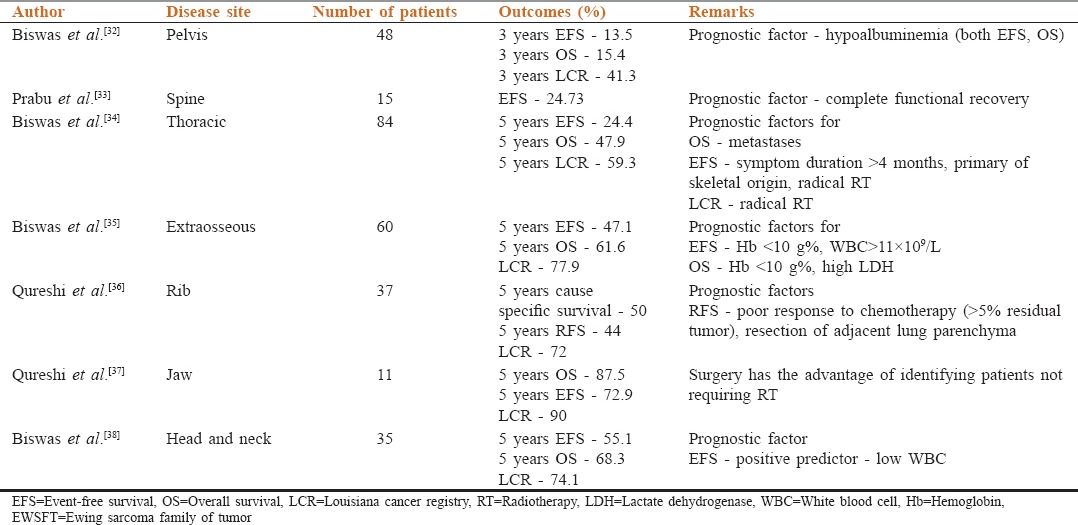
Multiple groups have published their data regarding outcomes in location specific EWSFT, which offers an understanding of the management modalities and responses in these rare tumors. These are tabulated in Table 2.[32,33,34,35,36,37,38] From this data, it can be seen that pelvic and spinal EWSFT seem to have poorer outcomes, compared to head and neck, extraosseous and jaw EWSFTs.
Table 2.
Site specific data in EWSFT from India
One of the major constraints in resource-limited settings is the cost and availability of prosthesis which is essential in allowing for good functional outcomes. In this direction, there has been significant work with a technique, namely, extracorporeal radiation therapy (ECRT). Puri et al. published their results on 32 patients (16 OGS and 16 EWSFT) who underwent this procedure of en bloc resection with preservation of the adjoining joints and reconstruction with reimplantation of sterilized tumor bone after extracorporeal radiation (50 Gy). The mean resected length of bone was 19 cm with a mean time to union for all osteotomy sites being 7.3 months. There were three LRs, all in soft-tissue away from irradiated graft. At the time of final follow-up, 19 patients were free-of-disease, a single patient was alive with disease, and 11 had died-of-disease. There were three LRs, all away from irradiated graft.[39]
Soft Tissue Sarcomas (STS)
Unlike bone sarcomas, STSs constitute a more heterogeneous group of tumors, with more than 50 different tumor entities that make these tumors fairly complex and diagnostically challenging. These tumors account for <1% of all adult tumors and 15% of pediatric tumors.[40] The most common types identified have been UPS, gastrointestinal stromal tumor (GIST), liposarcoma, leiomyosarcoma, and synovial sarcoma.[41] During the past 10 years, immunohistochemistry (IHC) has been a very helpful ancillary technique for exact categorization of the various STSs, namely synovial sarcoma, malignant peripheral nerve sheath tumor (MPNST), epithelioid sarcoma, dermatofibrosarcoma protuberans, to name, but a few. Nearly one-third sarcomas are associated with a specific translocation that has helped in their further accurate diagnosis which directly influences their management. Considering certain sarcomas such as synovial sarcoma and Ewing sarcoma/PNET are relatively more chemosensitive over their mimics such as MPNSTs, their exact recognition is vital. This section will concentrate on published work in non-GIST-STSs from various centers across India.
Diagnsotics
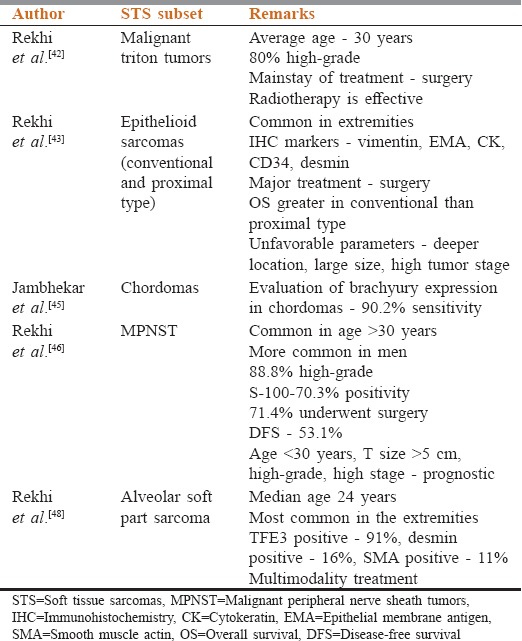
Various clinicopathological studies have been published regarding different subtypes of STSs[42,43,44,45,46,47,48,49] [Table 3]. At TMH, Mumbai, Maharashtra, India presently synovial sarcoma constitutes the most common adult STS, and rhabdomyosarcoma is the most common pediatric STS. As per hospital-based cancer registry, TMH, Mumbai, Maharashtra, India in the year 2001, the most common STS was spindle cell sarcoma.[50] This clearly shows the value of IHC, over these years that has been helpful in correct identification of certain “spindle cell sarcomas,” including certain fibrosarcomas into more definite subtypes, including synovial sarcomas.
Table 3.
Subsite specific pathology data in soft tissue sarcoma from India
Among all sarcomas, irrespective of adult or pediatric patients, the most frequently observed specific subtypes are Ewing sarcoma/PNET, followed by synovial sarcoma; both the entities require IHC for confirmation in all cases. Over the years, with applications of immunohistochemical stains, usage of the histopathological term “spindle cell sarcoma” has reduced, and is being replaced by rather specific histopathological subtypes. Earlier, as per 2001 cancer registry, among pediatric patients, Ewing sarcoma, followed by rhabdomyosarcoma were the most common histopathological subtypes.
Diagnosis of STS definitely requires ancillary techniques such as IHC in significant number of cases, and molecular diagnostics in certain cases, including sarcomas such as synovial sarcoma, Ewing sarcoma, and alveolar rhabdomyosarcoma that have specific underlying molecular signatures. Rekhi et al., at TMH, Mumbai, Maharashtra, India evaluated the optimal diagnostic IHC panel for synovial sarcoma, including transducing like enhancer of split-1 (TLE1) a useful IHC marker for diagnosing synovial sarcoma. In a cohort of 42 tumors, they found that its sensitivity for the diagnosis of synovial sarcomas was 95.2%, while specificity was 63.7%, with tumors, such as schwannomas, neurofibromas, MPNST, and EWSFT also expressing TLE1, though to varying degrees. They concluded that TLE1, in view of its high sensitivity might be a useful marker within the optimal IHC panel comprising EMA, BCL-2, MIC2, CD34, and CK7 for substantiating a diagnosis of synovial sarcoma.[51] In another Indo-German study, Rekhi and Vogel analyzed a characteristic expression of INI1/SMARCB1in synovial sarcomas. They concluded that a unique “weak to absent” IHC expression of INI1 is highly sensitive (88.2%) and specific (97.3%) for synovial sarcomas, irrespective of its subtypes and types of biopsies and can be used an adjunct in diagnosis, especially in settings lacking molecular/cytogenetic analysis.[52]
Prognosis and management
Metastatic STSs are a heterogeneous entity with limited data on treatment modalities and outcomes, especially in India. Iqbal et al. analyzed 119 patients with metastatic STS treated between 2003 and 2012 atavisms. They observed the most common histological type was synovial sarcoma and leiomyosarcoma. As a group with a median follow-up of 10 months, these tumors had poor outcomes with an EFS and OS of 6 and 10 months, respectively. They identified Hb <10 g%, tumor size >10 cm and single modality treatment as poor prognostic factors, in terms of OS.[53]
Another group of STSs with manifestations are retroperitoneal sarcomas (RPS). Panda et al. retrospectively analyzed 23 patients for treatment modalities and prognostic factors. In their study, surgery was performed in 20 (87%) patients for primary RPS and 3 (13%) for recurrent RPS at initial presentation. They observed that completeness of resection, tumor grade, and sex were prognostic factors for RPS.[54] In a study on dedifferentiated liposarcomas (DDLPS), including RPS DDLPS, Rekhi et al. observed surgery as the main treatment modality for these tumors.[55]
Radiotherapy is an essential component of adjuvant therapy in certain high-grade, extremity STS treated with surgery, followed by chemotherapy. Brachytherapy (BRT) alone or with external beam radiotherapy (EBRT) has been evaluated in these adjuvant settings by Indian investigators as well.
Jamema et al., from TMH, Mumbai, Maharashtra, India conducted a dosimetric study quantitatively comparing electron arc (EA) versus photon intensity modulated radiotherapy (IMRT) for Askin’s tumors and concluded that IMRT resulted in superior planning target volume coverage and sparing of organs at risk compared to EA plans. They added the caveat that low dose regions associated with IMRT need to be kept in mind.[56] The same group conducted a three-dimensional image-based dosimetric study to quantitatively compare geometric versus dose-point optimization in combination with graphical optimization for interstitial BRT of STS.[57] They found that optimization with dose points defined away from the implant plane and on target results in superior target coverage with optimal values of other indices.
Laskar et al. performed a retrospective analysis of fifty pediatric patients with STSs, who received BRT as part of loco-regional treatment. Most patients had synovial sarcomas (32%). After a median follow-up of 51 months, the LC, DFS, and OS were 82%, 68%, and 71%, respectively. LC was superior in patients with tumor size <5 cm, symptom duration <2 months, and lower histopathological tumor grade. There were no differences in LC rates between HDR and LDR BRT.[58] Earlier, in 1998, the same group from TMH also published their experience with BRT in the adult STS patients. In a cohort of 151 patients, spindle cell sarcoma was found to be the most common STS (30%), with the extremities involved in 75% patients. In patients receiving BRT only, 25 out of 33 (75%) were locally controlled after a median follow-up of 30 months. After successful salvage of local failures, the overall LC improved to 82%. Treatment-related complications were <1%. They concluded that the sequential combination of limited surgery and tumor bed BRT with or without EBRT provided an effective alternative to more morbid procedures.[59]
In a recent study, Sharma et al. evaluated the role of perioperative high-dose-rate interstitial brachytherapy (PHDRIBT) in combination with EBRT in patients with localized STS. In their study, PHDRIBT was started on the 3rd postoperative day to an HDR dose of 16 Gy over 4 fractions in 2 days, and then 4 weeks later, followed by EBRT. They observed a 5-year OS and DFS of 67% and 63%, respectively, with no LRs. Major toxicity observed in this study was delayed wound healing in 5.7% of patients with common late toxicity being chronic skin/subcutaneous fibrosis in 9.6% patients.[60]
Conclusions
As per hospital registry data, OGS is the most common bone sarcoma, followed by Ewing sarcoma and chondrosarcoma
Percentage of post-NACT tumor necrosis is significantly associated with survival in conventional high-grade OGSs
VEGF expression has been shown to correlate with histological necrosis, pre- and post-chemotherapy in OGS. DCE-MRI might serve as a surrogate for tumor angiogenesis in cases of OGS
Nonmethotrexate-based multiagent chemotherapy regimens seem to be economic and as efficacious as methotrexate-based chemotherapy in OGS with less complex pharmacokinetic monitoring, and acceptable toxicity as per studies from TMH and AIIMS
Compliance is an important component of treatment in OGS. Early data from TMH have shown that compliance correlates with histological necrosis
EWSR1 gene rearrangement detected by FISH testing is useful in diagnosis of Ewing sarcomas, especially in cases with tumors occurring at unusual sites, in uncommon age-groups, and in cases with equivocal immunohistochemical features. EWSR1 testing was found to be more sensitive than PCR testing, the latter tests for specific fusion transcripts. Molecular results should be correlated with clinicopathological and immunohistochemical results in all cases
Increased FDG uptake on FDG-PET-CT, as measured by mean SUVmax, was indicative of metastatic potential of the tumor in Ewing sarcoma. FDG-PET-CT might also correlate with response after chemotherapy
Multiple prognostic factors in Ewing sarcomas include WBC, albumin levels, symptom duration >4 months, tumor diameter >8 cm; these require further validation as prognostic markers
ECRT seems like a viable alternate option to artificial prosthesis
STSs are a heterogeneous group of tumors with IHC and molecular diagnostics being increasingly used to differentiate the different subtypes, especially the ones that are chemosensitive, such as Ewing sarcoma/PNET and syovial sarcoma. Several clinicopathological studies have been published on various STSs. Synovial sarcoma and Ewing sarcoma/PNET are the most common types of STSs as per Hospital registry
Optimal diagnostic panel for synovial sarcoma includes EMA, BCL-2, MIC2, and CD34, with the addition of TLE1, a new sensitive marker for diagnosis of synovial sarcoma, as per study from TMH. A unique “weak to absent” expression of INI1/SMARCB1, is fairly specific for diagnosis of synovial sarcoma and can be useful especially in settings lacking molecular diagnostics
BRT alone or with EBRT has been evaluated in adjuvant settings in the treatment of STSs by Indian investigators with heartening results
On the evaluation of outcomes when BRT has been used for adjuvant treatment in pediatric STSs, LC has been found to be superior in patients with tumor size <5 cm, symptom duration <2 months, and lower histopathological tumor grade.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, et al., editors. SEER Cancer Statistics Review. Bethesda, MD: National Cancer Institute; 1975-2013. [Last accessed on 2016 Jun 01]. Available from: http://seer.cancer.gov/csr/1975_2013/ [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Yeole BB, Jussawalla DJ. Descriptive epidemiology of bone cancer in greater Bombay. Indian J Cancer. 1998;35:101–6. [PubMed] [Google Scholar]
- 4.Dinshaw KA, Ganesh B. Hospital Based Cancer registry. Annual Report 2001. Mumbai: Tata Memorial Hospital; 2005. pp. 90–1. [Google Scholar]
- 5.Rao VS, Pai MR, Rao RC, Adhikary MM. Incidence of primary bone tumours and tumour like lesions in and around Dakshina Kannada district of Karnataka. J Indian Med Assoc. 1996;94:103–4, 121. [PubMed] [Google Scholar]
- 6.Jain K, Sunila, Ravishankar R, Mruthyunjaya, Rupakumar CS, Gadiyar HB, et al. Bone tumors in a tertiary care hospital of South India: A review 117 cases. Indian J Med Paediatr Oncol. 2011;32:82–5. doi: 10.4103/0971-5851.89778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosen G, Caparros B, Huvos AG, Kosloff C, Nirenberg A, Cacavio A, et al. Preoperative chemotherapy for osteogenic sarcoma: Selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer. 1982;49:1221–30. doi: 10.1002/1097-0142(19820315)49:6<1221::aid-cncr2820490625>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 8.Shirke-Satpute S, Rekhi B, Desai SS. Correlation of MDM2, P53, MIB1 expression with response to neoadjuvant chemotherapy in high-grade extremity osteosarcoma and clinical outcome. Mod Pathol. 2013;26(Suppl 2):20A. [Google Scholar]
- 9.Kumar C, Vats K, Lohar SP, Korde A, Samuel G. Camptothecin enhances cell death induced by (177) Lu-EDTMP in osteosarcoma cells. Cancer Biother Radiopharm. 2014;29:317–22. doi: 10.1089/cbr.2014.1663. [DOI] [PubMed] [Google Scholar]
- 10.Bajpai J, Sharma M, Sreenivas V, Kumar R, Gamnagatti S, Khan SA, et al. VEGF expression as a prognostic marker in osteosarcoma. Pediatr Blood Cancer. 2009;53:1035–9. doi: 10.1002/pbc.22178. [DOI] [PubMed] [Google Scholar]
- 11.Bajpai J, Gamanagatti S, Sharma MC, Kumar R, Vishnubhatla S, Khan SA, et al. Noninvasive imaging surrogate of angiogenesis in osteosarcoma. Pediatr Blood Cancer. 2010;54:526–31. doi: 10.1002/pbc.22328. [DOI] [PubMed] [Google Scholar]
- 12.Amit P, Patro DK, Basu D, Elangovan S, Parathasarathy V. Role of dynamic MRI and clinical assessment in predicting histologic response to neoadjuvant chemotherapy in bone sarcomas. Am J Clin Oncol. 2014;37:384–90. doi: 10.1097/COC.0b013e31827b4f6f. [DOI] [PubMed] [Google Scholar]
- 13.Amit P, Malhotra A, Kumar R, Kumar L, Patro DK, Elangovan S. Evaluation of static and dynamic MRI for assessing response of bone sarcomas to preoperative chemotherapy: Correlation with histological necrosis. Indian J Radiol Imaging. 2015;25:269–75. doi: 10.4103/0971-3026.161452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai J, Kumar R, Sreenivas V, Sharma MC, Khan SA, Rastogi S, et al. Prediction of chemotherapy response by PET-CT in osteosarcoma: Correlation with histologic necrosis. J Pediatr Hematol Oncol. 2011;33:e271–8. doi: 10.1097/MPH.0b013e31820ff78e. [DOI] [PubMed] [Google Scholar]
- 15.Pathak AB, Advani SH, Iyer RS, Pai SK, Gopal R, Nadkarni KS, et al. Adjuvant chemotherapy for osteogenic sarcoma of the extremity with sequential adriamycin and cisplatin. J Surg Oncol. 1993;52:181–4. doi: 10.1002/jso.2930520313. [DOI] [PubMed] [Google Scholar]
- 16.Nataraj V, Batra A, Rastogi S, Khan SA, Sharma MC, Vishnubhatla S, et al. Developing a prognostic model for patients with localized osteosarcoma treated with uniform chemotherapy protocol without high dose methotrexate: A single-center experience of 237 patients. J Surg Oncol. 2015;112:662–8. doi: 10.1002/jso.24045. [DOI] [PubMed] [Google Scholar]
- 17.Poudel RR, Kumar VS, Bakhshi S, Gamanagatti S, Rastogi S, Khan SA. High tumor volume and local recurrence following surgery in osteosarcoma: A retrospective study. Indian J Orthop. 2014;48:285–8. doi: 10.4103/0019-5413.132520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bajpai J, Chandrashekharan A, Pandey N. Capetown: Presented at International Society of Pediatric Oncology Congress; 2015. Oct, Non-high Dose – Methotrexate (HD-MTX) Based, Dose-Dense (DD) Chemotherapy (CT) in Osteosarcoma: Is it Effective, Economic and Easy? [Google Scholar]
- 19.Bajpai J, Puri A, Shah K, Susan D, Jambhekar N, Rekhi B, et al. Chemotherapy compliance in patients with osteosarcoma. Pediatr Blood Cancer. 2013;60:41–4. doi: 10.1002/pbc.24155. [DOI] [PubMed] [Google Scholar]
- 20.Laskar S, Basu A, Muckaden MA, D’Cruz A, Pai S, Jambhekar N, et al. Osteosarcoma of the head and neck region: Lessons learned from a single-institution experience of 50 patients. Head Neck. 2008;30:1020–6. doi: 10.1002/hed.20820. [DOI] [PubMed] [Google Scholar]
- 21.Puri A, Gulia A, Pruthi M. Outcome of surgical resection of pelvic osteosarcoma. Indian J Orthop. 2014;48:273–8. doi: 10.4103/0019-5413.132515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gulia A, Puri A, Pruthi M, Desai S. Oncological and functional outcome of periosteal osteosarcoma. Indian J Orthop. 2014;48:279–84. doi: 10.4103/0019-5413.132518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puri A, Gulia A, Hawaldar R, Ranganathan P, Badwe RA. Does intensity of surveillance affect survival after surgery for sarcomas. Results of a randomized noninferiority trial? Clin Orthop Relat Res. 2014;472:1568–75. doi: 10.1007/s11999-013-3385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rekhi B, Vogel U, Basak R, Desai SB, Jambhekar NA. Clinicopathological and molecular spectrum of Ewing sarcomas/PNETs, including validation of EWSR1 rearrangement by conventional and array FISH technique in certain cases. Pathol Oncol Res. 2014;20:503–16. doi: 10.1007/s12253-013-9721-2. [DOI] [PubMed] [Google Scholar]
- 25.Tilak TV, Sharawat S, Gupta R, Agarwala S, Vishnubhatla S, Bakhshi S. Circulating T-regulatory cells in PNET: A prospective study. Pediatr Blood Cancer. 2014;61:228–32. doi: 10.1002/pbc.24734. [DOI] [PubMed] [Google Scholar]
- 26.Jully B, Vijayalakshmi R, Gopal G, Sabitha K, Rajkumar T. Junction region of EWS-FLI1 fusion protein has a dominant negative effect in Ewing’s sarcoma in vitro . BMC Cancer. 2012;12:513. doi: 10.1186/1471-2407-12-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswas B, Sharma MC, Mridha AR, Bakhshi S. Expression of cathepsin L in tumor cells and tumor-associated macrophages in patients with Ewing sarcoma family of tumors: A pilot study. Indian J Pathol Microbiol. 2015;58:170–4. doi: 10.4103/0377-4929.155307. [DOI] [PubMed] [Google Scholar]
- 28.Gupta K, Pawaskar A, Basu S, Rajan MG, Asopa RV, Arora B, et al. Potential role of FDG PET imaging in predicting metastatic potential and assessment of therapeutic response to neoadjuvant chemotherapy in Ewing sarcoma family of tumors. Clin Nucl Med. 2011;36:973–7. doi: 10.1097/RLU.0b013e31822f684b. [DOI] [PubMed] [Google Scholar]
- 29.Biswas B, Rastogi S, Khan SA, Shukla NK, Deo SV, Agarwala S, et al. Developing a prognostic model for localized Ewing sarcoma family of tumors: A single institutional experience of 224 cases treated with uniform chemotherapy protocol. J Surg Oncol. 2015;111:683–9. doi: 10.1002/jso.23861. [DOI] [PubMed] [Google Scholar]
- 30.Biswas B, Rastogi S, Khan SA, Shukla NK, Deo SV, Agarwala S, et al. Hypoalbuminaemia is an independent predictor of poor outcome in metastatic Ewing’s sarcoma family of tumours: A single institutional experience of 150 cases treated with uniform chemotherapy protocol. Clin Oncol (R Coll Radiol) 2014;26:722–9. doi: 10.1016/j.clon.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Biswas B, Rastogi S, Khan SA, Mohanti BK, Sharma DN, Sharma MC, et al. Outcomes and prognostic factors for Ewing-family tumors of the extremities. J Bone Joint Surg Am. 2014;96:841–9. doi: 10.2106/JBJS.M.00411. [DOI] [PubMed] [Google Scholar]
- 32.Biswas B, Agarwala S, Rastogi S, Khan SA, Mohanti BK, Sharma DN, et al. High burden of metastases and poor outcome in pelvic PNET. Pediatr Blood Cancer. 2013;60:E97–9. doi: 10.1002/pbc.24552. [DOI] [PubMed] [Google Scholar]
- 33.Prabu R, Thulkar S, Chand Sharma M, Mohanti BK, Dhawan D, Bakhshi S. PNET spine: Morbid and mortal, but ignored till late. J Pediatr Hematol Oncol. 2012;34:e164–9. doi: 10.1097/MPH.0b013e31824414b2. [DOI] [PubMed] [Google Scholar]
- 34.Biswas B, Agarwala S, Shukla NK, Deo S, Sharma D, Thulkar S, et al. Evaluation of outcome and prognostic factors in thoracic primitive neuroectodermal tumor: A study of 84 cases. Ann Thorac Surg. 2013;96:2006–14. doi: 10.1016/j.athoracsur.2013.06.062. [DOI] [PubMed] [Google Scholar]
- 35.Biswas B, Shukla NK, Deo SV, Agarwala S, Sharma DN, Vishnubhatla S, et al. Evaluation of outcome and prognostic factors in extraosseous Ewing sarcoma. Pediatr Blood Cancer. 2014;61:1925–31. doi: 10.1002/pbc.25095. [DOI] [PubMed] [Google Scholar]
- 36.Qureshi SS, Kembhavi S, Vora T, Ramadwar M, Laskar S, Talole S, et al. Prognostic factors in primary nonmetastatic Ewing sarcoma of the rib in children and young adults. J Pediatr Surg. 2013;48:764–70. doi: 10.1016/j.jpedsurg.2012.07.049. [DOI] [PubMed] [Google Scholar]
- 37.Qureshi SS, Kembhavi S, Bhagat M, Laskar S, Chinnaswamy G, Vora T, et al. Primary non-metastatic Ewing sarcoma of the jaw in children: Results of surgical resection and primary reconstruction. J Surg Oncol. 2014;110:689–95. doi: 10.1002/jso.23698. [DOI] [PubMed] [Google Scholar]
- 38.Biswas B, Thakar A, Mohanti BK, Vishnubhatla S, Bakhshi S. Prognostic factors in head and neck Ewing sarcoma family of tumors. Laryngoscope. 2015;125:E112–7. doi: 10.1002/lary.24985. [DOI] [PubMed] [Google Scholar]
- 39.Puri A, Gulia A, Jambhekar N, Laskar S. The outcome of the treatment of diaphyseal primary bone sarcoma by resection, irradiation and re-implantation of the host bone: Extracorporeal irradiation as an option for reconstruction in diaphyseal bone sarcomas. J Bone Joint Surg Br. 2012;94:982–8. doi: 10.1302/0301-620X.94B7.28916. [DOI] [PubMed] [Google Scholar]
- 40.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 41.Coindre J, Terrier P, Guillou L, Le Doussal V, Collin F, Ranchère D, et al. Predictive value of grade for metastases development in the main histological types of adult soft tissue sarcomas: A study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91:1914–26. doi: 10.1002/1097-0142(20010515)91:10<1914::aid-cncr1214>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 42.Rekhi B, Jambhekar NA, Puri A, Agrawal M, Chinoy RF. Clinicomorphologic features of a series of 10 cases of malignant triton tumors diagnosed over 10 years at a tertiary cancer hospital in Mumbai, India. Ann Diagn Pathol. 2008;12:90–7. doi: 10.1016/j.anndiagpath.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Rekhi B, Gorad BD, Chinoy RF. Clinicopathological features with outcomes of a series of conventional and proximal-type epithelioid sarcomas, diagnosed over a period of 10 years at a tertiary cancer hospital in India. Virchows Arch. 2008;453:141–53. doi: 10.1007/s00428-008-0639-0. [DOI] [PubMed] [Google Scholar]
- 44.Kumar R, Rekhi B, Shirazi N, Pais A, Amare P, Gawde D, et al. Spectrum of cytomorphological features, including literature review, of an extraskeletal myxoid chondrosarcoma with t(9;22)(q22;q12) (TEC/EWS) results in one case. Diagn Cytopathol. 2008;36:868–75. doi: 10.1002/dc.20931. [DOI] [PubMed] [Google Scholar]
- 45.Jambhekar NA, Rekhi B, Thorat K, Dikshit R, Agrawal M, Puri A. Revisiting chordoma with brachyury, a “new age” marker: Analysis of a validation study on 51 cases. Arch Pathol Lab Med. 2010;134:1181–7. doi: 10.5858/2009-0476-OA.1. [DOI] [PubMed] [Google Scholar]
- 46.Rekhi B, Ingle A, Kumar R, DeSouza MA, Dikshit R, Jambhekar NA. Malignant peripheral nerve sheath tumors: Clinicopathological profile of 63 cases diagnosed at a tertiary cancer referral center in Mumbai, India. Indian J Pathol Microbiol. 2010;53:611–8. doi: 10.4103/0377-4929.71998. [DOI] [PubMed] [Google Scholar]
- 47.Rekhi B, Deshmukh M, Jambhekar NA. Low-grade fibromyxoid sarcoma – A clinicopathological study of 18 cases, including histopathological relationship with sclerosing epithelioid fibrosarcoma in a subset of cases. Ann Diagn Pathol. 2011;15:303–11. doi: 10.1016/j.anndiagpath.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Rekhi B, Ingle A, Agarwal M, Puri A, Laskar S, Jambhekar NA. Alveolar soft part sarcoma “revisited”: Clinicopathological review of 47 cases from a tertiary cancer referral centre, including immunohistochemical expression of TFE3 in 22 cases and 21 other tumours. Pathology. 2012;44:11–7. doi: 10.1097/PAT.0b013e32834d7ba4. [DOI] [PubMed] [Google Scholar]
- 49.Rekhi B, Ahmed S, Basak R, Qureshi SS, Desai SS, Ramadwar M, et al. Desmoplastic small round cell tumor-clinicopathological spectrum, including unusual features and immunohistochemical analysis of 45 tumors diagnosed at a tertiary cancer referral centre, with molecular results t(11; 22) (p13; q12) (EWS-WT1) in select cases. Pathol Oncol Res. 2012;18:917–27. doi: 10.1007/s12253-012-9522-z. [DOI] [PubMed] [Google Scholar]
- 50.Dinshaw KA, Ganesh B. Hospital Based Cancer Registry. Annual Report 2001. Mumbai: Tata Memorial Hospital; 2005. pp. 72–91. [Google Scholar]
- 51.Rekhi B, Basak R, Desai SB, Jambhekar NA. Immunohistochemical validation of TLE1, a novel marker, for synovial sarcomas. Indian J Med Res. 2012;136:766–75. [PMC free article] [PubMed] [Google Scholar]
- 52.Rekhi B, Vogel U. Utility of characteristic “eak to absent” INI1/SMARCB1/BAF47 expression in diagnosis of synovial sarcomas. APMIS. 2015;123:618–28. doi: 10.1111/apm.12395. [DOI] [PubMed] [Google Scholar]
- 53.Iqbal N, Shukla NK, Deo SV, Agarwala S, Sharma DN, Sharma MC, et al. Prognostic factors affecting survival in metastatic soft tissue sarcoma: An analysis of 110 patients. Clin Transl Oncol. 2016;18:310–6. doi: 10.1007/s12094-015-1369-9. [DOI] [PubMed] [Google Scholar]
- 54.Panda N, Das R, Banerjee S, Chatterjee S, Gumta M, Bandyopadhyay SK. Retroperitoneal Sarcoma. Outcome Analysis in a Teaching Hospital in Eastern India- a Perspective. Indian J Surg Oncol. 2015;6:99–105. doi: 10.1007/s13193-015-0404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rekhi B, Navale P, Jambhekar NA. Critical histopathological analysis of 25 dedifferentiated liposarcomas, including uncommon variants from a tertiary cancer referral centre. Indian J Pathol Microbiol. 2012;55:294–302. doi: 10.4103/0377-4929.101733. [DOI] [PubMed] [Google Scholar]
- 56.Jamema SV, Sharma PK, Laskar S, Deshpande DD, Shrivastava SK. Treatment planning comparison of electron arc therapy and photon intensity modulated radiotherapy for Askin’s tumor of chest wall. Radiother Oncol. 2007;84:257–65. doi: 10.1016/j.radonc.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 57.Jamema SV, Sharma PK, Sharma D, Laskar S, Deshpande DD, Shrivastava SK. Dose optimization of intra-operative high dose rate interstitial brachytherapy implants for soft tissue sarcoma. J Cancer Res Ther. 2009;5:240–6. doi: 10.4103/0973-1482.59893. [DOI] [PubMed] [Google Scholar]
- 58.Laskar S, Bahl G, Ann Muckaden M, Puri A, Agarwal MG, Patil N, et al. Interstitial brachytherapy for childhood soft tissue sarcoma. Pediatr Blood Cancer. 2007;49:649–55. doi: 10.1002/pbc.21118. [DOI] [PubMed] [Google Scholar]
- 59.Chaudhary AJ, Laskar S, Badhwar R. Interstitial brachytherapy in soft tissue sarcomas. The Tata Memorial Hospital experience. Strahlenther Onkol. 1998;174:522–8. doi: 10.1007/BF03038985. [DOI] [PubMed] [Google Scholar]
- 60.Sharma DN, Deo SV, Rath GK, Shukla NK, Bakhshi S, Gandhi AK, et al. Perioperative high-dose-rate interstitial brachytherapy combined with external beam radiation therapy for soft tissue sarcoma. Brachytherapy. 2015;14:571–7. doi: 10.1016/j.brachy.2015.03.002. [DOI] [PubMed] [Google Scholar]