

ABSTRACT
Crystal structure determination of an active polycomb repressive complex 2 (PRC2) from a thermophilic fungus, Chaetomium thermophilum, revealed some long-sought structural mechanisms for assembly, catalysis, and regulation of this important enzyme complex, responsible for trimethylation of histone H3K27 (H3K27me3) and silencing of developmentally regulated genes. In light of the crystal structures of the fungal PRC2 captured in the basal and H3K27me3-stimulated states as well as the structural analysis published previously,1 we examined surface conservation and electrostatic potential distribution to provide additional insights into functional similarity and divergence between the fungal and human PRC2 and for PRC2 binding by nucleic acids. Structure comparison indicated a conformational change of the catalytic SET domain within PRC2 during transition from the inactive to active state. This conserved structural mechanism is also used by another histone methyltransferase family associated with gene activation for enzyme regulation and may underlie the allosteric stimulation of PRC2 as well.
KEYWORDS: facultative heterochromatin, histone, H3K27 trimethylation (H3K27me3), Polycomb repressive complex 2 (PRC2)
Introduction
Histones package genomic DNA into nucleosomes and are considered as general repressors of transcription.2 Polycomb repressive complex 2 (PRC2), a major member of the polycomb-group (PcG) protein family mostly identified as gene repressors in Drosophila originally, modifies histone tails and, together with other PcG proteins, promotes formation of developmentally regulated facultative heterochromatin to stably repress gene expression and confer a repressive epigenetic memory of cell identity.3 PRC2-mediated gene silencing also plays an important role in some fundamental cellular processes, such as imprinting, X-chromosome inactivation, and genome defense against transposable elements.4,5
Specifically, PRC2 mediates histone H3 lysine 27 trimethylation (H3K27me3), a hallmark of repressed chromatin. A canonical PRC2 contains 4 core subunits, Ezh2 (enhancer of zeste homolog 2), Eed (embryonic ectoderm development), Suz12 (suppressor of zeste 12 protein homolog), and Rbbp4 (retinoblastoma binding protein 4) (Fig 1A). Ezh2 is the catalytic subunit, harboring a catalytic SET [su(var)3–9, enhancer-of-zeste and trithorax] domain that belongs to a unique lysine methyltransferase family. The Ezh2, Eed, and Suz12 subunits of PRC2 are minimally required to catalyze H3K27 methylation.6
Figure 1.
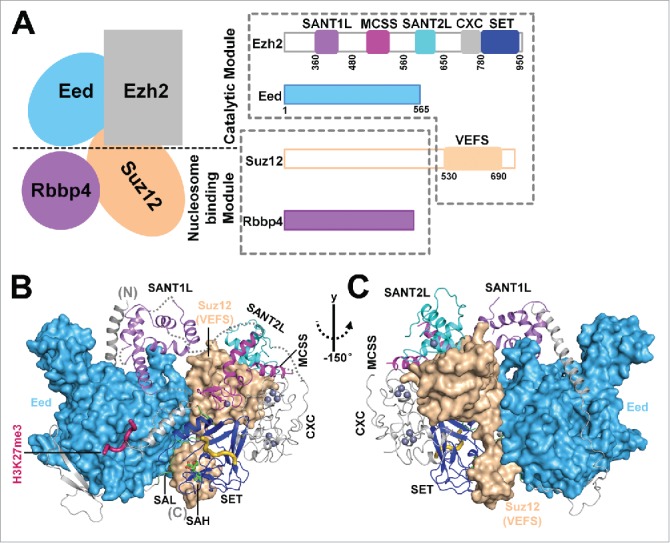
Overall view of the crystal structure of an active fungal PRC2. (A) Schematic composition and domain architecture of PRC2. Four core subunits of PRC2, Ezh2, Eed, Suz12, and Rbbp4, are shown. Two dotted boxes correspond to the catalytic module and the putative nucleosome-binding module. Ezh2 domains that are discussed in the text are highlighted by colored blocks. (B and C) Front and back views of the overall structure of the active Ezh2-Eed-Suz12(VEFS) ternary complex in the H3K27me3-stimulated state. Ezh2 is shown in cartoon representation with the individual domains colored as in Fig. 1A. Eed and Suz12(VEFS) are shown as surface representations. These two subunits form extensive interactions with different domains of Ezh2 and are important for the integrity and catalysis of PRC2.
Notably, the enzymatic activity of PRC2 is subjected to allosteric simulation by distinct cellular mechanisms, which act complementarily to establish functional H3K27me3 domains in a locus- and cell type-specific manner. The end product of PRC2 catalysis, H3K27me3, binds to the Eed subunit and enhances the catalytic activity of PRC2.7 This positive feedback mechanism is believed to account for propagation of the repressive H3K27me3 mark and spreading of the facultative heterochromatin.7 In addition, Jarid2 (jumonji/ARID domain-containing protein 2), an auxiliary subunit of PRC2, is methylated at lysine 116 by PRC2 and the product of this methylation reaction, Jarid2-K116me3, also contacts the Eed surface identical to that for H3K27me3 binding to stimulate PRC2 catalysis. This mechanism is thought to promote H3K27 trimethylation at loci devoid of the existing H3K27me3 mark and hence lack of the H3K27me3-mediated enzyme stimulation.8 Furthermore, a region of an unmodified N-terminal tail of histone H3 (residues 31–42) from neighboring nucleosomes in a dense chromatin environment interacts with the VEFS [Vrn2-Emf2-Fis2-Su(z)12] domain of Suz12 [Suz12(VEFS)] to stimulate the enzymatic activity of PRC2, which coincides with the observation that local chromatin compaction precedes H3K27 trimethylation during gene silencing in mouse embryonic stem cells.9
Aberrant PRC2 function is frequently associated with cancer and developmental disorder, and in particular Ezh2 mutations were linked to hematological malignancies and Weaver syndrome.10,11 Many cellular functions of PRC2 are governed by its intrinsic enzymatic activity toward H3K27me3. However, mechanistic understanding of PRC2 catalysis has been impeded, at least in part, by the lack of structural information of this enzyme complex. We determined the crystal structures of an active fungal PRC2 of 170KDa containing Ezh2, Eed, and Suz12(VEFS) [Ezh2-Eed-Suz12(VEFS)] captured in both basal and H3K27me3-stimulated states (Fig. 1B and 1C).1 In this Extra View, we carry out in-depth structural analysis of the fungal PRC2 and relate the structural information to some key aspects of human PRC2 function and regulation.
Conservation of PRC2 across species
PRC2 and H3K27 methylation are not conserved in the model yeast organisms S. cerevisiae and S. pombe.12 However, both the repressive H3K27me3 mark and PRC2-like enzyme complexes were recently identified in other yeast species, such as the filamentous fungus N. crassa (Neurospora crassa) and the pathogenic budding yeast C. neoformans (Cryptococcus neoformans).13,14 While N. crassa encodes homologs of all 4 core subunits of human PRC2, Suz12 is lost in C. neoformans. A Suz12 homolog is also absent in C. elegans, which together indicates a degree of compositional diversity behind the overall functional conservation.13-15 We identified PRC2 components in the genome of a thermophilic yeast, C. thermophilum (Chaetomium thermophilum), and showed that its encoded fungal Ezh2, Eed, and Suz12(VEFS) form a stable PRC2-like enzyme complex. We further found that, similar to its human PRC2 counterpart, this ternary complex carries out H3K27 trimethylation and is subjected to the H3K27me3-mediated allosteric enzyme stimulation, and hence serves as a suitable model for structural study of the conserved function and regulation of PRC2.1 In particular, this fungal PRC2 displays an overall structural similarity to its counterpart in a human holo-PRC2, based on a negative stain EM map of the latter.1,16
Ezh2 contains 10 structurally and functionally distinct regions and, from its N- to C-terminus, they are SBD (SANT1L-binding domain), EBD (Eed-binding domain), BAM (β-addition motif), SAL (SET activation loop), SRM (stimulation-responsive motif), SANT1L (SANT1-like), MCSS (motif connecting SANT1L and SANT2L), SANT2L (SANT2-like), CXC, and SET (Fig. 1A).1 The solvent-exposed surface of the catalytic SET domain and, to a lesser extent, the cysteine-rich CXC region of Ezh2, are conserved (Fig. 2A and 2B). Surfaces on Eed and Suz12(VEFS) that either interact with each other or account for Ezh2 binding are also conserved. In contrast, the SANT1L and SANT2L of Ezh2 represent the least conserved surfaces in the structure, implicating a functional divergence (Fig. 2A and 2B).
Figure 2.
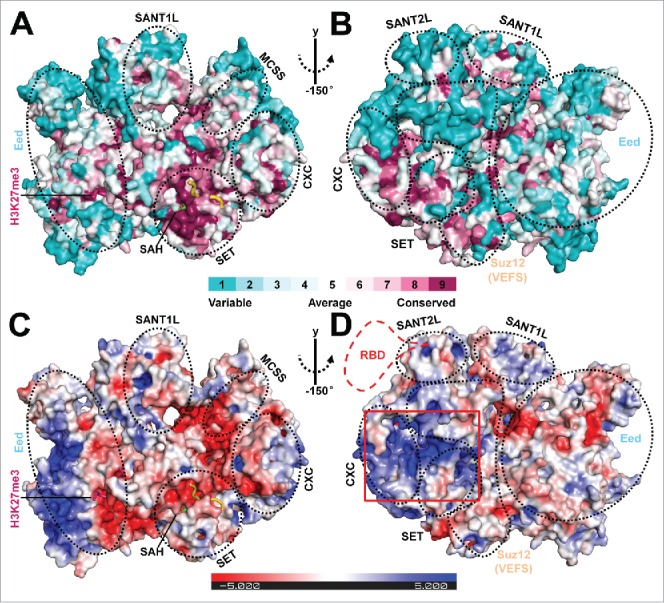
Structural analysis of the surface features of PRC2. (A and B) Front and back views of the structural conservation of the active fungal PRC2 in comparison to its human counterpart in surface representation. Surfaces are colored by ConSurf (http://consurf.tau.ac.il/).28 A complete structural model of PRC2 was obtained by adding the missing residues using Modeler 29 based on the PRC2 structure in the stimulated state. The new model was then used for conservation analysis. The scale bar indicates the conservation level of the residues on the surface. The individual domains in the complex are indicated by dotted black circles. The same model was used to generate Fig. 2C and 2D. (C and D) Front and back views of the electrostatic surface of the fungal PRC2 generated by Pymol using APBS calculation and colored by potential on solvent accessible surface.30 The scale bar (±5 kT/e) indicates the electrostatic potential on the surface. The predicted conserved RNA binding surface is highlighted by a red rectangular box. A putative human-specific RNA binding domain (RBD) is also illustrated by a dotted red line in Fig. 2D.
Notable electronegative potential was observed for surfaces corresponding to or contiguous with the binding sites for the H3 substrate and H3K27me3 stimulating peptide. Conceivably, these negatively charged surfaces function to capture the positively charged histone tails (Fig. 2C). Human and mouse PRC2 binds to RNAs including long non-coding RNAs (lncRNAs) and nascent RNAs promiscuously, a prominent feature thought to facilitate locus-specific recruitment of PRC2.3,17 Like for many other SET-containing histone methyltransferases, RNA binding markedly inhibits the enzymatic activity of PRC2.18 By examining both surface conservation and electrostatic potential distribution of the fungal PRC2 structure, we identified a conserved, positively charged concave surface formed by Suz12(VEFS) and the SANT2, CXC, and SET domain of Ezh2, which may mediate the observed enzyme inhibition by RNA binding (Fig. 2A–2D). Consistently, the single-stranded DNA and RNA binding surface on Drosophila E(z), a homolog of human Ezh2, was formerly mapped to its CXC domain by an in vitro pull-down assay.19 Our analysis purely based on the fungal PRC2 structure is unable to disclose the RNA binding surface specific to human PRC2,20 which is nonetheless intriguingly predicted to be located close to the conserved putative RNA binding surface on the fungal PRC2 (Fig. 2D). In addition, it may also be possible that these basic patches on the surface of PRC2 are involved in binding of nucleosomal DNA on a chromatin substrate.
Structural features of PRC2 that confer catalysis
The ten structural regions of Ezh2 do not all pack against each other and instead some of them are intimately associated with Eed and Suz12(VEFS), making PRC2 an obligate protein complex of a catalytically active Ezh2 (Fig. 1B and 1C).1 Indeed, extensive protein-protein interaction was observed among Eed, Suz12(VEFS) and individual domains of Ezh2 (Fig. 3A–3E). Mutations of some of the residues on the binding interface were previously found in human disease, implicating an impaired enzyme activity for these PRC2 mutants in cells.1
Figure 3.
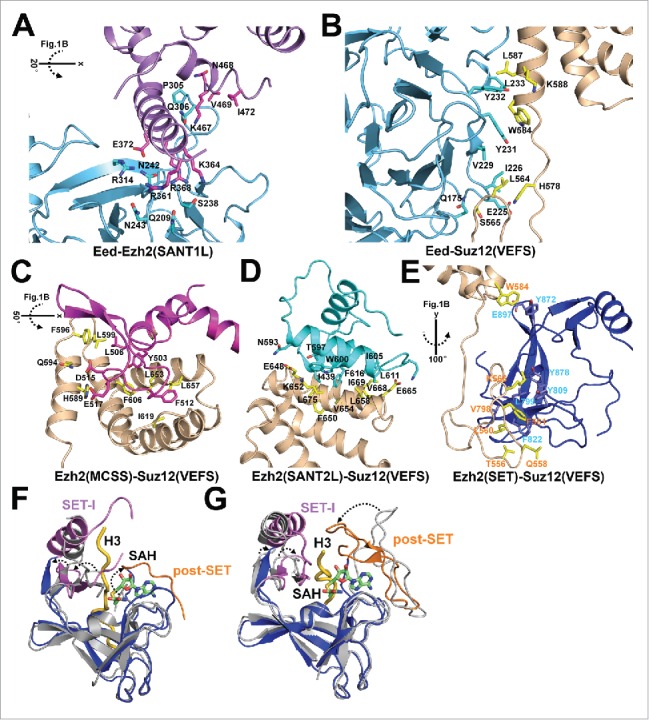
Close-up views of the intermolecular interactions within PRC2 and structural comparison of Ezh2 and MLL histone methyltransferases. (A) Interaction between Ezh2(SANT1L) and Eed. Ezh2(SANT1L) interacts with a binding groove on Eed mainly through electrostatic interactions. Residues on the binding interface were selected with distances no larger than 4Å and shown as sticks. The same selection criterion and representation scheme were used below in Fig. 3B through Fig. 3E. (B) Interaction between Eed and Suz12(VEFS). Eed interacts with Suz12(VEFS) mainly through hydrophobic interactions. In particular, W584 from Suz12(VEFS) is inserted into a hydrophobic pocket formed by Y231 and Y232 from Eed. (C, D and E) Interactions between Ezh2(MCSS, SANT2L, and SET) and Suz12(VEFS). The latter interacts with and facilitates positioning of these 3 Ezh2 domains through extensive hydrophobic interactions, ranging from the helix bundle interaction [Ezh2(SANTL2)-Suz12(VEFS)] and the helix-to-loop interaction [Ezh2(MCSS)-Suz12(VEFS)] to the loop-to-strand interaction [Ezh2(SET)-Suz12(VEFS)]. (F) Structural comparison of the active fungal Ezh2(SET) and the inactive human Ezh2(SET) (from PDB:4MI0). The SET-I and post-SET subdomains of the fungal Ezh2(SET) are shown in magenta and orange, respectively, with the rest colored in blue. The human Ezh2(SET) shown in gray is aligned with the fungal Ezh2(SET) with an R.M.SD of 1.7Å. Compared with the isolated inactive SET from human Ezh2, the SET-I and post-SET subdomains of the active SET from the fungal PRC2 structure are rotated to open up the histone H3 substrate binding groove and to form a complete SAM cofactor binding pocket. The H3 substrate shown in yellow was modeled in from the crystal structure of a plant H3K27 methyltransferase (PDB:4O30) by structural alignment. (G) Structural comparison of human MLL1(SET) (from PDB: 2W5Z) and human MLL4(SET) (from PDB:4Z4P). The same as in Fig. 3F, the SET-I and post-SET subdomains of the MLL4(SET) are shown in magenta and orange, respectively, with the rest colored in blue. The MLL1(SET) shown in gray is superimposed with the MLL4(SET) with an R.M.SD of 1.4Å. Compared with the MLL1(SET), which displays weak catalytic activity in the absence of other COMPASS components, the SET-I and post-SET subdomains of the MLL4(SET), which retains a considerably higher activity in isolation, move toward each other to form a closed substrate binding groove to enhance catalysis.
The CXC-SET region of Ezh2 folds independently but is unable to catalyze the chemical reaction in part due to an autoinhibited SET conformation and an incomplete cofactor-binding pocket.21,22 By comparing the active fungal PRC2 and the inactive CXC-SET region of human Ezh2, we revealed 2 notable, possibly interconnected structural features that might explain the conversion between the inactive and active SET conformation. First, the autoinhibited SET conformation is relieved by structure movement of 2 subdomains of the SET, SET-I and post-SET. They move away from each other to open up the otherwise blocked substrate-binding groove and to form the cofactor binding pocket (Fig. 3F). Second, Ezh2 contains a split catalytic domain, where the SAL region from the N-terminal portion of Ezh2 is inserted onto the back of the SET at the C-terminus of Ezh2, not contributing to catalysis per se but bridging the SET-I region to Eed, Suz12(VEFS), and some other Ezh2 domains at the periphery of the SET. This unique structural architecture of the complex renders Eed and Suz12(VEFS) indispensable components for Ezh2 catalysis.
Such a structural rearrangement of the SET domain of Ezh2 from the autoinhibited to active conformation in PRC2 is reminiscent of activation of an isolated MLL1 (mixed lineage leukemia 1) histone methyltransferase by other members of COMPASS (complex of proteins associated with Set1), including Wdr5, Ash2l, Dpy30, and Rbbp5.23 MLL1/COMPASS mediates H3K4 trimethylation correlated with active transcription and belongs to the Trithorax group (TrxG) proteins, which functionally antagonizes PcG proteins during development. In a stark contrast to the case for the isolated Ezh2, the SET-I and post-SET subdomains of the MLL1 SET are located too far away from each other to stabilize substrate binding, and the relative position of these 2 subdomains was thought to be readjusted in COMPASS to confer an activated enzyme (Fig. 3G).23,24 Indeed, a recent structural study on the isolated MLL1/3 SET domains and the MLL1/3–Rbbp5–Ash2l ternary complexes suggested that association of Rbbp5 and Ash2l activated the MLL1/3 SET by suppressing the dynamic motion of the SET-I subdomain.25 The 2 antagonistic histone methylation enzyme complexes central for transcriptional control, PRC2 and COMPASS, thus appear to utilize a similar mechanism to control methyltransferase activity within their respective core complexes. It is intriguing to speculate that such a structural mechanism involving conformational change of the SET-I subdomain may also serve as the basis, at least to some extent, for enzyme regulation by other auxiliary factors associated these 2 enzyme complexes.
Structural basis of PRC2 enzyme stimulation
Many PRC2-mediated cellular transactions including H3K27 methylation occur on chromatin. The exiting histone marks and in particular those on the histone H3 tail exert profound impact on PRC2 catalysis. The solved crystal structures of an active fungal PRC2 in both H3K27me3-free and H3K27me3-bound states provide a structural mechanism for the allosteric stimulation of PRC2 by H3K27me3.1 H3K27me3 interacts concomitantly with both Eed, as previously reported,7 and the flexible SRM region of Ezh2 to juxtapose the latter to the SET-I subdomain of the SET. Interactions, mostly hydrophobic ones, are established to control conformation of the active site residues through the conserved salt bridge within the SET-I subdomain. At least 3 central aspects of PRC2 catalysis are potentially influenced by such a mechanism, including binding affinity of the substrate and cofactor, rate of catalysis, and methylation multiplicity (see below). PRC2 enzyme stimulation by Jarid2-K116me3 may use the same mechanism.8 The enzymatic activity of PRC2 is also stimulated by an unmodified H3 (residues 31-42) in a dense chromatin environment,9 for which the binding residues on Suz12(VEFS) are not conserved in the fungal PRC2 structure. It is nonetheless clear from the structural analysis that such a allosteric regulation may be achieved through the Suz12(VEFS)→Ezh2(SAL)→Ezh2(SET-I) pathway, distinct from the Ezh2(SRM)→Ezh2(SET-I) pathway used by H3K27me3 (Fig. 4A).
Figure 4.
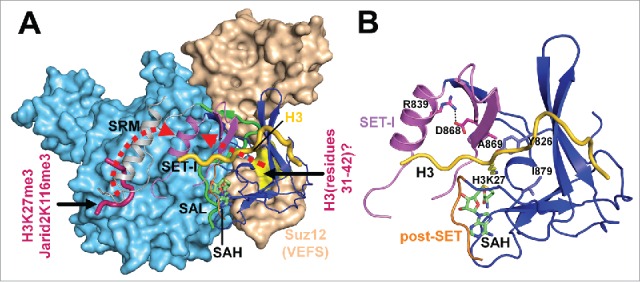
Structural analysis of the allosteric stimulation of PRC2. (A) Illustration of the 2 potential mechanistic pathways for the enzyme stimulation by H3K27me3 and Jarid2-K116me3 and by H3 (residues 31–42), respectively. Eed and Suz12(VEFS) are shown in surface representation. The SRM, SAL and SET domains of Ezh2 are shown in cartoon representation. The two dotted red arrows indicate the 2 distinct potential stimulation pathways, specifically Ezh2(SRM)→Ezh2(SET-I) for the H3K27me3 or Jarid2-K116me3-mediated enzyme stimulation and Suz12(VEFS)→Ezh2(SAL)→Ezh2(SET-I) for the H3 (residues 31–42)-mediated enzyme stimulation. As in Fig. 3G, the H3 substrate shown in yellow was modeled in by structural alignment. (B) Close-up view of the active site of the fungal Ezh2(SET). The conserved salt bridge within the SET-I subdomain is formed by residues R839 and D868 and their interaction is indicated by a dotted black line. The conserved active site residues of the fungal Ezh2 (Y826, A869 and I879) corresponding to the sites of gain-of-function disease mutations of human Ezh2, Y645F/N/H/C, A677G and A687V, are shown as sticks. The modeled residue H3K27 is also shown in sticks to indicate the location of the active site.
The lysine access channel lies between the SET-I and post-SET subdomains and an obvious consequence of the structural rearrangement of the SET-I subdomain is conformational change of the active site. For the successive methylation reaction catalyzed by Ezh2, a compromise must be reached for size of the active site to favor either a low or high methylation multiplicity. While robust monomethylation entails a tightly packed active site to facilitate lysine deprotonation or to stabilize substrate binding, dimethylation or trimethylation requires rotation of their respective methylated substrates that are slightly larger in size to realign the nitrogen lone pair to the methyl donor SAM. The fact that the active site of the wild type Ezh2 is evolved to disfavor H3K27 trimethylation in the basal state makes intricate regulation of the H3K27me3 patterns on chromatin possible. Accordingly, the structural mechanism discussed here involving conformational change of the active site may underlie many regulatory pathways for modulating H3K27 trimethylation. In line with this prediction, 3 activating mutations of Ezh2 that lead to H3K27 hypertrimethylation in lymphoid neoplasms, Y641F/N/H/C, A677G, and A687V, are all located at the active site of Ezh2 (Fig. 4B).1,26 Indeed, we suggested that the H3K27me3-mediated enzyme stimulation may employ a structural mechanism analogous to that used by the A677G cancer mutant of human Ezh2 to promote H3K27 trimethylation, since in both cases the size of the active site may be slightly expanded.1
Finally, no specific structural mechanism is available for regulation of the post-SET subdomain, which may nonetheless be a hotspot for cellular control of PRC2 catalysis. First, the post-SET subdomain, exposed to the solvent and important for both substrate and cofactor binding, may serve as a docking site for other cellular factors for enzyme regulation. Second, the post-SET subdomain is subjected to posttranslational modification such as serine phosphorylation,27 which may also result in either conformational change of itself or recruitment of other cellular factors for regulating PRC2 catalysis.
Summary and perspective
Crystal structure determination of the active fungal PRC2 enzyme complex shed light on some long-standing mysteries regarding PRC2 assembly, catalysis, and regulation. Analysis of the surface conservation and electrostatic potential distribution was informative for predicting functional similarity and divergence including RNA binding by the fungal and human PRC2. Ezh2 depends on Eed and Suz12(VFES) to adopt a split, catalytically active SET conformation for substrate and cofactor binding, likely by maintaining properly positioned SET-I and post-SET subdomains.1 The MLL family of methyltransferases uses a mechanistically similar strategy to achieve enzyme regulation by other non-catalytic core members of a holo COMPASS.23-25 In addition, we proposed that such a structural mechanism involving movement of the SET-I and post-SET subdomains toward an ultimate structural rearrangement of the active site residues may also underlie the allosteric stimulation of PRC2 by some different cellular pathways.
Some predicted conformational changes of the active site residues associated with enzyme regulation might be too dynamic or too subtle to be reliably assessed by the crystal structures at current resolution. In this regard, in silico simulation of PRC2 dynamics at different functional states may provide additional insights for the catalytic mechanism of H3K27 methylation and in particular for the control of methylation multiplicity. Furthermore, the 4-subunit PRC2 core complex as well as that bound to additional auxiliary cellular factors and nucleosomal substrates represent some of the most rewarding targets for future structural studies for understanding the mechanism of gene regulation by PRC2.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Liu lab for comments on the manuscript. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. DOE under contract no. DE-AC02-05CH11231. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Funding
This research was supported by the Welch Foundation research grant I-1790, CPRIT research grant R1119, Rita Allen Foundation research grant, UT Southwestern Medical Center Endowed Scholar fund, and NIH grant GM114576 to X.L. X.L. is a W. W. Caruth, Jr. Scholar in Biomedical Research. This research also received support from the Cecil H. and Ida Green Center Training Program in Reproductive Biology Sciences Research.
References
- [1].Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015; 350:aac4383; PMID:26472914; http://dx.doi.org/ 10.1126/science.aab2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999; 98:285-94; PMID:10458604; http://dx.doi.org/ 10.1016/S0092-8674(00)81958-3 [DOI] [PubMed] [Google Scholar]
- [3].Simon JA, Kingston RE. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell 2013; 49:808-24; PMID:23473600; http://dx.doi.org/ 10.1016/j.molcel.2013.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet 2011; 12:123-35; PMID:21221116; http://dx.doi.org/ 10.1038/nrg2932 [DOI] [PubMed] [Google Scholar]
- [5].Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev 2010; 24:265-76; PMID:20123906; http://dx.doi.org/ 10.1101/gad.544410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 2004; 15:57-67; PMID:15225548; http://dx.doi.org/ 10.1016/j.molcel.2004.06.020 [DOI] [PubMed] [Google Scholar]
- [7].Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al.. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009; 461:762-7; PMID:19767730; http://dx.doi.org/ 10.1038/nature08398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M, Michaud A, Lombard B, da Rocha ST, Offer J, et al.. Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell 2015; 57:769-83; PMID:25620564; http://dx.doi.org/ 10.1016/j.molcel.2014.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al.. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 2012; 337:971-5; PMID:22923582; http://dx.doi.org/ 10.1126/science.1225237 [DOI] [PubMed] [Google Scholar]
- [10].Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, et al.. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010; 42:665-7; PMID:20601954; http://dx.doi.org/ 10.1038/ng.620 [DOI] [PubMed] [Google Scholar]
- [11].Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, et al.. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 2012; 90:110-8; PMID:22177091; http://dx.doi.org/ 10.1016/j.ajhg.2011.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shaver S, Casas-Mollano JA, Cerny RL, Cerutti H. Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics: Off J DNA Methylation Soc 2010; 5:301-12; PMID:20421736; http://dx.doi.org/ 10.4161/epi.5.4.11608 [DOI] [PubMed] [Google Scholar]
- [13].Jamieson K, Rountree MR, Lewis ZA, Stajich JE, Selker EU. Regional control of histone H3 lysine 27 methylation in Neurospora. Proc Natl Acad Sci U S A 2013; 110:6027-32; PMID:23530226; http://dx.doi.org/ 10.1073/pnas.1303750110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dumesic PA, Homer CM, Moresco JJ, Pack LR, Shanle EK, Coyle SM, Strahl BD, Fujimori DG, Yates JR 3rd, Madhani HD. Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 2015; 160:204-18; PMID:25533783; http://dx.doi.org/ 10.1016/j.cell.2014.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bender LB, Cao R, Zhang Y, Strome S. The MES-2/MES-3/MES-6 complex and regulation of histone H3 methylation in C. elegans. Curr Biol: CB 2004; 14:1639-43; PMID:15380065; http://dx.doi.org/ 10.1016/j.cub.2004.08.062 [DOI] [PubMed] [Google Scholar]
- [16].Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. Molecular architecture of human polycomb repressive complex 2. eLife 2012; 1:e00005; PMID:23110252; http://dx.doi.org/ 10.7554/eLife.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol 2013; 20:1250-7; PMID:24077223; http://dx.doi.org/ 10.1038/nsmb.2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev 2014; PMID:25170018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Krajewski WA, Nakamura T, Mazo A, Canaani E. A motif within SET-domain proteins binds single-stranded nucleic acids and transcribed and supercoiled DNAs and can interfere with assembly of nucleosomes. Mol Cell Biol 2005; 25:1891-9; PMID:15713643; http://dx.doi.org/ 10.1128/MCB.25.5.1891-1899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 2010; 24:2615-20; PMID:21123648; http://dx.doi.org/ 10.1101/gad.1983810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, MacEwan I, Zhang A, Ashok S, Rodgers L, et al.. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PloS One 2013; 8:e84147; PMID:24367637; http://dx.doi.org/ 10.1371/journal.pone.0084147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, Seitova A, Duan S, Brown PJ, Vedadi M, et al.. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PloS One 2013; 8:e83737; PMID:24367611; http://dx.doi.org/ 10.1371/journal.pone.0083737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell 2009; 33:181-91; PMID:19187761; http://dx.doi.org/ 10.1016/j.molcel.2008.12.029 [DOI] [PubMed] [Google Scholar]
- [24].Zhang Y, Mittal A, Reid J, Reich S, Gamblin SJ, Wilson JR. Evolving catalytic properties of the MLL family SET domain. Structure 2015; 23:1921-33; PMID:26320581; http://dx.doi.org/ 10.1016/j.str.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Y, Han J, Zhang Y, Cao F, Liu Z, Li S, Wu J, Hu C, Wang Y, Shuai J, et al.. Structural basis for activity regulation of MLL family methyltransferases. Nature 2016; 530:447-52; PMID:26886794; http://dx.doi.org/ 10.1038/nature16952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell 2014; 5:265-96; PMID:24622842; http://dx.doi.org/ 10.1007/s13238-014-0031-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ruse CI, McClatchy DB, Lu B, Cociorva D, Motoyama A, Park SK, Yates JR 3rd. Motif-specific sampling of phosphoproteomes. J Proteome Res 2008; 7:2140-50; PMID:18452278; http://dx.doi.org/ 10.1021/pr800147u [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 2010; 38:W529-33; PMID:20478830; http://dx.doi.org/ 10.1093/nar/gkq399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234:779-815; PMID:8254673; http://dx.doi.org/ 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- [30].Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2010. [Google Scholar]