

Summary
Hereditary angioedema (HAE) is a rare disease associated with either a quantitative or qualitative deficiency in C1‐inhibitor (C1‐INH) or normal C1‐INH. HAE with normal C1‐INH is associated in 20% of cases with mutations in the gene for factor XII (FXII) or FXII‐HAE. A recent review described 41 families, including 14 German and 15 Spanish families. We have constructed a register of French patients and their characteristics. A national survey was launched through the French National Center of Reference for Angioedema (CREAK) to study the clinical, biological and therapeutic characteristics of patients with HAE linked to a mutation of FXII gene. Fifty‐seven patients were identified from 24 different families. In most cases they were young women (mean age at diagnosis: 31 years, mean age at first symptom: 21 years, female/male ratio: 76%). Twenty‐one per cent of the patients experienced angioedema attacks only during pregnancy or when on oestrogen contraception. Sixty‐three per cent had attacks at all times, but they were more severe during these same periods. Male carriers of the mutation were more frequently asymptomatic than females (P = 0·003). C1‐INH concentrate and icatibant were both effective for treating attacks. The prophylactic use of tranexamic acid led to a 64% decrease in the number of attacks. This is one of the largest series reported of HAE patients with FXII mutation. The therapeutic management appeared to be identical to that of HAE with C1‐INH deficiency.
Keywords: bradykinin, C1 inhibitor, factor XII, hereditary angioedema
Introduction
Hereditary angioedema (HAE) is a genetic disease linked most often to a mutation of the gene SERPING1 encoding the C1‐inhibitor (C1‐INH), with type I HAE being quantitative C1‐INH deficiency, and type II functional C1‐INH deficiency. Since 2000, a new form of HAE has been identified with normal C1‐INH which has a similar clinical phenotype to HAE with C1‐INH deficiency (C1‐INH‐HAE). Bork et al. 1 and Binkley and Davis 2 described this type III HAE as simultaneously affecting primarily women and very sensitive to oestrogenic changes (pregnancy, oestrogen–progestin contraception, puberty, etc.). Bradykinin is one of the main mediators implicated in these pathologies 3.
In 2006 two missense mutations (p.Thr309Lys and p.Thr309Arg) were detected for the first time in the gene for factor XII (FXII), encoding for the ‘Hageman factor’ in a German population 4, 5. Subsequently, several family studies reported the presence of this symptomatic mutation 4, 6, 7, 8, 9, corresponding to approximately 150 reported cases of HAE with FXII mutations (FXII‐HAE). Depending on the series, the prevalence of the FXII mutation within HAE families without C1‐INH deficiency would be in the order of 20–24·5% 10, 11. Other FXII mutations have been described recently in Turkish (deletion c.971_1018 + 24del72) and Caucasian (c.892_909dup) families 12. Nevertheless, the p.Thr309Lys mutation remains the most common 6.
Several diagnostic and therapeutic aspects need to be clarified in the management of this type of HAE. The aim of this study was to describe the clinical and laboratory characteristics and the therapeutic management of patients with FXII‐HAE followed by the French National Centre of Reference for Angioedema (CREAK).
Methods
Study design
All members of CREAK were contacted to register retrospectively and describe all patients followed for FXII‐HAE since 2006. All patients have or had recurrent angioedema unresponsive to anti‐histamines and without cutaneous urticaria. They all had normal C1‐INH laboratory results (activity and concentration), along with the identification of a mutation in the FXII gene. We collected data regarding age, gender, family history and index case, ethnicity, age at first symptoms and age at diagnosis, delay in diagnosis, location, frequency and intensity of acute angioedema episodes (called ‘attacks’, characterized by AE described by the patient or seen by a members of CREAK), treatment of attacks and preventive treatment, treatment response and any link with oestrogens.
Biochemical assays
Nephelometry was used to assay serum C1‐INH and complement C4 fraction using an automated analyser (BNII; Siemens, Saint‐Denis, France) and specific anti‐sera (Siemens). The laboratory results considered as normal were: C1‐INH concentration: 210–355 mg/l; C4 fraction: 100–380 mg/l; C1‐INH: activity: 17·2–27·4 U/ml.
Genetic study
All patients were genotyped at the time of diagnosis with standardized methods. After DNA extraction and polymerase chain reaction (PCR), amplification direct sequencing was performed. Detection of the p.Thr328Lys mutation (Sanger sequencing of exon 9 and flanking introns) confirmed the diagnosis.
Statistical analysis
Data were analysed with xlstat 2015 version 1·01 software using non‐parametric tests due to low sample size. Comparisons of continuous quantitative variables were made using a Mann–Whitney U‐test and correlations were determined with Pearson's correlation coefficient. A P‐value less than 0·05 was considered as statistically significant.
Results
We found 57 patients, belonging to 24 different families, with a mutation in the FXII gene (Table 1). The main families are described in Fig. 1. The majority were women (46 of 57 patients, 76%) and among the symptomatic cases 36 of 38 (95%) were female. Most patients (91%) described attacks affecting the face, the extremities (87%) and/or the abdominal region (80%). Seventy‐four per cent of patients reported episodes affecting the ears, nose and throat (ENT). Episodes affecting the urinary system were less common (1·9%). When possible, the genetic study of the family of the index case found the mutation in 42% (33 subjects) of tested subjects. However, the presence of the mutation in these subjects was frequently asymptomatic (19 of 33 subjects, 56%). For eight patients no family history of angioedema was found (sporadic cases in patients 3, 12, 13, 14, 16, 19, 21 and 23, Table 1) (eight of 24, 33%).
Table 1.
Clinical characteristics of the 57 patients with FXII gene mutation
| Patient no. | Sex/age (years) | Familial origin | Age at diagnosis (years) | Delay in diagnosis (years) | Family history of AE | Localization of attacks | Annual frequency of attacks | Sensitivity to oestrogen | Oestrogen‐dependent |
|---|---|---|---|---|---|---|---|---|---|
| 1‐1 | F/38 | North African | 36 | 14 | Yes | Face, ENT, abdominal | 4 | Yes | No |
| 1‐2 | F/16 | North African | 4 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 1‐3 | M/12 | North African | 4 | n.a. | Yes | Abdominal | 1 | No | No |
| 1‐4 | M/6 | North African | 3 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 1‐5 | F/36 | North African | 34 | 25 | Yes | ENT, face, abdominal | 6 | Yes | No |
| 1‐6 | F/32 | North African | 30 | 15 | Yes | ENT, face, abdominal | 4 | Yes | No |
| 1‐7 | M/12 | North African | 10 | 4 | Yes | Abdominal | 1 | No | No |
| 2 | F/37 | North African | 31 | 2 | yes, not tested | Face, cutaneous, abdominal | 1 | Yes | No |
| 3 | F/70 | Caucasian | 66 | 39 | no | Face, ENT, cutaneous | 3 | No | Yes |
| 4‐1 | F/25 | Caucasian | 18 | 0 | Yes | Face, ENT, cutaneous, | 8 | No | Yes |
| 4‐2 | M/51 | Caucasian | 50 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 4‐3 | M/2 | Caucasian | 1 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 5‐1 | F/34 | Caucasian | 29 | 0 | Yes | Face, ENT, cutaneous, abdominal | 4 | Yes | Yes |
| 5‐2 | F/56 | Caucasian | 52 | 27 | Yes | ENT, cutaneous, abdominal | 13 | Yes | No |
| 5‐3 | F/31 | Caucasian | 27 | 5 | Yes | Abdominal | 4 | Yes | No |
| 5‐4 | F/27 | Caucasian | 25 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 5‐5 | M/45 | Caucasian | 43 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 5‐6 | F/22 | Caucasian | 20 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 6‐1 | F/36 | North African | 33 | 13 | Yes | Face, ENT, cutaneous, abdominal, urinary | 28 | Yes | No |
| 6‐2 | F/31 | North African | 27 | 10 | Yes | Face, ENT, cutaneous, abdominal | 3 | Yes | No |
| 7‐1 | F/35 | Caucasian | 31 | 23 | Yes | Face, cutaneous, abdominal | 2 | Yes | Yes |
| 7‐2 | F/67 | Caucasian | 65 | 40 | Yes | Extremities, face | 0 | No | No |
| 7‐3 | F/6 | Caucasian | 6 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 8 | F/35 | North African | 33 | 8 | Yes, not tested | Face, ENT, cutaneous, abdominal | 1 | Yes | No |
| 9‐1 | F/34 | North African | 28 | 1 | Yes | Face, cutaneous, abdominal | 3 | No | No |
| 9‐2 | M/31 | North African | 29 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 9‐3 | M/62 | North African | 60 | n.a. | Yes | asymptomatic | 0 | n.a. | n.a. |
| 10‐1 | F/39 | North African | 32 | 7 | Yes | Face, cutaneous | 1 | Yes | No |
| 10‐2 | M/66 | North African | 64 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 10‐3 | M/34 | North African | 32 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 10‐4 | F/36 | North African | 29 | 4 | Yes | Face, cutaneous, abdominal | 0 | Yes | No |
| 11‐1 | F/50 | North African | 45 | 5 | Yes | Face, ENT, cutaneous | 1 | Yes | No |
| 11‐2 | M/46 | North African | 44 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 11‐3 | F/72 | North African | 70 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 12 | F/29 | North African | 26 | 1 | no | ENT, face, abdominal, cutaneous, bladder | 40 | Yes | No |
| 13 | F/54 | Caucasian | 48 | 34 | no | Face, ENT, cutaneous, abdominal, bladder | 3 | Yes | No |
| 14 | F/38 | Caucasian | 37 | 8 | no | Face, ENT | 1 | Yes | No |
| 15‐1 | F/25 | Caucasian | 19 | 1 | Yes | Face, bladder, abdominal | 1 | Yes | Yes |
| 15‐2 | F/22 | Caucasian | 18 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 16 | F/25 | North African | 24 | 8 | no | Face, cutaneous, ENT, abdominal | 2 | Yes | No |
| 17‐1 | F/43 | Caucasian | 38 | 15 | Yes | Face, ENT, cutaneous, abdominal | 0 | Yes | No |
| 17‐2 | F/38 | Caucasian | 36 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 18‐1 | F/41 | Caucasian | 33 | 4 | Yes | Face, ENT, cutaneous, abdominal | 6 | Yes | No |
| 18‐2 | F/14 | Caucasian | 12 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 18‐3 | F/11 | Caucasian | 10 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 18‐4 | F/38 | Caucasian | 36 | n.a. | Yes | Asymptomatic | 0 | n.a. | n.a. |
| 19 | F/30 | Caucasian | 26 | 14 | no | Face, ENT, cutaneous, abdominal | 2 | Yes | No |
| 20‐1 | F/35 | Caucasian | 33 | 14 | Yes | Face, ENT, cutaneous | 0 | Yes | Yes |
| 20‐2 | F/40 | Caucasian | 38 | 3 | Yes | ENT, abdominal | 0 | Yes | Yes |
| 21 | F/28 | Caucasian | 27 | 10 | No | Face, cutaneous | 3 | Yes | Yes |
| 22‐1 | F/19 | North African | 18 | 0 | Yes | Face, cutaneous, ENT | 3 | Yes | Yes |
| 22‐2 | F/52 | North African | 51 | 28 | Yes | Face, cutaneous | 0 | Yes | Yes |
| 22‐3 | F/48 | North African | 47 | 25 | Yes | Face, cutaneous | 2 | Yes | Yes |
| 22‐4 | F/22 | North African | 21 | 4 | Yes | Face, cutaneous | 3 | Yes | No |
| 23 | F/41 | North African | 40 | 22 | no | Face, ENT, cutaneous, abdominal | 4 | Yes | Yes |
| 24‐1 | F/30 | North African | 25 | 12 | Yes | Face, ENT, cutaneous, abdominal | 4 | Yes | No |
| 24‐2 | F/50 | North African | 46 | 25 | Yes | Face, bladder, abdominal | 0 | Yes | No |
ENT = ears, nose and throat; n.a. = not available; F = female; M= male.
Figure 1.
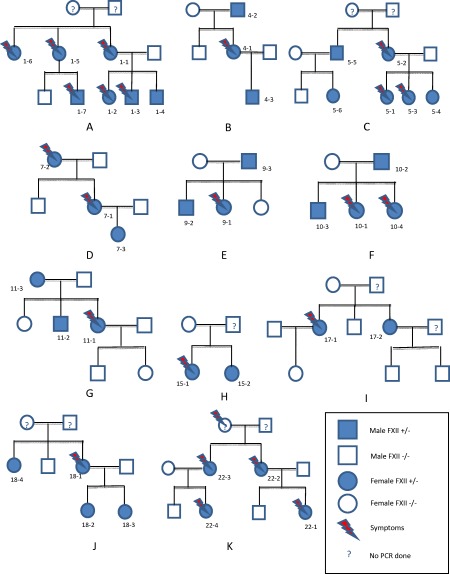
Main families with mutation in the gene encoding for FXII. (a) Family 01; (b) family 04; (c) family 05; (d) family 07; (e) family 09; (f) family 10; (g) family 11; (h) family 15; (i) family 17; (j) family 18; (k) family 22.
The average age of the first angioedema attack in the index case was 21 years (range 8–40 years) with an average delay in diagnosis of 10·6 years (range = 0–39 years). From the systematic genetic investigation, the average age at diagnosis in family members, excluding the index case, was older than the index cases (31·4 years versus 21·6 years, P < 0·05). The annual number of attacks varied considerably from one subject to another (average 5·1 attacks in symptomatic subjects, range = 0–40). There was no difference in the frequency of attacks between the index cases and symptomatic family members. The three patients who presented the highest frequency of attacks were pregnant during this period (patients 5–2, 6–1, 12). No deaths were reported in this cohort. Men carrying the mutation were more often asymptomatic than women (nine of 11 men versus 10 of 46 women were asymptomatic, P = 0·002). No patient reported the appearance of erythema marginatum prior to an attack.
Angioedema attacks were associated frequently with hormonal status (36 of 38 symptomatic patients, 95%). Eight patients (21%) experienced attacks only during pregnancy or when on oestrogen plus progestin contraception (hormone dependence). While 24 patients (67%) had attacks outside these periods, the pregnancy and/or oestrogen plus progestin contraception exacerbated the frequency and/or intensity of attacks (hormone sensitivity). Four patients (two men, 1‐3 and 1‐7 and two women, 7‐2 and 9‐1) experienced attacks unrelated to oestrogens.
The main treatments for acute episodes of angioedema were: tranexamic acid (TA) (six patients, 13 attacks), C1‐INH concentrate (Berinert®) (eight patients, 16 attacks) and icatibant (eight patients, 18 attacks), with comparable efficacy. Few patients received long term prophylaxis, although 10 patients (26%) received prophylactic doses of TA that seemed to be effective, as the frequency of attacks decreased by 64% (average number attacks prior to starting prophylactic treatment: 8·7/year versus 3·8/year during treatment, P = 0·07). Two patients (8; 6‐1) received long‐term C1‐INH concentrate (Berinert®) as a prophylactic.
Discussion
To our knowledge, this is one of the largest series of FXII‐HAE patients. A recent recommendation 13 described 41 families with mutations in the FXII gene, including 14 Spanish and 15 German families.
As described previously in the literature, we observed a variable penetrance of this missense mutation 11, 33·3% of our subjects with the mutation were asymptomatic (mainly men or prepubescent women), compared to between 66 and 33%, depending on the study 5, 13. We noted the absence of a family history in 14·0% of cases in our series, similar to the series of Bork et al. 5 (five of 23 patients, 21·7%). The phenotype of FXII‐HAE in our study is close to that described in the literature (Table 2) 11, except for angioedema affecting the extremities, which was present less often in our cohort than in that of Bork et al. (31 versus 62·2%, respectively, P < 0·01). In 2013, we reported the clinical characteristics of 193 French patients with C1‐INH‐HAE 14. A comparison of FXII‐HAE and C1‐INH‐HAE cohorts showed that the localization of the angioedema was different (Table 2). The face was affected more frequently in FXII‐HAE (38·1 versus 7% in C1‐INH‐HAE, P < 0·05), while the abdomen and ENT seem to be affected more often in C1‐INH‐HAE (57·1 versus 27·1% and 8 versus 1·9%, P = 0·02 and 0·004, respectively). Also the age of onset of symptoms was older in our C1‐INH‐HAE series of symptomatic patients (21 versus 14 years, P = 0·02), in line with other C1‐INH‐HAE series 9, 10, 11. The absence of erythema marginatum could also suggest FXII gene mutation rather than C1‐INH deficiency 15. We found an important proportion of families from North Africa (30 of 57 patients, 52·6%) which has not been described previously 16 and differs from C1‐INH‐HAE, where no ethnic predilection was found 14, 16.
Table 2.
Localization of angioedema (AE) attacks in the 57 patients over 1 year, compared to literature data for AE with FXII mutation 11 and AE with C1‐inhibitor (C1‐INH) deficiency from the French National Center of Reference for Angioedema (CREAK) database published in 2013 14
| Sites | FXII‐HAE in this study | FXII‐HAE in the German study 11 | C1‐INH‐HAE in the French study 14 |
|---|---|---|---|
| Face | 123 (38·1%) | 1506 (37·0%) | 39 (7%) |
| ENT | 6 (1·9%) | 167 (2·6%) | 45 (8%) |
| Abdomen | 88 (27·1%) | 2267 (34·6%) | 324 (57·1%) |
| Extremities | 100 (31%) | 2530 (62·2%) | 241(42·5%) |
| Bladder | 6 (1·9%) | 27 (0·7%) | n.a. |
| Total of all attacks | 323 (100%) | 6547 (100%) | 568 (100%) |
ENT = ears, nose and throat; n.a. = not available.
The oestrogen‐dependent nature of FXII‐HAE is striking. This corresponds with the observation that, among symptomatic subjects, only 5% were males compared with 95% females (P < 0·001). Most attacks (89·5%) were associated with hormonal changes (pregnancy, menstruation, menopause, oestrogen‐progestin contraception). Bork et al. 11 demonstrated the influence of oestrogens in the pathogenesis of FXII‐HAE; the first attack was triggered by an oestrogen plus progestin contraceptive, taken in 63% of cases, or during pregnancy (12%). Nevertheless, patients can develop symptoms outside the context of hyperoestrogenism 15. The obstetric complications described in three families with FXII mutations 17, 18 were reported in one of our patients with the birth of a stillborn during a second pregnancy.
Regarding prophylactic therapy, we noted the efficacy of tranexamic acid (median dose of 2 g/day) in the prevention of recurrent attacks of FXII‐HAE (64% reduction in the number of attacks, P = 0·07) without reported side effects. Tranexamic acid is a competitive inhibitor of plasmin activation and of plasminogen, leading to a decrease in the degradation of fibrin and thus of natural fibrinolysis. Joseph et al. 19 showed recently that FXII‐HAE patients were deficient in PAI‐2, an inhibitor of plasminogen cleavage. We plan to analyse the PAI‐2 levels in our FXII‐HAE cohort. Furthermore, in a previous retrospective study we showed that progestin was an interesting background therapy for patients with all types of HAE 20 (the introduction of progestin in 10 FXII‐HAE patients resulted in the disappearance of AE attacks). During an acute attack in our FXII‐HAE patients, treatment with C1‐INH concentrate appears to be as effective as icatibant, despite normal levels of C1‐INH. The control of the kallikrein–kinin pathway by C1‐INH appears to be concentration‐dependent and without a plateau effect. This is in line with reports in the literature and is reflected in the international recommendations 13, 21. Moreover, C1‐INH concentrate has been validated in pregnant women and for minor patients whereas icatibant was not validated.
Conclusion
We describe one of the largest cohorts, to our knowledge, of FXII‐HAE patients. The specific clinical features are predominant expression in women, major sensitivity to oestrogens and attacks that often affect the face and ENT. Specific treatments of bradykinic angioedema (icatibant, C1‐INH concentrates and tranexamic acid) and non‐specific treatments (such as progestin) are effective in these patients.
Author contributions
A. D., I. B. G. and L. B. conceived and designed the study, A. D. and I. B. G. collected data, A. D., I. B. G. and L. B. wrote the paper and all authors critically revised the manuscript. All authors read and approved the final version of the manuscript.
Disclosure
None of the authors have disclosures with respect to this work.
Acknowledgements
We thank Dr Alison Foote (Grenoble Alpes University Hospital) for translation of the manuscript. No funding was received for this study.
References
- 1. Bork K, Barnstedt S, Koch P, Traupe P. Hereditary angioedema with normal C1‐inhibitor activity in women. Lancet 2000; 356:213–7. [DOI] [PubMed] [Google Scholar]
- 2. Binkley KE, Davis A. Clinical, biochemical, and genetic characterization of a novel estrogen‐dependent inherited form of angioedema. J Allergy Clin Immunol 2000; 106:546–50. [DOI] [PubMed] [Google Scholar]
- 3. Cugno M, Nussberger J, Cicardi M, Agostoni A. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol 2003; 3:311–7. [DOI] [PubMed] [Google Scholar]
- 4. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun 2006; 343:1286–9. [DOI] [PubMed] [Google Scholar]
- 5. Bork K, Gul D, Hardt J, Dewald G. Hereditary angioedema with normal C1 inhibitor: clinical symptoms and course. Am J Med 2007; 120:987–92. [DOI] [PubMed] [Google Scholar]
- 6. Cichon S, Martin L, Hennies HC et al Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet 2006; 79:1098–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martin L, Raison‐Peyron N, Nothen MM, Cichon S, Drouet C. Hereditary angioedema with normal C1 inhibitor gene in a family with affected women and men is associated with the p.Thr328Lys mutation in the F12 gene. J Allergy Clin Immunol 2007; 120:975–7. [DOI] [PubMed] [Google Scholar]
- 8. Prieto A, Tornero P, Rubio M, Fernandez‐Cruz E, Rodriguez‐Sainz C. Missense mutation Thr309Lys in the coagulation factor XII gene in a Spanish family with hereditary angioedema type III. Allergy 2009; 64:284–6. [DOI] [PubMed] [Google Scholar]
- 9. Nagy N, Greaves MW, Tanaka A, McGrath JA, Grattan CE. Recurrent European missense mutation in the F12 gene in a British family with type III hereditary angioedema. J Dermatol Sci 2009; 56:62–4. [DOI] [PubMed] [Google Scholar]
- 10. Vitrat‐Hincky V, Gompel A, Dumestre‐Perard C et al Type III hereditary angiooedema: clinical and biological features in a French cohort. Allergy 2010; 65:1331–6. [DOI] [PubMed] [Google Scholar]
- 11. Bork K, Wulff K, Hardt J, Witzke G, Staubach P. Hereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors and therapy. J Allergy Clin Immunol 2009; 124:129–34. [DOI] [PubMed] [Google Scholar]
- 12. Kiss N, Barabas E, Varnai K et al Novel duplication in the F12 gene in a patient with recurrent angioedema. Clin Immunol 2013; 149:142–5. [DOI] [PubMed] [Google Scholar]
- 13. Cicardi M, Bork K, Caballero T et al (Hereditary Angioedema International Working Group). Evidence‐based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–57. [DOI] [PubMed] [Google Scholar]
- 14. Bouillet L, Launay D, Fain O et al French National Reference Center for Hereditary Angioedema (CREAK). Hereditary angioedema with C1 inhibitor deficiency: clinical presentation and quality of life of 193 French patients. Ann Allergy Asthma Immunol 2013; 111:290. [DOI] [PubMed] [Google Scholar]
- 15. Firinu D, Bafunno V, Vecchione G et al Characterization of patients with angioedema without wheals: the importance of F12 gene screening. Clin Immunol 2015; 157:239–48. [DOI] [PubMed] [Google Scholar]
- 16. Cicardi M, Aberer W, Banerji A et al Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 2014; 69:602–16. [DOI] [PubMed] [Google Scholar]
- 17. Picone O, Donnadieu AC, Brivet FG, Boyer‐Neumann C, Fremeaux‐Bacchi V, Frydman R. Obstetrical complications and outcome in two families with hereditary angioedema due to mutation in the F12 gene. Obstet Gynecol Int 2010; 2010:957507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bouillet L, Ponard D, Rousset H, Cichon S, Drouet C. A case of hereditary angioedema type III presenting with C1‐inhibitor cleavage and a missense mutation in the F12 gene. Br J Dermatol 2007; 156:1063–5. [DOI] [PubMed] [Google Scholar]
- 19. Joseph K, Tholanikunnel BG, Wolf B, Bork K, Kaplan AP. Deficiency of plasminogen activator inhibitor 2 in plasma of patients with hereditary angioedema with normal C1 inhibitor levels. J Allergy Clin Immunol 2016; 137:1822–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saule C, Boccon‐Gibod I, Fain O et al Benefits of progestin contraception in non‐allergic angioedema. Clin Exp Allergy 2013; 43:475–82. [DOI] [PubMed] [Google Scholar]
- 21. Xu YY, Buyantseva LV, Agarwal NS, Olivieri K, Zhi YX, Craig TJ. Update on treatment of hereditary angioedema. Clin Exp Allergy 2013; 43:395–405. [DOI] [PubMed] [Google Scholar]