

Summary
A single determinant factor for autoimmunity does not exist; disease development probably involves contributions from genetics, the environment and immune dysfunction. Type 1 diabetes is no exception. Genomewide‐associated studies (GWAS) analysis in T1D has proved disappointing in revealing contributors to disease prediction; the only reliable marker has been human leucocyte antigen (HLA). Specific HLAs include DR3/DR4/DQ2/DQ8, for example. Because HLA molecules present antigen to T cells, it is reasonable that certain HLA molecules have a higher affinity to present self‐antigen. Recent studies have shown that additional polymorphisms in HLA that are restricted to autoimmune conditions are further contributory. A caveat is that not all individuals with the appropriate ‘pro‐autoimmune’ HLA develop an autoimmune disease. Another crucial component is autoaggressive T cells. Finding a biomarker to discriminate autoaggressive T cells has been elusive. However, a subset of CD4 helper cells that express the CD40 receptor have been described as becoming pathogenic. An interesting function of CD40 on T cells is to induce the recombination‐activating gene (RAG)1/RAG2 T cell receptor recombination machinery. This observation is contrary to immunology paradigms that changes in TCR molecules cannot take place outside the thymic microenvironment. Alteration in TCR, called TCR revision, not only occurs, but may help to account for the development of autoaggressive T cells. Another interesting facet is that type 1 diabetes (T1D) may be more than a single disease; that is, multiple cellular components contribute uniquely, but result ultimately in the same clinical outcome, T1D. This review considers the process of T cell maturation and how that could favor auto‐aggressive T cell development in T1D. The potential contribution of TCR revision to autoimmunity is also considered.
Keywords: autoimmune diabetes, immune dysfunction, TCR revision
Introduction
Type 1 diabetes (T1D), unlike its far more prevalent counterpart, type 2 diabetes, is a classic autoimmune disease characterized by immune cell attacks of pancreatic islets resulting in hyperglycaemia subsequent to insulin loss. Islets are composed of five cell types that produce hormones and enzymes associated with digestion and nutrient uptake. Beta cells constitute approximately 80% of islets and produce insulin; alpha cells produce glucagon, delta cells produce somatostatin‐D, gamma cells produce pancreatic polypeptide and epsilon cells produce ghrelin, the hunger hormone 1. During diabetogenesis immune cells infiltrate islets creating an inflammatory condition called insulitis. This was first described more than 100 years ago in a 6‐year‐old girl who died from complications of ketoacidosis 2. The first cells to infiltrate islets are probably neutrophils 3, followed by antigen‐presenting cells (APC), including macrophages and dendritic cells (DC) 4, 5. In fact, merocytic DC that mediate cross‐presentation of islet antigens to CD8+ and CD4+ cells in pancreatic lymph nodes could be instrumental in initiating disease 6, 7. Eventually, lymphocytes including CD4+ and CD8+, as well as B cells, infiltrate the islets and are presumed to be the primary players in loss of insulin. The cellular phenotypes in animal models have been determined, and some of the cellular phenotypes in human islets are known. However, the aetiology of T1D remains a mystery: why do cells traffic to the islets? Why do cells infiltrate the islets? By what mechanism do these invaders cause loss of insulin?
Development of type 1 diabetes (T1D) requires a series of unfortunate events involving alignment of genetic, environmental and immunological contributors. This argument, in fact, can be made for any autoimmune disease. Developmental parameters for all autoimmune diseases are similar in many ways, although the symptoms and clinical outcomes vary dramatically. It has become clear that no individual contributor can cause diabetes, but each contributor acting in concert establishes danger and ultimately causes disease. Interestingly, rates vary greatly, depending upon geography. For example, incidence is high in Finland and Sardinia at approximately one in 250 8; in the United States the incidence is currently one in 300 by 18 years of age 9; and in Canada, Australia and New Zealand the incidence is much lower, at one per 1750. In China and Venezuela the incidence is approximately one in 100 000. A 2012 study of T1D trends in Europe during a 20‐year period ending in 2008 reported that 22 of 23 centres in 19 countries showed significant incidence increases, specifically reporting a 3·4% annual increase over the entire period 10. Another study reports that incidence in Europe saw a yearly increase rate of 2·8% from 1990 to 1998, increasing to 3·2% and then 3·9% in 2010 11. The increases are being reported in the very young and those with moderate genetic susceptibility 11. More and more environmental factors are believed to be contributory, but major determinants have yet to be defined.
Genetics
Surprisingly, while type 2 diabetes is reaching epidemic levels in the United States and other developed countries, T1D incidence is increasing similarly. Epidemiological patterns indicate that the worldwide incidence has increased by 2–5% during the last 20 years 9. T1D occurs in familial clusters, yet examination of monozygotic twins shows, surprisingly, that concordance is less than 40% 12. The mouse model of T1D, the non‐obese diabetic or NOD mouse, develops intrinsically the same disease characteristics as human T1D, including lymphocytic infiltrates in islets, loss of insulin secretion and hyperglycaemia. NOD colonies are bred to maintain high genetic susceptibility loci multi‐generationally to ensure disease, yet within any given NOD colony disease incidence ranges from 50 to 90% in females and 20 to 50% in males 13, 14, 15. Male and female NOD mice are genetically identical, thus gender bias, perhaps hormonal‐driven, would appear to contribute to disease. Studies show that early‐life microbial exposures determine sex hormone levels that modify progression to autoimmunity in NOD mice 16, 17. Early studies in NOD mice indicate that hormonal imprinting in neonates had an influence on diabetes 18. The situation for human disease is different; gender bias does not occur in human disease, and female to male disease ratios are virtually 1 : 1 19, 20.
GWAS studies
A concerted effort to address genetic contributions to T1D has been attempted using genomewide analytical studies (GWAS). In T1D, greater than 40 susceptibility loci have been identified thus far 21. The most significant indicator is the human leucocyte antigen (HLA). HLA haplotypes vary within diseases such that a specific HLA haplotype associates with a particular autoimmune disease. Many of the associated haplotypes are class II; HLA DRB1*1501 (referred to as DR2 or DR15) and DQB1*0601 (DQ6) associate with multiple sclerosis 22, DRB1*0401 (DR4) and DRB1*0101 (DR1) or DQB1*0302 (DQ8) associate with rheumatoid arthritis 23, 24, etc. The T1D‐associated molecules include DRB1*0301 (DR3), DR4, DQB1*0201 (DQ2) and DQ8 25. Class I alleles in addition to, and independently of, class II alleles associate with T1D. For example A*2402, A*0201, B*1801 and C*0501 were considered predisposing; and A*1101, A*3201, A*6601, B*0702, B*4403, B*3502, C*1601 and C*0401 appear to be protective 26. An intriguing observation was that while DQ2 and DQ8 are linked independently to T1D development, when subjects carry both DQ2 and DQ8 alleles the disease incidence increases drastically 25. Further study demonstrates that a transposition of the DQ2 α chain to associate with the DQ8 β chain occurs, creating a unique HLA‐DQ referred to as trans‐DQ8 25. Thus autoimmune conditions reflect the presence of unique polymorphisms in MHC genes that further promote disease.
GWAS has generated numerous other indicators including, but not limited to, interleukin (IL)‐2R, cytotoxic T lymphocyte antigen (CTLA)‐4, IL‐27, IL‐2, IL‐10, signal transducer and activator of transcription (STAT)‐4, C‐C chemokine receptor type 5 (CCR5) and the lymphocyte marker, CD69 27. Typically, CD69 has been identified as a lymphocyte ‘activation’ marker, but recent studies show that CD69 plays an important decisional role in lymph node migration, cell retention and memory formation 28. It was determined that CD69 and sphingosine‐1‐phosphate receptor 1 (S1PR) expressions are intertwined inversely. S1PR expression is required for lymphocyte egress from secondary lymphoid tissues and thymus 29, and CD69 expression increases as S1PR expression decreases 28, 29. GWAS has been performed on a number of autoimmune diseases in addition to T1D, including lupus, multiple sclerosis, rheumatoid arthritis, coeliac disease and Crohn's colitis 30. After all these studies, thus far the only discriminating factor for disease prediction is the HLA haplotype, which is not absolute. The hope was that GWAS would discover a single gene or gene cluster that would predict likelihood to develop T1D in much the way that BRCA has done for breast cancer. This has not happened. Given that no clearly predictive genes have yet been discovered, one review summarized the question of ‘Have GWASs been a failure?’ with a qualified ‘yes’ 31. The best conclusion from the information provided by GWAS is that autoimmune diseases require immune dysfunction.
Immune contribution
The reason that HLA is linked with autoimmunity is probably because of its function, the presentation of antigen(s) to T cells. As such, HLA molecules dictate immune response outcomes. HLA class I (HLA‐A, B and C) molecules are expressed on almost all cell types while HLA class II molecules are restricted to what are known as professional APC, including B cells, macrophages and DCs 32, and in humans, activated T cells express HLA class II 33. Once professional APCs encounter exogenous antigens, classically a non‐viral antigen, the antigen is taken up via phagocytosis involving membrane engulfment or by receptor mediated uptake; for example, a B cell receptor binding an antigen and the receptor/antigen becoming internalized through membrane invagination. Once internalized the antigen associates with an endosome to be broken down to constituent parts, peptide fragments, nucleotide fragments and lipids, etc. Peptides associate chemically with HLA to be transported to the cell surface for external presentation. HLA molecules are polymorphic, and those individuals unfortunate enough to express HLA haplotypes that can present self‐antigens preferentially are clearly much more at risk of developing disease. In addition it is possible that the autoimmune background generates unique autoimmune‐favouring HLA haplotypes, such as trans‐DQ8 in T1D 25, 34. The physics of antigen presentation leading to a T cell activation event that leads further to inflammation requires that the antigen/HLA association be sufficient to stimulate appropriate T cells. Thus, appropriate T cells carrying T cell receptor (TCR) molecules that can respond to self‐antigens must be available, adding the next ingredient in autoimmunity. Another layer of complexity that could contribute to autoimmunity is post‐translational modification (PTM) of proteins that may generate novel self‐antigens. As much as 90% of proteins produced by mammals undergo some form of post‐translational modification 35. Modifications include glycosylation, phosphorylation, citrullination, acetylation, peroxidation and deamination, etc. 35. Each of these processes occur in the periphery, thus creating an environment for generation of novel antigens; in other words, proteins and peptides that have not been seen in the thymus by developing T cells. Further complicating this mechanism, it is also possible that autoimmune backgrounds have altered PTM mechanisms compared to non‐autoimmune backgrounds. For example, protein glycosylation patterns were different when the protein was isolated from an autoimmune background than when the same protein was examined from a non‐autoimmune background 36, 37. A recently described mechanism for creation of novel epitopes is the hybrid insulin peptides (HIP) model, which is acutely appropriate for T1D. Insulin peptides have been considered potential self‐antigens in T1D for several years 38. The HIP model suggests that during type 1 diabetes insulin peptides hybridize with other proteins, creating a unique set of epitopes that may be capable of activating autoaggressive T cells 39.
Generation of rogue T cells and central tolerance
The immunological rules of inflammation are identical for foreign antigen removal response to self‐antigen‐driven autoaggressive responses: T cells carrying a TCR that have affinity for antigen(s) recognize and react to the presented antigen, leading to production and release of proinflammatory cytokines that establish inflammation. The process by which T cells are generated creates the possibility for development of autoaggression. Pluripotent, immature lymphocytes (designated as double‐negative, DN) migrate to the thymus from the bone marrow. Developing T cells in the thymus acquire TCR molecules after recombination‐activating gene (RAG)1 and RAG2 recombination proteins become induced during the DN stage of development 40, and this action takes place physically in the cortical region (Fig. 1). The architecture of the thymus is similar to that of secondary lymphoid organs, including lymph nodes, composed of a cortical and a medullary region. Once RAGs are induced, the TCR‐β gene is rearranged and the resultant molecule is expressed, associating with a prefabricated α chain. This constitutes the pre‐TCR 41. By as‐yet unknown mechanisms, RAG1 and RAG2 proteins are induced once again and the α locus is rearranged, removing the delta gene locus 42. The newly rearranged α gene is transcribed, translated and the protein derived displaces the pre‐α protein and associates with the β protein to create a potentially functional TCR (αβ). This process happens at least somewhat randomly within each developing T cell, therefore any given T cell is as likely to express a self‐antigen reactive TCR as it is likely to express a foreign‐antigen reactive TCR. At this developmental stage cells have become double‐positive (DP) for expression of CD4 and CD8 and remain in the cortical region (Fig. 1). The newly expressed TCR molecules on DP cells interact with major histocompatibility complex (MHC) molecules, including HLA‐A, B and C, class I molecules, or HLA‐D, class II molecules that are located at the cortico–medullary junction (Fig. 1), to undergo positive selection. Cells that interact successfully are primed for survival; cells that cannot interact with the available HLA molecules undergo death‐by‐neglect (Fig. 1). Positive selection assures that only T cells that are responsive to the available HLA are maintained 43, 44.
Figure 1.
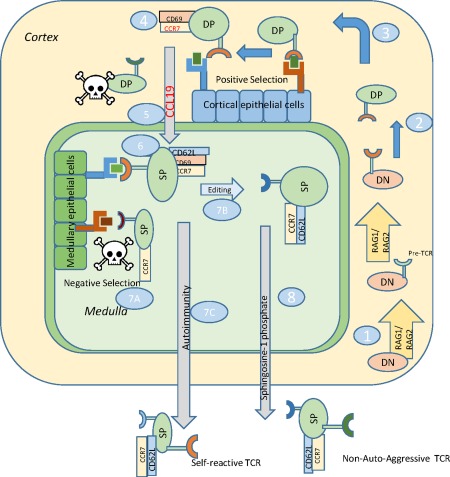
Autoaggressive T cell development in the thymus. 1, Double‐negative (DN) cells enter the thymus and recombination‐activating gene (RAG) proteins become activated to rearrange T cell receptor (TCR) beta locus, cells express pre‐TCR, RAGs become induced again to generate a mature TCR. 2, Developing cells progress to double‐positive (DP) state. 3, DP cells interact with cortical epithelium to undergo positive selection in the thymic cortex. 4, Following TCR interaction with cortical thymic epithelial cells (cTEC), DP cells express CCR7 and CD69. 5, CCL19 is released from the medulla to attract DP cells based on CCR7 expression. 6, DP cells become single‐positive for CD4 or CD8, interact with medullary thymic epithelial cells (mTEC) to undergo negative selection. Cells express CD62L. 7a, cells with high affinity, self‐reactive TCRs are deleted; 7b, some cells undergo TCR editing to alter TCR V alpha expression; 7c, Self‐reactive T cells may escape negative selection or be generated from faulty TCR editing. 8, SP cells with appropriate TCRs exit to the periphery following sphingosine‐1‐phosphate (S1P) concentration gradients.
Successful interactions between TCR and MHC cause the cells to express CD69 and CCR7, a chemokine receptor that interacts with CCL19 45, 46, 47, 48. Chemokine receptors allow cells to migrate towards the source of chemokine, following an upslope concentration gradient. CCR7 expression allows the cells to migrate to the medulla 49. Mice that are deficient in CCR7 or if CCL19 is blocked accumulate immature thymocytes in the cortex 48. CD69 has long been considered a very early ‘activation’ marker, in that TCR engagement induces CD69 rapidly in the periphery as well as in the thymus 50. Therefore, CD69 expression has been associated with successful positive selection 51. While CD69 function is not understood fully, it is now known to be involved in tissue retention 28. When CD69 was over‐expressed in mice, thymocyte development and positive or negative selection were not hampered but cells accumulated in the medulla, much the same as CCR7‐deficient mice, and failed to be exported from the thymus, creating systemic lymphopenia 52. CD69 helps to retain developing cells within the cortical region but CCL19, released from the medulla, over‐rides CD69, allowing cells to migrate to the medullary region 53, 54. Once in the medulla, DP cells complete maturation to the single‐positive, CD4+ or CD8+ stage. In the medulla, cells begin to express CD62L, a selectin involved in lymph node homing 55. While the mechanism of CD62L induction is not yet defined, expression of CD62L is associated with more mature thymocytes 56. Once cells reach the medullary region, epithelial cells (medullary thymic epithelial cells or mTEC) located there express MHC molecules that are presumed to carry a series of self‐antigens. The now positively selected thymocytes interact once again with HLA class I and class II molecules where self‐antigen reactive T cells are presumably removed by activation‐induced cell death if they encounter an antigen to which they have high affinity 57, 58. A transcription factor called AIRE (autoimmune regulator) regulates this process 58. Severe autoimmunity develops in AIRE deficiency 59, 60. Humans with AIRE deficiency experience a multitude of autoimmune diseases 59. As cells complete maturation in the medulla expression of CD69 is lost, while retaining CCR7 and CD62L expression. These phenotypically mature cells can migrate to populate lymph nodes through CCL19 and other signals, and be retained in the node through CD62L interactions.
During the process of negative selection some thymocytes destined for failure have an opportunity for redemption, utilizing a process called TCR editing 61, 62, 63, 64, 65, 66. Rather than a single attempt at TCR generation, which was the prevailing paradigm for many years, self‐reactive thymocytes can be induced to re‐express the RAG proteins, which will once again rearrange TCR genes 64. Again, the mechanism of RAG induction in the thymus is unknown. Receptor editing was first described in B cells, taking place during development in bone marrow 67. TCR editing helps to account for repertoire expansion in the thymus and argues in favour of cell conservation, thus reducing the need for continual migration of bone marrow cells. The particulars of negative selection have not yet been explained fully. How are cells able to interact positively with MHC to undergo positive selection and then the same TCR again interacts with MHC to undergo negative selection? One explanation is that positional kinetics, i.e. positive selection, occurring in the cortex while negative selection occurs in the medulla, create differential signals to the developing T cell, although this hypothesis has not been proven. Another viable option is that different affinity or avidity thresholds between TCR molecules on developing T cells and HLA/MHC promote positive versus negative selection. Autoimmunity can certainly create permissive conditions allowing autoaggressive T cells to escape thymic negative selection known as central tolerance, which has been proposed 68, 69. One concern about central tolerance failure being the only means of generating autoaggression is that the vast majority of thymic output occurs early in life. In mammals, thymic involution, loss of thymic architecture and volume, occurs at or close to puberty. If autoaggressive T cells arise solely by escaping negative selection, then logically autoimmune disease would only onset prior to or soon after puberty. This, however, is not always the case.
In T1D the majority of disease onsets occur in juvenile subjects; however, an ever‐growing population is experiencing onset during the 3rd, 4th, 5th and even 6th decade of life 70. To account for this, either peripheral mechanisms of tolerance are in place that become dysfunctional over time, or an alternative mechanism of autoaggressive T cell development occurs. Another intriguing option is that early in life diabetes onset constitutes one type of disease, perhaps associated closely with central tolerance failure, while disease onset later in life constitutes a different type of disease. The latter case would involve mechanisms to develop autoaggressive T cells independently of thymic control. It has been presumed that TCR editing is the final point in TCR development. To the contrary, we and others demonstrated that T cells are capable of inducing RAG1 and RAG2 proteins in the periphery and, subsequent to that, alter TCR expression 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86. This process is known as TCR revision. While it has not yet been determined what induces RAGs in the thymus, the mechanisms of revision are the same as those for editing; the locale of the T cell, in the periphery as opposed to the thymus, has dictated the name change.
TCR revision
It has been shown that a subset of T cells, both in the thymus and in the periphery, express the CD40 molecule 36, 37, 71, 72, 73, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98. This was somewhat surprising, given that CD40 expression has long been associated with only APC. However, more extensive research demonstrated that CD40 expression is ubiquitous, being expressed on all identified APC, on neural cells including microglia, on adipocytes, on endothelial cells and on T cells, including CD4+ and CD8+ cells 36, 37, 71, 72, 73, 80, 88, 89, 90, 91, 92, 93, 94, 99, 100, 101. CD40‐expressing CD4 cells are referred to as Th40 cells, and have been shown to become highly pathogenic in autoimmune disease models 36, 37, 71, 72, 73, 87, 88, 89, 90, 91, 92, 93, 94, 95, 99. Among its functions, CD40 acts as a co‐stimulus on T cells 37, 87, 88, 91, 92, 93, 94, 99. This indicates that alternative, and heretofore under‐considered, co‐stimulatory molecules occur on T cells. Identifying these molecules could reshape the understanding of T cell biology significantly.
An intriguing and surprising discovery was that CD40 engagement on Th40 cells induced the RAG1/RAG2 TCR recombination machinery 71, 73. This was the first ever demonstration in a primary T cell of a mechanism to induce RAG proteins. RAG1 and RAG2 form heterodimers that interact further with Ku proteins, DNA polymerases and helicases, etc., leading to alteration of TCR expression 71, 72, 73, 91, 102, 103. In the periphery, at least, CD40 interacts directly with the RAG(s) complex in the nucleus 71. Following induction of RAGs, altered expression of TCR‐α 73, 104 and TCR‐β 83, 84, 105 molecules on long‐standing peripheral T cells occurs 71, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 85, 102, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119. A central paradigm of immunology holds that once T cells exit the thymus TCR molecules do not undergo alteration. To the contrary, several laboratories have shown that peripheral T cells re‐express RAG1 and RAG2 proteins and subsequently alter TCR expression 81, 83, 85, 109, 120, 121. Importantly, revised T cells are distinct from recent thymic emigrants 81. There is an evolutionary advantage to TCR revision, which is to generate an expansive T cell repertoire able to respond to a universe of foreign antigens (Fig. 2). Viruses are extremely numerous, exhibiting a survival advantage by being highly mutable. Success of any organism, in this case mammals, at controlling viral infection will rely upon a highly adaptive immune system. The estimated number of individual TCR molecules achievable through RAG‐mediated recombination is vast, approximately 1013 distinct molecules 122. Maintaining and storing such a number of T cells would constitute a Herculean task. A more efficient means of adaptive immunity than generating an enormous number of individual T cells that must be stored and maintained for later use could include TCR revision. This process also could generate a localized subset of responsive T cells. For example, a viral infection at localized tissue would generate several antigens (Fig. 2). TCR revision occurring only on the alpha locus while maintaining the original beta chain could create a selectively diverse subset of T cells (Fig. 2) to facilitate antigen recognition and attack of the infection more effectively. The downside to a highly adaptive immune system is that some individuals, those with the appropriate MHC/HLA, would have an increased risk for autoimmunity. Alteration of TCR in the thymus could facilitate escape of self‐reactive T cells, including cells capable of responding to improperly post‐translationally modified proteins or responding to hybrid protein epitopes. For example, if TCR editing (TCR changes in the thymus are referred to as editing, while alteration of TCR expression in the periphery is called revision) occurs in the wrong compartment, thereby thwarting positional kinetics, i.e. in the medulla after negative selection takes place, then a self‐reactive TCR bearing T cell could escape the thymus. Similarly, a T cell that successfully positively selects, and has low enough affinity or avidity to pass through negative selection, is then revised to possess a high‐affinity self‐reactive TCR cell that poses new danger potential by being able to escape into the periphery.
Figure 2.
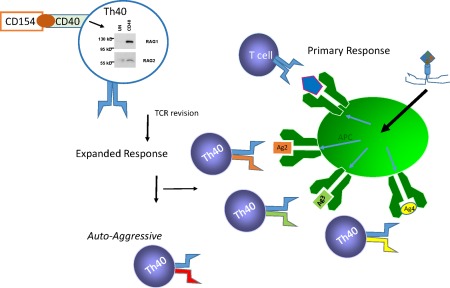
T cell receptor (TCR) revision generates expanded adaptive immunity but may create autoaggressive TCRs. Th40 cells are induced through CD40 engagement to express recombination‐activating genes (RAG)1 and RAG2. Because viruses have multiple antigens and a broader TCR response would facilitate viral clearance, TCR revision provides an economical means to expand the immune response. A negative consequence would be the potential to generate a non‐negatively selected, autoaggressive T cell.
TCR revision opens new avenues for consideration by possibly contributing to the process of autoaggressive T cell generation, but also by providing new potential therapeutic options. Because revision is not restricted to a single event, but rather one Th40 cell can undergo two or even three revisions 71, 72, 73, 107, this process could constitute an unexplored tolerance mechanism. Thus, while revision could promote the generation of a ‘new’ autoaggressive T cell that responds to self‐antigen, additional revision might alter the autoaggressive T cells' TCR expression, making that cell tolerant of self‐antigens.
T1D disease model
The initiation of T1D minimally requires (1) the generation of beta islet self‐antigens (sAg); (2) the presence of appropriate MHC (HLA); and (3) T cells bearing autoaggressive TCR molecules. In prodiabetic conditions neutrophils, which are capable of generating oxidative stress and hence tissue damage, infiltrate the islets early during postnatal development 3. In rodents and humans waves of beta cell death occur, due probably to islet reconstruction during pancreatic development 123, 124, thus creating additional islet sAg sources. While this process occurs under normal conditions, it is probable that the process is exaggerated under autoimmune conditions. (Model: trauma to the islet, including neutrophil action, initiates sAg creation.) Plasmacytoid DC (pDC) and macrophages are recruited to the islet and take up sAg then migrate to pancreatic lymph nodes (LN). Th40 cell numbers in spleen and peripheral LN of young NOD mice are equivalent to non‐autoimmune mice, but in pancreatic LNs Th40 cell numbers are expanded significantly as early as 3 weeks of age 72. Pathogenicity of Th40 cells is demonstrated by their ability to transfer T1D to NOD.severe combined immunodeficient (SCID) recipients 37, 72, 73, 90, 91. Purportedly then, Th40 cells are stimulated in the pancreatic LN and are then recruited to infiltrate islets. Because Th40 cells are capable of TCR revision, the odds of increasing sAg‐reactive T cells on site would be increased dramatically. Th40 cells produce IL‐17 87, 91, 94 that can act as a neutrophil attractant 125, 126 and produce interferon (IFN)‐γ that drives diabetogenesis.
An issue in T1D is why is disease onset so disparate? Some individuals experience onset as young children or juveniles, while others do not experience onset until adulthood. Potential explanations are that all conditions for disease onset, including pre‐existing autoaggressive T cells, are present in juveniles, but not in adults. We hypothesize that certain T cells, specifically Th40, alter TCR usage over time to become autoaggressive (TCR revision). While the conditions to create such cells would necessarily be determined genetically, in some individuals those T cells either do not yet exist or are regulated successfully through tolerance mechanisms that eventually fail. A central paradigm of tolerance is the control of the CD40–CD154 dyad 127, 128. Multiple studies demonstrate that controlling CD40‐mediated signals is tolerogenic, including in T1D 72, 90, 129. Consequently, disruption of the CD40/CD154 balance becomes pathogenic. Induced increases in CD40‐bearing cells, specifically Th40 cells, would necessarily promote pathogenesis. When the balance of CD40 was broken by adoptive transfer of high numbers of Th40 cells, diabetes developed 72, 73, 95, 97. Such increases could be mediated by any of a number of infectious agents. A major element to explain why disease onset is so varied may focus upon the generation of autoaggressive T cells. Even with all the other disease‐specific criteria, e.g. sufficient sAg, appropriate HLA for presenting antigen, etc., until autoaggressive T cells develop in sufficient numbers or are able to break tolerance, disease will not onset. If CD40 were engaged purposefully on autoaggressive T cells, at the proper time it may be possible to forestall T1D onset by altering autoaggressive TCR expression.
Disclosure
D. H. W. is Chief Scientific Officer for Op‐T‐Mune, Inc., Denver, CO, USA and owns stock in the company Op‐T‐Mune, Inc., Denver, CO. The current value of the company is less than US$10 000. D. H. W. has received funding for research but only from Federal and State Agencies, not from private companies.
Acknowledgements
The author declares grant support from the following: the American Diabetes Association and NIDDK.
References
- 1. Elayat AA, el‐Naggar MM, Tahir M. An immunocytochemical and morphometric study of the rat pancreatic islets. J Anat 1995; 186:629–37. [PMC free article] [PubMed] [Google Scholar]
- 2. In't Veld P. Insulitis in human type 1 diabetes: the quest for an elusive lesion. Islets 2011; 3:131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diana J et al Crosstalk between neutrophils, B‐1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med 2013; 19:65–73. [DOI] [PubMed] [Google Scholar]
- 4. Hanenberg H et al Macrophage infiltration precedes and is a prerequisite for lymphocytic insulitis in pancreatic islets of pre‐diabetic BB rats. Diabetologia 1989; 32:126–34. [DOI] [PubMed] [Google Scholar]
- 5. Uno S et al Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor‐alpha in patients with recent‐onset type 1 diabetes. Diabetologia 2007; 50:596–601. [DOI] [PubMed] [Google Scholar]
- 6. Katz JD, Janssen EM. Breaking T cell tolerance to beta cell antigens by merocytic dendritic cells. Cell Mol Life Sci 2011; 68:2873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katz JD et al Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in nonobese diabetic mouse diabetes. J Immunol 2010; 185:1999–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borchers AT, Uibo R, Gershwin ME. The geoepidemiology of type 1 diabetes. Autoimmun Rev 2010; 9:A355–65. [DOI] [PubMed] [Google Scholar]
- 9. Maahs D et al Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 2010; 39:481–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patterson CC et al Trends in childhood type 1 diabetes incidence in Europe during 1989‐2008: evidence of non‐uniformity over time in rates of increase. Diabetologia 2012; 55:2142–7. [DOI] [PubMed] [Google Scholar]
- 11. Vehik K, Dabelea D. The changing epidemiology of type 1 diabetes: why is it going through the roof? Diabetes Metab Res Rev 2011; 27:3–13. [DOI] [PubMed] [Google Scholar]
- 12. Knip M et al Environmental triggers and determinants of type 1 diabetes. Diabetes 2005; 54 Suppl 2:S125–36. [DOI] [PubMed] [Google Scholar]
- 13. Grattan M et al Congenic mapping of the diabetogenic locus Idd4 to a 5.2‐cM region of chromosome 11 in NOD mice: identification of two potential candidate subloci. Diabetes 2002; 51:215–23. [DOI] [PubMed] [Google Scholar]
- 14. Casteels KM et al Sex difference in resistance to dexamethasone‐induced apoptosis in NOD mice: treatment with 1,25(OH)2D3 restores defect. Diabetes 1998; 47:1033–7. [DOI] [PubMed] [Google Scholar]
- 15. Mathieu C et al Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia 1994; 37:552–8. [DOI] [PubMed] [Google Scholar]
- 16. Markle JG et al Microbiome manipulation modifies sex‐specific risk for autoimmunity. Gut Microbes 2014; 5:485–93. [DOI] [PubMed] [Google Scholar]
- 17. Markle JG et al Sex differences in the gut microbiome drive hormone‐dependent regulation of autoimmunity. Science 2013; 339:1084–8. [DOI] [PubMed] [Google Scholar]
- 18. Hawkins T, Gala RR, Dunbar JC. The effect of neonatal sex hormone manipulation on the incidence of diabetes in nonobese diabetic mice. Proc Soc Exp Biol Med 1993; 202:201–5. [DOI] [PubMed] [Google Scholar]
- 19. Harjutsalo V, Forsblom C, Groop PH. Time trends in mortality in patients with type 1 diabetes: nationwide population based cohort study. BMJ 2011; 343:d5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet 2008; 371:1777–82. [DOI] [PubMed] [Google Scholar]
- 21. Bergholdt R et al Identification of novel type 1 diabetes candidate genes by integrating genome‐wide association data, protein‐protein interactions, and human pancreatic islet gene expression. Diabetes 2012; 61:954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hillert J, Olerup O. Multiple sclerosis is associated with genes within or close to the HLA‐DR‐DQ subregion on a normal DR15,DQ6,Dw2 haplotype. Neurology 1993; 43:163–8. [DOI] [PubMed] [Google Scholar]
- 23. Angelini G et al Analysis of HLA DP, DQ, and DR alleles in adult Italian rheumatoid arthritis patients. Hum Immunol 1992; 34:135–41. [DOI] [PubMed] [Google Scholar]
- 24. Wordsworth BP et al HLA‐DR4 subtype frequencies in rheumatoid arthritis indicate that DRB1 is the major susceptibility locus within the HLA class II region. Proc Natl Acad Sci USA 1989; 86:10049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Lummel M et al Type 1 diabetes‐associated HLA‐DQ8 transdimer accommodates a unique peptide repertoire. J Biol Chem 2012; 287:9514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noble JA et al HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes 2010; 59:2972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bakay M, Pandey R, Hakonarson H. Genes involved in type 1 diabetes: an update. Genes (Basel) 2013; 4:499–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mackay LK et al Cutting edge: CD69 interference with sphingosine‐1‐phosphate receptor function regulates peripheral T cell retention. J Immunol 2015; 194:2059–63. [DOI] [PubMed] [Google Scholar]
- 29. Shiow LR et al CD69 acts downstream of interferon‐alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006; 440:540–4. [DOI] [PubMed] [Google Scholar]
- 30. Lettre G, Rioux JD. Autoimmune diseases: insights from genome‐wide association studies. Hum Mol Genet 2008; 17:R116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Visscher PM et al Five years of GWAS discovery. Am J Hum Genet 2012; 90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parker DC. T cell‐dependent B cell activation. Annu Rev Immunol 1993; 11:331–60. [DOI] [PubMed] [Google Scholar]
- 33. Casati C et al Human lymphocyte activation gene‐3 molecules expressed by activated T cells deliver costimulation signal for dendritic cell activation. J Immunol 2008; 180:3782–8. [DOI] [PubMed] [Google Scholar]
- 34. Michels AW et al Structure‐based selection of small molecules to alter allele‐specific MHC class II antigen presentation. J Immunol 2011; 187:5921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol 2012; 24:112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vaitaitis GM, Wagner DH Jr. Galectin‐9 controls CD40 signaling through a Tim‐3 independent mechanism and redirects the cytokine profile of pathogenic T cells in autoimmunity. PLOS ONE 2012; 7:e38708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vaitaitis GM, Wagner DH Jr. CD40 glycoforms and TNF‐receptors 1 and 2 in the formation of CD40 receptor(s) in autoimmunity. Mol Immunol 2010; 47:2303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol 2008; 20:111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Delong T et al Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016; 351:711–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yui MA, Rothenberg EV. Deranged early T cell development in immunodeficient strains of nonobese diabetic mice. J Immunol 2004; 173:5381–91. [DOI] [PubMed] [Google Scholar]
- 41. von Boehmer H et al Thymic selection revisited: how essential is it? Immunol Rev 2003; 191:62–78. [DOI] [PubMed] [Google Scholar]
- 42. Krangel MS. Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol 2009; 21:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matsuki Y et al Different role of Apaf‐1 in positive selection, negative selection and death by neglect in foetal thymic organ culture. Scand J Immunol 2002; 56:174–84. [DOI] [PubMed] [Google Scholar]
- 44. Cilio CM et al Cytotoxic T lymphocyte antigen 4 is induced in the thymus upon in vivo activation and its blockade prevents anti‐CD3‐mediated depletion of thymocytes. J Exp Med 1998; 188:1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nitta T et al CCR7‐mediated migration of developing thymocytes to the medulla is essential for negative selection to tissue‐restricted antigens. Proc Natl Acad Sci USA 2009; 106:17129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nitta T et al Thymic microenvironments for T‐cell repertoire formation. Adv Immunol 2008; 99:59–94. [DOI] [PubMed] [Google Scholar]
- 47. Kurobe H et al CCR7‐dependent cortex‐to‐medulla migration of positively selected thymocytes is essential for establishing central tolerance. Immunity 2006; 24:165–77. [DOI] [PubMed] [Google Scholar]
- 48. Ueno T et al CCR7 signals are essential for cortex‐medulla migration of developing thymocytes. J Exp Med 2004; 200:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ueno T et al Role for CCR7 ligands in the emigration of newly generated T lymphocytes from the neonatal thymus. Immunity 2002; 16:205–18. [DOI] [PubMed] [Google Scholar]
- 50. Paz Morante M et al Activation‐associated phenotype of CD3 T cells in acute graft‐versus‐host disease. Clin Exp Immunol 2006; 145:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guyden JC, Pezzano M. Thymic nurse cells: a microenvironment for thymocyte development and selection. Int Rev Cytol 2003; 223:1–37. [DOI] [PubMed] [Google Scholar]
- 52. Feng C et al A potential role for CD69 in thymocyte emigration. Int Immunol 2002; 14:535–44. [DOI] [PubMed] [Google Scholar]
- 53. Yin X et al CCR7 expression in developing thymocytes is linked to the CD4 versus CD8 lineage decision. J Immunol 2007; 179:7358–64. [DOI] [PubMed] [Google Scholar]
- 54. Kwan J, Killeen N. CCR7 directs the migration of thymocytesinto the thymic medulla. J Immunol 2004; 172:3999–4007. [DOI] [PubMed] [Google Scholar]
- 55. Brinkman CC et al Peripheral tissue homing receptors enable T cell entry into lymph nodes and affect the anatomical distribution of memory cells. J Immunol 2013; 191:2412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carlson CM et al Kruppel‐like factor 2 regulates thymocyte and T‐cell migration. Nature 2006; 442:299–302. [DOI] [PubMed] [Google Scholar]
- 57. Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol 2003; 21:139–76. [DOI] [PubMed] [Google Scholar]
- 58. Shi Y, Zhu M. Medullary thymic epithelial cells, the indispensable player in central tolerance. Sci China Life Sci 2013; 56:392–8. [DOI] [PubMed] [Google Scholar]
- 59. Michels AW, Gottlieb PA. Autoimmune polyglandular syndromes. Nat Rev Endocrinol 2010; 6:270–7. [DOI] [PubMed] [Google Scholar]
- 60. Matsumoto M. The role of autoimmune regulator (Aire) in the development of the immune system. Microbes Infect 2009; 11:928–34. [DOI] [PubMed] [Google Scholar]
- 61. Yang Y, Jacoby E, Fry TJ. Challenges and opportunities of allogeneic donor‐derived CAR T cells. Curr Opin Hematol 2015; 22:509–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Holman PO, Walsh ER, Hogquist KA. The central tolerance response to male antigen in normal mice is deletion and not receptor editing. J Immunol 2003; 171:4048–53. [DOI] [PubMed] [Google Scholar]
- 63. McGargill MA et al A spontaneous CD8 T cell‐dependent autoimmune disease to an antigen expressed under the human keratin 14 promoter. J Immunol 2002; 169:2141–7. [DOI] [PubMed] [Google Scholar]
- 64. McGargill MA, Derbinski JM, Hogquist KA. Receptor editing in developing T cells. Nat Immunol 2000; 1:336–41. [DOI] [PubMed] [Google Scholar]
- 65. Osborn MJ et al Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9 and megaTAL nucleases. Mol Ther 2016; 24:570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lugassy J et al Modulation of TCR responsiveness by the Grb2‐family adaptor, Gads. Cell Signal 2015; 27:125–34. [DOI] [PubMed] [Google Scholar]
- 67. von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol 2010; 11:14–20. [DOI] [PubMed] [Google Scholar]
- 68. Kishimoto H, Sprent J. A defect in central tolerance in NOD mice. Nat Immunol 2001; 2:1025–31. [DOI] [PubMed] [Google Scholar]
- 69. Kishimoto H, Sprent J. The thymus and negative selection. Immunol. Res 2000; 21:315–23. [DOI] [PubMed] [Google Scholar]
- 70. Schwartz SS et al The time is right for a new classification system for diabetes: rationale and implications of the beta‐cell‐centric classification schema. Diabetes Care 2016; 39:179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vaitaitis GM, Wagner DH Jr. CD40 interacts directly with RAG1 and RAG2 in autoaggressive T cells and Fas prevents CD40‐induced RAG expression. Cell Mol Immunol 2013; 10:483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Waid DM, Vaitaitis GM, Wagner DH Jr. Peripheral CD4loCD40+ auto‐aggressive T cell expansion during insulin‐dependent diabetes mellitus. Eur J Immunol 2004; 34:1488–97. [DOI] [PubMed] [Google Scholar]
- 73. Vaitaitis GM et al Cutting edge: CD40‐induced expression of recombination activating gene (RAG) 1 and RAG2: a mechanism for the generation of autoaggressive T cells in the periphery. J Immunol 2003; 170:3455–9. [DOI] [PubMed] [Google Scholar]
- 74. Higdon LE et al Receptor revision in CD4 T cells is influenced by follicular helper T cell formation and germinal‐center interactions. Proc Natl Acad Sci USA 2014; 111:5652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Simmons KB et al Modulation of TCRbeta surface expression during TCR revision. Cell Immunol 2012; 272:124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hale JS et al Bcl‐2‐interacting mediator of cell death influences autoantigen‐driven deletion and TCR revision. J Immunol 2011; 186:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hale JS, Wubeshet M, Fink PJ. TCR revision generates functional CD4+ T cells. J Immunol 2010; 185:6528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hale JS, Fink PJ. T‐cell receptor revision: friend or foe? Immunology 2010; 129:467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hale JS et al Cutting edge: Rag deletion in peripheral T cells blocks TCR revision. J Immunol 2010; 184:5964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cooper CJ et al Cutting edge: TCR revision occurs in germinal centers. J Immunol 2004; 173:6532–6. [DOI] [PubMed] [Google Scholar]
- 81. Cooper CJ et al T cell receptor revision does not solely target recent thymic emigrants. J Immunol 2003; 171:226–33. [DOI] [PubMed] [Google Scholar]
- 82. Ali M et al Differential regulation of peripheral CD4+ T cell tolerance induced by deletion and TCR revision. J Immunol 2003; 171:6290–6. [DOI] [PubMed] [Google Scholar]
- 83. McMahan CJ, Fink PJ. Receptor revision in peripheral T cells creates a diverse V beta repertoire. J Immunol 2000; 165:6902–7. [DOI] [PubMed] [Google Scholar]
- 84. Fink PJ, McMahan CJ. Lymphocytes rearrange, edit and revise their antigen receptors to be useful yet safe. Immunol Today 2000; 21:561–6. [DOI] [PubMed] [Google Scholar]
- 85. McMahan CJ, Fink PJ. RAG reexpression and DNA recombination at T cell receptor loci in peripheral CD4+ T cells. Immunity 1998; 9:637–47. [DOI] [PubMed] [Google Scholar]
- 86. Amrani A et al CD154‐dependent priming of diabetogenic CD4(+) T cells dissociated from activation of antigen‐presenting cells. Immunity 2002; 16:719–32. [DOI] [PubMed] [Google Scholar]
- 87. Waid DM et al Defining a new biomarker for the autoimmune component of Multiple Sclerosis: Th40 cells. J Neuroimmunol 2014; 270:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vaitaitis GM et al A CD40‐targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia 2014; 57:2366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vaitaitis GM et al An alternative role for Foxp3 as an effector T cell regulator controlled through CD40. J Immunol 2013; 191:717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Carter J et al CD40 engagement of CD4+ CD40+ T cells in a neo‐self antigen disease model ablates CTLA‐4 expression and indirectly impacts tolerance. Eur J Immunol 2012; 42:424–35. [DOI] [PubMed] [Google Scholar]
- 91. Vaitaitis G, Waid DM, Wagner D Jr. The expanding role of TNF‐receptor super family member CD40 (tnfrsf5) in autoimmune disease: focus on Th40 cells. Curr Immunol Rev 2010; 6:130–7. [Google Scholar]
- 92. Waid DM et al Disruption of the homeostatic balance between autoaggressive (CD4+CD40+) and regulatory (CD4+CD25+FoxP3+) T cells promotes diabetes. J Leukoc Biol 2008; 84:431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vaitaitis GM, Wagner DH Jr. High distribution of CD40 and TRAF2 in Th40 T cell rafts leads to preferential survival of this auto‐aggressive population in autoimmunity. PLOS ONE 2008; 3:e2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Waid DM et al A unique T cell subset described as CD4loCD40+ T cells (TCD40) in human type 1 diabetes. Clin Immunol 2007; 124:138–48. [DOI] [PubMed] [Google Scholar]
- 95. Wagner DH Jr. et al Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc Natl Acad Sci USA 2002; 99:3782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wagner DH Jr. et al Increased expression of CD40 on thymocytes and peripheral T cells in autoimmunity: a mechanism for acquiring changes in the peripheral T cell receptor repertoire. Int J Mol Med 1999; 4:231–42. [DOI] [PubMed] [Google Scholar]
- 97. Baker RL, Wagner DH Jr. Haskins K. CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J Autoimmun 2008; 31:385–92. [DOI] [PubMed] [Google Scholar]
- 98. Deng G et al Pro‐inflammatory T‐lymphocytes rapidly infiltrate into the brain and contribute to neuronal injury following cardiac arrest and cardiopulmonary resuscitation. J Neuroimmunol 2014; 274:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Siebert JC et al An analytical workflow for investigating cytokine profiles. Cytometry a 2008; 73:289–98. [DOI] [PubMed] [Google Scholar]
- 100. Kobayashi T et al TRAF6 is required for generation of the B‐1a B cell compartment as well as T cell‐dependent and ‐independent humoral immune responses. PLOS ONE 2009; 4:e4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Munroe ME, Bishop GA. A costimulatory function for T cell CD40. J Immunol 2007; 178:671–82. [DOI] [PubMed] [Google Scholar]
- 102. Wagner DH Jr. Re‐shaping the T cell repertoire: TCR editing and TCR revision for good and for bad. Clin Immunol 2007; 123:1–6. [DOI] [PubMed] [Google Scholar]
- 103. Wagner DH Jr. The specific antigen approach in multiple sclerosis: can it ever be enough? Clin Immunol 2012; 144:139–41. [DOI] [PubMed] [Google Scholar]
- 104. Waid DM, Vaitaitis GM, Wagner DH Jr, Peripheral expansion of CD4loCD40+ auto‐aggressive T cells during insulin‐dependent diabetes mellitus. Eur J Immunol 2004; 34:1488–97. [DOI] [PubMed] [Google Scholar]
- 105. Blish C et al Chronic modulation of the TCR repertoire in the lymphoid periphery. J Immunol 1999; 162:3131–40. [PubMed] [Google Scholar]
- 106. Tsumiyama K, Miyazaki Y, Shiozawa S. Self‐organized criticality theory of autoimmunity. PLoS One 2009; 4:e8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lantelme E et al An in vitro model of T cell receptor revision in mature human CD8+ T cells. Mol Immunol 2008; 45:328–37. [DOI] [PubMed] [Google Scholar]
- 108. Zehn D, Bevan MJ, Fink PJ. Cutting edge: TCR revision affects predominantly Foxp3 cells and skews them toward the Th17 lineage. J Immunol 2007; 179:5653–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Takase M, Kanagawa EM, Kanagawa O. Age‐dependent TCR revision mediated by interaction between alphabeta TCR and self‐antigens. J Immunol 2007; 179:2163–9. [DOI] [PubMed] [Google Scholar]
- 110. Kuklina EM. Revision of the antigen receptor of T‐lymphocytes. Biochemistry (Mosc) 2006; 71:827–37. [DOI] [PubMed] [Google Scholar]
- 111. Huang CY et al Superantigen‐induced TCR alpha locus secondary rearrangement: role in tolerance induction. J Immunol 2002; 168:3259–65. [DOI] [PubMed] [Google Scholar]
- 112. Bohluli B et al Trigeminocardiac reflex during Le Fort I osteotomy: a case‐crossover study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010; 110:178–81. [DOI] [PubMed] [Google Scholar]
- 113. Lazic J et al Immunoglobulin genes and T‐cell receptors as molecular markers in children with acute lymphoblastic leukaemia. Srp Arh Celok Lek 2009; 137:384–90. [DOI] [PubMed] [Google Scholar]
- 114. Zou HY et al Expression of recombination‐activating genes and T cell receptor gene recombination in the human T cell leukemia cell line. Chin Med J (Engl) 2007; 120:410–5. [PubMed] [Google Scholar]
- 115. Huang CY, Sleckman BP, Kanagawa O. Revision of T cell receptor {alpha} chain genes is required for normal T lymphocyte development. Proc Natl Acad Sci USA 2005; 102:14356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bynoe MS et al T cells from epicutaneously immunized mice are prone to T cell receptor revision. Proc Natl Acad Sci USA 2005; 102:2898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rosette C et al The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity 2001; 15:59–70. [DOI] [PubMed] [Google Scholar]
- 118. Lantelme E et al Cutting edge: recombinase‐activating gene expression and V(D)J recombination in CD4+CD3low mature T lymphocytes. J Immunol 2000; 164:3455–9. [DOI] [PubMed] [Google Scholar]
- 119. Rosat JP et al CD1‐restricted microbial lipid antigen‐specific recognition found in the CD8+ alpha beta T cell pool. J Immunol 1999; 162:366–71. [PubMed] [Google Scholar]
- 120. Giachino C, Padovan E, Lanzavecchia A. Re‐expression of RAG‐1 and RAG‐2 genes and evidence for secondary rearrangements in human germinal center B lymphocytes. Eur J Immunol 1998; 28:3506–13. [DOI] [PubMed] [Google Scholar]
- 121. Serra P et al RAG‐dependent peripheral T cell receptor diversification in CD8+ T lymphocytes. Proc Natl Acad Sci USA 2002; 99:15566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nikolich‐Zugich J, Slifka MK, Messaoudi I. The many important facets of T‐cell repertoire diversity. Nat Rev Immunol 2004; 4:123–32. [DOI] [PubMed] [Google Scholar]
- 123. Mathis D, Vence L, Benoist C. beta‐Cell death during progression to diabetes. Nature 2001; 414:792–8. [DOI] [PubMed] [Google Scholar]
- 124. Kassem SA et al Beta‐cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 2000; 49:1325–33. [DOI] [PubMed] [Google Scholar]
- 125. Feinen B et al Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal Immunol 2010; 3:312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Watanabe H et al Functional characterization of IL‐17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol 2009; 129:650–6. [DOI] [PubMed] [Google Scholar]
- 127. Karimi MH, Pourfathollah AA. CD40 and tolerance induction. Iran J Allergy Asthma Immunol 2012; 11:1–13. [PubMed] [Google Scholar]
- 128. Quezada SA et al CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol 2004; 22:307–28. [DOI] [PubMed] [Google Scholar]
- 129. Rigby MR et al CD28/CD154 blockade prevents autoimmune diabetes by inducing nondeletional tolerance after effector T‐cell inhibition and regulatory T‐cell expansion. Diabetes 2008; 57:2672–83. [DOI] [PMC free article] [PubMed] [Google Scholar]