

Abstract
In this report, a method to multiplex fluorescence anisotropy measurements is described using frequency encoding. As a demonstration of the method, simultaneous competitive immunoassays for insulin and glucagon were performed by measuring the ratio of bound and free Cy5-insulin and FITC-glucagon in the presence of their respective antibodies. A vertically polarized 635 nm laser was pulsed at 73 Hz and used to excite Cy5-insulin, while a vertically polarized 488 nm laser pulsed at 137 Hz excited FITC-glucagon. The total emission was split into parallel and perpendicular polarizations and collected onto separate photomultiplier tubes. The signals from each channel were demodulated using a fast Fourier transform, resolving the contributions from each fluorophore. Anisotropy calculations were carried out using the magnitude of the peaks in the frequency domain. The method produced the expected shape of the calibration curves with limits of detection of 0.6 and 5 nM for insulin and glucagon, respectively. This methodology could readily be expanded to other biological systems and further multiplexed to monitor increased numbers of analytes.
Keywords: color-blind, immunoassay, binding, polarization, high-throughput
Graphical abstract

Introduction
The availability of a multitude of immunoassay methods has enabled the measurement of peptides and other small molecules in complex mixtures, expanding the number of biological questions that can be posed.1,2 With the advent of microfluidics, these methods have further been adapted for the online measurement of peptide release directly from cells.3–7 In most immunoassay implementations, quantitation is achieved by determining the amount of bound (B) and free (F) antigen (Ag) or labeled antigen (Ag*). Conventionally, most immunoassay methods rely on a separation step to determine the fractions of B and F; however, the separation increases the number of processing steps and the complexity of the assay. To circumvent this step, an alternative scheme based on fluorescence anisotropy (or polarization) can be performed.
Fluorescence anisotropy is a measure of the rotational diffusion of fluorophores during their fluorescence lifetime.8,9 Since the degree of rotation is dependent on the size of the fluorophore, it can be used to obtain the absolute, or relative, change in the molar volume of the fluorescent species. This information is especially valuable in binding assays, where the relative size of a fluorescently-labeled probe changes upon binding. These methods have been used for decades for the quantitation of clinically relevant species via fluorescence polarization or anisotropy ligand binding assays.9–17 In most of these, a competitive immunoassay is performed where the ratio of B/F Ag* is determined, which is indirectly proportional to the amount of Ag. The advantage of using anisotropy immunoassays is that the B/F ratio can be determined without separating the two species.9,12 This is possible because the total anisotropy of the solution is due to the weighted fraction of each species:
| (1) |
here rtot is the total anisotropy of the solution, fB and fF are the fraction of B and F, respectively, and rB and rF are the anisotropy of the B and F species, respectively. Since fB and fF are unique for each concentration of unlabeled Ag, rtot is unique for each concentration of Ag.
To multiplex anisotropy binding assays, different assays are physically separated into parallel wells or channels.7,18,19 As a recent example, fluorescence anisotropy was used with a commercially available microfluidic system that contained 2304 separate chambers to examine the binding of up to 48 protein samples with 48 ligands.18 In another example, an anisotropy imaging system was developed and used to examine 5 parallel assays.19 While the use of microfluidic devices makes these parallel analyses advantageous over conventional multi-well plates, in some cases, it is more desirable to perform the multiplexed assays simultaneously, i.e. multiple assays within a single sample.
In recent years, simultaneous immunoassays have been performed to increase the information content that can be obtained at one time.20–23 These simultaneous assays are performed using multiple antibodies (Ab), each specific to a different Ag, but require a separation step to resolve the various species present. This need for a separation creates a difficulty when using fluorescence anisotropy since no separation is performed. One way to mitigate this difficulty would be to optically separate the individual assays using different fluorophores for each Ag*. With this multi-color approach, further optical components would be required: either increased numbers of detectors, or a rotatable filter wheel. As the number of wavelength channels increase, the system will scale considerably in cost and complexity. In contrast, color-blind detection systems have been used in other multi-wavelength systems to reduce the number of optical components.24–27 These detection systems utilize wavelength-independent methods to discriminate multiple optical signals. Although multiple modes of color-blind detections systems have been described and may be applicable to multi-color fluorescence anisotropy, frequency-encoded detection, which has been employed for DNA sequencing25,26 and multi-color, quantitative infrared-mediated PCR27, was used in this report.
In frequency-encoded detection, lasers specific to the excitation of individual fluorophores are pulsed at non-harmonic frequencies resulting in the emission of the fluorophores at the same frequency.25–27 The total emission from all wavelengths is collected simultaneously on a single photodetector and is then demodulated using Fourier analysis to reveal the magnitude of the individual fluorophore signals. An optical chopper, or a laser capable of being modulated at a particular frequency, is the only optical component that is required to use this methodology. Besides the advantage of utilizing fewer optical components than traditional detection systems, this method also benefits from an increased signal to noise (S/N) due to the inherent filtering during demodulation since the frequency of interest can be isolated from other frequencies that may be present in the frequency domain.
In this report we demonstrate that frequency encoding is a powerful method for multiplexing fluorescence anisotropy detection. The method was tested by performing a simultaneous immunoassay for insulin and glucagon, two hormones involved in blood glucose regulation. In future applications, this method can be extended for online peptide measurements, readily modified for the measurement of other peptides at different wavelengths, or be used for measurement of other species pertinent to different biological systems.
Experimental section
Descriptions of the chemicals and reagents used, the immunoassay conditions, and device fabrication are found in the Supplementary Information.
Instrumentation
The schematic for the instrumental arrangement is provided in the Supporting Information (Figure S-1). The microfluidic device was placed on the stage of an Eclipse TS-100 microscope (Nikon Instruments, Inc., Melville, NY). Excitation was provided by a 635 nm laser (Coherent Inc., Santa Clara, CA) and a 488 nm laser (Melles Griot, Carlsbad, CA). The 635 nm laser was used to excite Cy5-insulin (Ins*) and the 488 nm laser was used to excite FITC-glucagon (Glu*). A variable neutral density filter (ThorLabs, Inc., Newton, NJ) reduced the power of the 635 nm laser from 25 to 2.5 mW and a neutral density filter (0.5 OD) reduced the power of the 488 nm laser from 28 to 8.8 mW. An optical chopper (ThorLabs) was used to modulate the 635 nm light at 73 Hz, and the 488 nm laser was modulated at 137 Hz using a TTL pulse train from a NI-PCIe 6321 card using LabVIEW (National Instruments, Austin, TX) software. The lasers were made collinear by a 505 nm dichroic mirror (ThorLabs) and brought into the back of the microscope by a series of mirrors. A linear polarizer (ThorLabs) placed in the vertical direction was placed at the entrance of the microscope. The incoming polarized excitation light was made incident on a triple band dichroic mirror (XF2054, Omega Optical, Inc., Brattleboro VT), which reflected the light into a 40×, 0.60 NA objective (CFI S Plan Fluor ELWD, Nikon, Melville, NY). The emitted light was collected via the same objective and passed through the dichroic, a quad-bandpass filter (446-523-600-677 Brightline, Semrock, Inc., Rochester, NY), spatial filter, a 635 nm notch filter (ZET635NF, Chroma Technology Corp., Bellow Falls VT), and was split into vertical and horizontal components by a polarizing beam splitter (ThorLabs). The horizontal and vertical portions of the emission where then sent through horizontal and vertical linear polarizers, respectively, before being detected by separate photomultiplier tubes (PMT, R928, Hamamatsu Photonics, Middlesex, NJ). The spatial filter, 635 nm notch, polarizing beam splitter, vertical and horizontal polarizers, and PMTs were housed in a 2-PMT photometer (Photon Technologies, Inc., Birmingham, NJ). Data from the PMTs were acquired at an acquisition rate of 1000 Hz with a NI-PCIe 6321 card using a LabView program written in-house.
Data Analysis
Data was analyzed using Origin (OriginLab, Northampton, MA). A single-sided, fast Fourier transform (FFT) using a rectangular window was performed on 4 discrete, equally spaced segments, that consisted of 2048 data points from each PMT. The magnitude of the peaks of interest (73 Hz and 137 Hz) were then extracted and used to calculate the anisotropy of each analyte by the equation:
| (2) |
where ri is the anisotropy of the 73 or 137 Hz signal, IV is the magnitude of either the 73 or 137 Hz peak in the frequency domain from the PMT that collected the vertical component of the polarization, and IH is the magnitude of either the 73 or 137 Hz peak in the frequency domain from the PMT that collected the horizontal component of the polarization. The average of the anisotropy values collected from the 4 sets of 2048 points are reported with error bars corresponding to +/− 1 standard deviation. Equation 2 does not include a G-factor because only changes in anisotropy (Δr) due to ligand binding were of interest. Immunoassay calibration curves were fitted with a four-parameter logistic model.28,29 Limits of detection were determined by the concentration of insulin or glucagon that would be required to produce an anisotropy value equivalent to the anisotropy value of the blank minus three times the standard deviation of the blank signal.
Results and Discussion
When developing an assay for monitoring temporal signatures, such as would be expected for peptide hormone secretion, it is ideal to measure the parallel and perpendicular components of emission simultaneously. This necessitates the use of multiple detectors, which becomes cumbersome as the number of wavelengths that are monitored is increased. The growth in complexity has repercussions with respect to cost and losses due to inefficient optics. We sought out an alternative colorblind method, frequency-encoded fluorescence detection, to perform simultaneous fluorescence anisotropy immunoassays.
Experimental plan
Competitive immunoassays were performed by mixing a limiting amount of insulin antibody (AbIns) and glucagon antibody (AbGlu) with Ins*, Glu*, and differing amounts of unlabeled insulin and glucagon. The B/F of Ins* and Glu* is then indirectly proportional to the amount of unlabeled peptide in solution. The workflow for determination of Ins* and Glu* B/F is illustrated in Figure 1. Both Glu* and Ins* are present in solution at a particular B/F depending on the concentrations of unlabeled glucagon and insulin. When these concentrations are low, (Figure 1A), Glu* and Ins* exist predominantly in the large, Ab-bound forms and a high B/F is produced. In contrast, the presence of a large amount of unlabeled insulin and glucagon outcompetes Glu* and Ins* for the binding sites, resulting in a low B/F (Figure 1B). To discern these compositional differences, a vertically polarized 488 nm laser (blue arrows) is pulsed at 137 Hz, resulting in emission of Glu* at this same frequency (blue arrows). Simultaneously, a vertically polarized 635 nm laser (red arrows) is pulsed at 73 Hz, prompting emission of Ins* at the same frequency (red arrows). The polarization of the emission light depends on the composition of the immunoassay mixture. A high B/F (Figure 1A) results in retention of the incident polarization of the excitation light, while a low B/F (Figure 1B) results in more depolarized emission light. The total emission is collected, split into vertical and horizontal components, and detected with separate PMTs. The signal from each PMT is deconvoluted and the magnitudes at each frequency are extracted and used to calculate anisotropy of the solution using equation 2. The total anisotropy value is then related to the B/F ratio through equation 1 and can be used to calculate the amount of unlabeled insulin or glucagon.
Figure 1. Frequency-encoded fluorescence anisotropy simultaneous immunoassays.
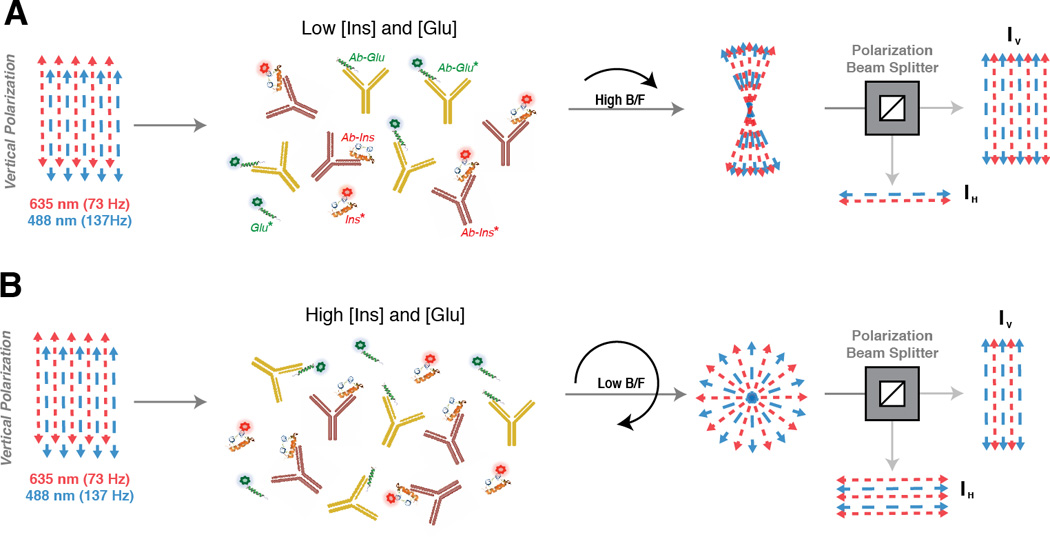
The excitation light is vertically polarized prior to sample excitation and separate channels encoded at different frequencies (red = 73 Hz, blue = 137 Hz), illustrated by different dashed lines. Following emission from the bulk solution, the vertical and horizontal polarizations are split and detected by two PMTs. An illustration of the results expected for low [insulin] and [glucagon] is shown in (A) and high [insulin] and [glucagon] is shown in (B). The extent of depolarization of the bulk emission light is determined by the rotational diffusion of the fluorescent probe, which is dependent on the size of the fluorescent probe. A high B/F occurs in (A) and a low B/F in (B) resulting in different extents of depolarization of the emission light, yielding distinct rtot values upon analysis.
The frequencies of 73 Hz and 137 Hz were selected for both necessary and practical reasons. For example, for frequency encoded fluorescence, the periods of the laser pulses must be longer than the fluorescence lifetimes of the fluorophores, and the pulse frequency must be below the Nyquist frequency of the detection system. To facilitate the measurements, prime numbers were chosen to reduce the possibility of harmonics from elsewhere in the detection system interfering with the signals of interest, high prime numbers were chosen to leave a large gap in the frequency domain between harmonics, and modulating the signal of interest to a higher frequency region avoided low frequency noise.
Since the signals from both fluorophores were collected on the same detector, the dynamic range can limit the range of signals measured. This problem was mitigated somewhat by the choice of fluorophores that were assigned to each peptide. As the concentration of Ins* in the immunoassay experiments was higher than Glu*, due to expected differences in the secretory levels of the peptides, insulin was labeled with Cy5 which has a lower quantum yield than FITC. This allowed both fluorophores to be collected with sufficient signal and without exceeding the dynamic range of the detector.
Characterization of the system
Examples of raw signals from both PMTs are shown in Figure 2A where the signal from the horizontal polarization is colored in black and the vertical polarization in grey. The signal is composed of large magnitude fluctuations from the Glu* and Ins* present and are non-discernable without data processing. The data shown was acquired over a 10 s period. Subsequently, 2048-point analysis windows were analyzed. The windows were centered at time points of 1.03, 3.68, 6.23 and 8.98 seconds. The FFTs obtained from these analysis windows from both the horizontal and vertical channels are shown in Figure 2B and 2C, respectively. As previously mentioned, the excitation channels were encoded at 73 and 137 Hz producing peaks in the frequency domain corresponding to the Ins* and Glu* signals, respectively. The 73 Hz band is slightly wider than the 137 Hz band due to the use of an optical chopper instead of TTL modulation. There were additional peaks at low frequencies (< 10 Hz) and another peak at 219 Hz that was a higher-order harmonic of the 73 Hz signal (not shown).
Figure 2. Data acquisition and analysis flow.
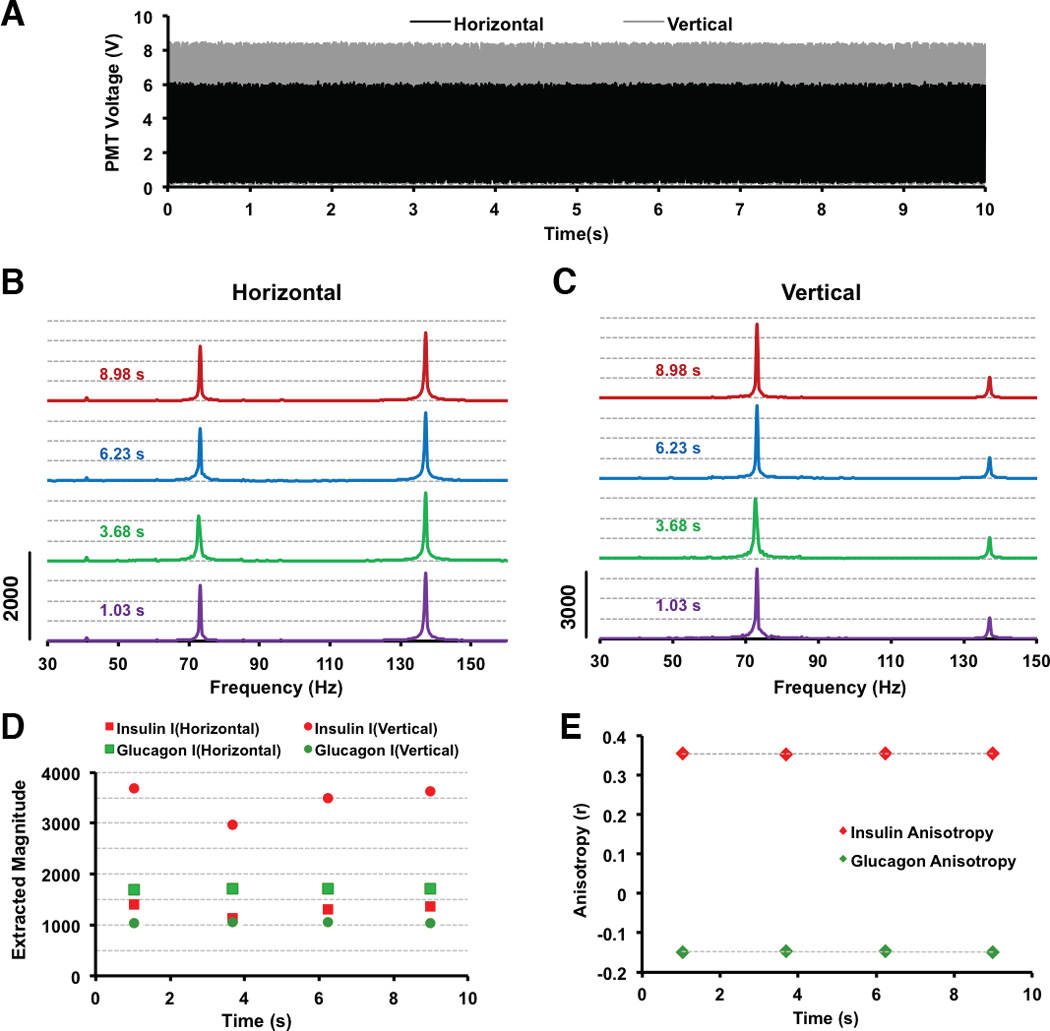
In the data shown, the sample contained Ins* and AbIns at 25 nM each, and Glu* and AbGlu at 7.2 and 22.5 nM, respectively. (A) Raw signal from both PMTs are shown with the signal from the horizontal polarization channel in black and the signal from the vertical polarization channel in grey. (B and C) The Fourier transforms from 2048 points of data acquired in A is shown with the horizontal (B) and vertical (C) channels plotted. The traces are offset for clarity and a scale bar showing the magnitude is on the left. The FFTs were performed at the centered time points annotated above the traces. (D) The individual, extracted magnitudes from the data shown in B and C are plotted as a function of time. (E) The calculated anisotropy values from the data shown in (D) are plotted as a function of time.
Following deconvolution, the magnitude of each peak in the vertical and horizontal channels was plotted as a function of time in Figure 2D and the extracted anisotropy values are shown in Figure 2E. It was found that the magnitudes of the individual fluorophores in the separate channels fluctuated considerably. Across four points shown in Figure 2D, the relative standard deviation (RSD) of the insulin assay was 8.5 and 8.7% for the horizontal and vertical channels, respectively, and 9.1 and 9.3% for the horizontal and vertical channels, respectively, of the glucagon assay. However, the RSD of the anisotropy values (Figure 2E) were 0.43 and 0.87% for insulin and glucagon, respectively. The reason for the lowered error is attributed to the ratiometric nature of the anisotropy measurement. By normalizing the extent of the depolarization to the total signal, any systemic fluctuations during the duration of the acquisition is canceled out. Possible causes for fluctuations in the fluorescence signal include changes in the flow rate or temperature fluctuations. This ratiometric measurement is an effective means to produce robust assays.
The cross-talk of the two dyes was examined and found to be negligible (Figure S-2). A minor note is that the anisotropy values for the glucagon assay are negative, while those for insulin are positive (Figure 2E). We believe this is due to a wavelength-dependent difference in the sensitivities of the two detection PMTs. A G-factor is often used in Equation 2 to account for this difference; however, since we did not use a G-factor we cannot account for these differences in sensitivities. Nevertheless, a decrease in the anisotropy upon an increase in the unlabeled [insulin] or [glucagon] is of the utmost importance, and as will be shown later, this relative change in anisotropy is still observed regardless of a positive or negative initial anisotropy value.
Frequency-encoded multiplexed immunoassay
Prior to examining the use of this system for multiplexing immunoassays, we set out to examine how implementation of frequency encoding may improve the S/N of anisotropy measurements. When extracting the amplitudes for calculation of anisotropy, noise signals are excluded, resulting in a potential improvement in the S/N. To quantify this improvement, a solution containing 25 nM Abins and 25 nM insulin* was delivered to the device and 10 s of data collected using conventional excitation and frequency-encoded excitation. We could only interrogate a single analyte because of the use of the conventional excitation format. For the conventional excitation, anisotropy was calculated for each time point (10,000 data points) and averaged producing a relative standard deviation (RSD) of 2.8%. On another 10 s of data, the lasers were pulsed and four anisotropy values were determined using the frequency-encoded method outlined above. The average of these 4 data points gave an RSD of 0.5%, which is ~5-fold lower than the conventional method and highlights the improved S/N of this method.
To test the ability of frequency encoded fluorescence detection to multiplex these assays, simultaneous immunoassays of insulin and glucagon were performed. In contrast to most multiplexed immunoassays, here a fixed concentration of AbIns, AbGlu, Ins*, and Glu* were mixed into a single solution containing varying concentrations of insulin and glucagon. This mixture was then perfused through the device and the process described in Figures 1 and 2 was performed, producing a measured anisotropy value at each concentration of [insulin] and [glucagon]. The calibration curve shown in Figure 3 was obtained by plotting the measured change of insulin (red) or glucagon (green) anisotropy at each unlabeled [insulin] and [glucagon]. The LODs were 0.6 and 5 nM for insulin and glucagon, respectively, which are comparable to what has been obtained using more conventional methods for insulin or glucagon.3,22,23,30–32
Figure 3. Multiplexed fluorescence anisotropy immunoassay of insulin and glucagon.
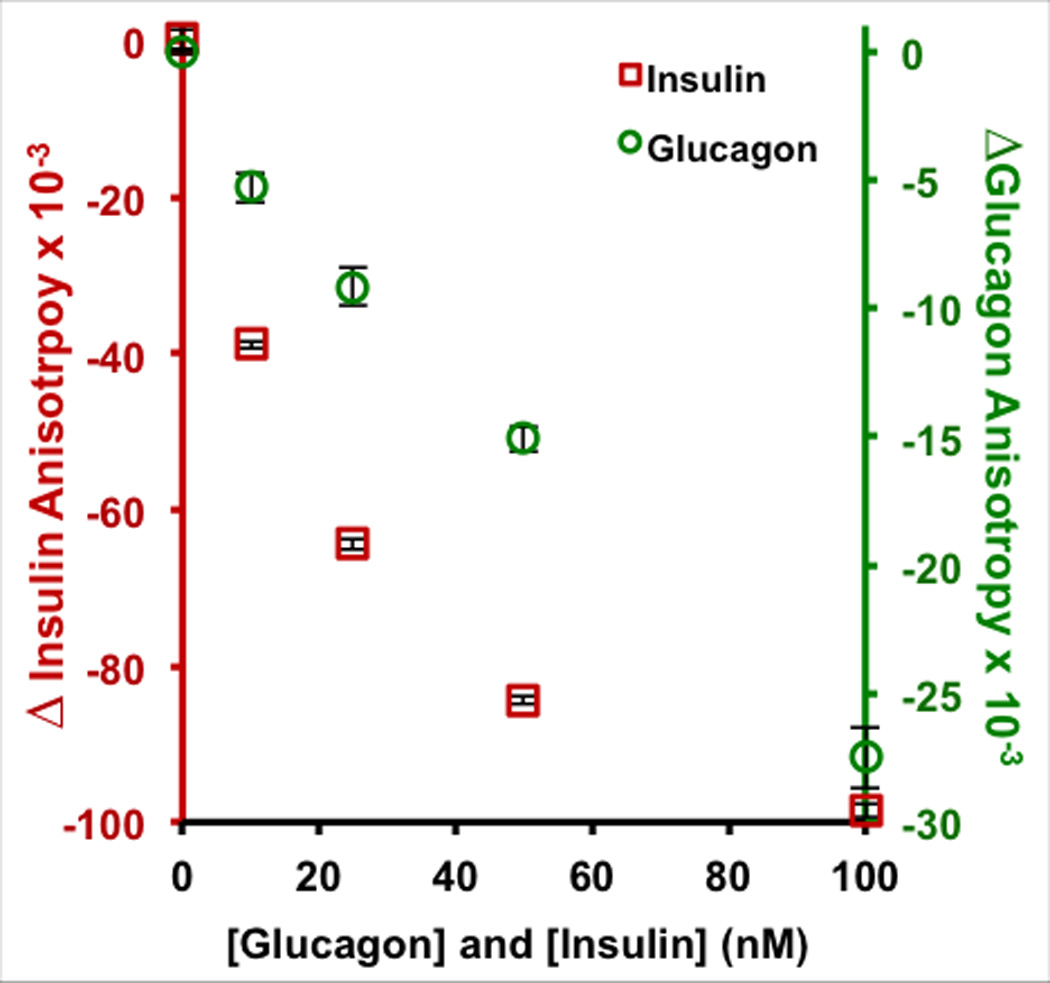
A calibration plot of insulin and glucagon was made simultaneous measuring both signals using standards of 0, 10, 25, 50, and 100 nM insulin and glucagon. The anisotropy values are reported as relative changes (Δr) with insulin plotted in red on the left axis and glucagon in green on the right axis.
Conclusion
In this work, frequency modulation was demonstrated as a method to perform simultaneous fluorescence anisotropy immunoassays. While the application was applied for the possible interfacing with online measurements, in the future, the detection scheme should be readily translatable to numerous other applications, and the number of detection channels assayed can be further increased by adding additional lasers, pulsed at different frequencies for each fluorophore. Particularly, the method could be readily implemented in multi-well plate systems by modulating the control voltage of LED excitation sources with less cost than rapid wavelength scanning systems. Finally, due to the inherent frequency filtering, this methodology results in an increased S/N than typical assays. In this application, the capabilities of this assay were demonstrated by performing a two-analyte immunoassay for insulin and glucagon, but in future iterations, the platform could be applied for the direct measurement of cellular secretions on-chip.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01 DK080714).
Footnotes
Associated Content
The Supporting Information is available free of charge on the ACS Publications website.
Descriptions of the chemicals and reagents, device fabrication, immunoassay conditions, and cross-talk experiments; optical arrangement; FFTs from cross-talk experiments (PDF).
References
- 1.Gosling JP. Clin. Chem. 1990;36:1408–1427. [PubMed] [Google Scholar]
- 2.Bange A, Halsall HB, Heineman WR. Biosens. Bioelectron. 2005;20:2488–2503. doi: 10.1016/j.bios.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Yi L, Wang X, Dhumpa R, Schrell AM, Mukhitov N, Roper MG. Lab Chip. 2015;15:823–832. doi: 10.1039/c4lc01360c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matharu Z, Patel D, Gao Y, Hague A, Zhou Q, Revzin A. Anal. Chem. 2014;86:8865–8872. doi: 10.1021/ac502383e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roper MG, Shackman JG, Dahlgren GM, Kennedy RT. Anal. Chem. 2003;75:4711–4717. doi: 10.1021/ac0346813. [DOI] [PubMed] [Google Scholar]
- 6.Dishinger JF, Kennedy RT. Anal. Chem. 2007;79:947–954. doi: 10.1021/ac061425s. [DOI] [PubMed] [Google Scholar]
- 7.Dishinger JF, Kennedy RT. Electrophoresis. 2008;29:3296–3305. doi: 10.1002/elps.200800067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd. New York: Springer; 2006. pp. 353–382. [Google Scholar]
- 9.Jameson DM, Ross JA. Chem. Rev. 2010;110:2685–2708. doi: 10.1021/cr900267p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gradinaru CC, Marushchak DO, Samim M, Krull UJ. Analyst. 2010;135:452–459. doi: 10.1039/b920242k. [DOI] [PubMed] [Google Scholar]
- 11.Burke TJ, Loniello KR, Beebe JA, Ervin KM. Comb. Chem. High Throughput Screen. 2003;6:183–194. doi: 10.2174/138620703106298365. [DOI] [PubMed] [Google Scholar]
- 12.Dandliker WB, Kelly RJ, Dandliker J, Farquahar J, Levin J. Immunochemistry. 1973;10:219–227. doi: 10.1016/0019-2791(73)90198-5. [DOI] [PubMed] [Google Scholar]
- 13.Urios P, Cittanova N, Jayle MF. FEBS Lett. 1978;94:54–58. doi: 10.1016/0014-5793(78)80905-3. [DOI] [PubMed] [Google Scholar]
- 14.Cobb M, Gotcher S. Am. J. Med. Technol. 1982;48:671–677. [PubMed] [Google Scholar]
- 15.Jolley ME, Stroupe SD, Wang C-HJ, Panas HN, Keegan CL, Schmidt RL, Schwenzer KS. Clin. Chem. 1981;27:1190–1197. [PubMed] [Google Scholar]
- 16.Wang H, Lu M, Tang MS, Van Houten B, Ross JBA, Weinfeld M, Le XC. Proc. Natl. Acad. Sci. USA. 2009;106:12849–12854. doi: 10.1073/pnas.0902281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Song Y, Song M, Wang Z, Li T, Wang H. Anal. Chem. 2009;81:7885–7891. doi: 10.1021/ac901681k. [DOI] [PubMed] [Google Scholar]
- 18.Cheow LF, Viswanathan R, Chin CS, Jennifer N, Jones RC, Guccione E, Quake SR, Burkholder WF. Anal. Chem. 2014;86:9901–9908. doi: 10.1021/ac502605f. [DOI] [PubMed] [Google Scholar]
- 19.Wakao O, Fujii Y, Maeki M, Ishida A, Tani H, Hibara A, Tokeshi M. Anal. Chem. 2015;87:9647–9652. doi: 10.1021/acs.analchem.5b01164. [DOI] [PubMed] [Google Scholar]
- 20.Caulum MM, Henry CS. Analyst. 2006;131:1091–1093. doi: 10.1039/b608722a. [DOI] [PubMed] [Google Scholar]
- 21.Caulum MM, Murphy BM, Ramsay LM, Henry CS. Anal. Chem. 2007;79:5249–5256. doi: 10.1021/ac070452v. [DOI] [PubMed] [Google Scholar]
- 22.Guillo C, Roper MG. Electrophoresis. 2008;29:410–416. doi: 10.1002/elps.200700399. [DOI] [PubMed] [Google Scholar]
- 23.Guillo C, Truong TM, Roper MG. J. Chromatogr. A. 2011;1218:4059–4064. doi: 10.1016/j.chroma.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis EK, Haaland WC, Nguyen F, Heller DA, Allen MJ, MacGregor RR, Berger CS, Willingham B, Burns LA, Scott GBI, Kittrell C, Johnson BR, Curl RF, Metzker ML. Proc. Natl. Acad. Sci. USA. 2005;102:5346–5351. doi: 10.1073/pnas.0501606102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alaverdian L, Alaverdian S, Bilenko O, Bogdanov I, Filippova E, Gavrilov D, Gorbovitski B, Gouzman M, Gudkov G, Domratchev S, Kosobokova O, Lifshitz N, Luryi S, Ruskovoloshin V, Stepoukhovitch A, Tcherevishnick M, Tyshko G, Gorfinkel V. Electrophoresis. 2002;23:2804–2817. doi: 10.1002/1522-2683(200208)23:16<2804::AID-ELPS2804>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 26.Dongre C, van Weerd J, Besselink GAJ, Vazquez RM, Osellame R, Cerullo G, van Weeghel R, van den Vlekkert HH, Hoekstra HJWM, Pollnau M. Lab Chip. 2011;11:679–683. doi: 10.1039/c0lc00449a. [DOI] [PubMed] [Google Scholar]
- 27.Schrell AM, Roper MG. Analyst. 2014;139:2695–2701. doi: 10.1039/c3an02334f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lomasney AR, Yi L, Roper MG. Anal. Chem. 2013;85:7919–7925. doi: 10.1021/ac401625g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Findlay JWA, Dillard RF. AAPS J. 2007;9:E260–E267. doi: 10.1208/aapsj0902029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shackman JG, Reid KR, Dugan CE, Kennedy RT. Anal. Bioanal. Chem. 2012;402:2797–2803. doi: 10.1007/s00216-012-5755-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomasney AR, Guillo C, Sidebottom AM, Roper MG. Anal. Bioanal. Chem. 2009;394:313–319. doi: 10.1007/s00216-009-2622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukhitov N, Yi L, Schrell AM, Roper MG. J. Chromatogr. A. 2014;1367:154–160. doi: 10.1016/j.chroma.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.