

Abstract
Objective:
To identify the causative gene mutation in a 5-generation Belgian family with dominantly inherited spinocerebellar ataxia and polyneuropathy, in which known genetic etiologies had been excluded.
Methods:
We collected DNA samples of 28 family members, including 7 living affected individuals, whose clinical records were reviewed by a neurologist experienced in ataxia. We combined linkage data of 21 family members with whole exome sequencing in 2 affected individuals to identify shared heterozygous variants mapping to potentially linked regions. Variants were screened for rarity and for predicted damaging effect. A candidate mutation was confirmed by Sanger sequencing and tested for cosegregation with the disease.
Results:
Affected individuals presented with late-onset sensorimotor axonal polyneuropathy; all but one also had cerebellar ataxia. We identified a variant in the MME gene, p.C143Y, that was absent from control databases, cosegregated with the phenotype, and was predicted to have a strong damaging effect on the encoded protein by all algorithms we used.
Conclusions:
MME encodes neprilysin (NEP), a zinc-dependent metalloprotease expressed in most tissues, including the central and peripheral nervous systems. The mutated cysteine 143 forms a disulfide bridge, which is 100% conserved in NEP and in similar enzymes. The recent identification of recessive MME mutations in 10 unrelated individuals from Japan with axonal polyneuropathy further supports the causality of the mutation, despite the dominant mode of inheritance and the presence of cerebellar involvement in our study family. Functional studies are needed to identify the mechanisms underlying these differences.
Autosomal dominant spinocerebellar ataxias (SCAs) are progressive disabling diseases characterized by dysfunction and neuronal loss in the cerebellum, the spinocerebellar tracts, or the sensory tracts (posterior columns) of the spinal cord.1 There are many genetic subtypes of SCAs, numbered from SCA1 to SCA42.
The most common SCAs are due to repeat expansions in the respective genes, resulting in expanded polyglutamine (polyQ) tracts in the encoded proteins. Some SCAs are associated with noncoding repeat expansion, and the remaining ones are due to point mutations or insertions/deletions. Mutated genes encode proteins whose functions are relevant for the cerebellar system, including calcium homeostasis, intracellular signaling, cytoskeleton, mitochondria, neuropeptides, and membrane ion channels.1
Many SCA subtypes are characterized by pathology affecting other structures in addition to the cerebellum, with associated weakness, pyramidal signs, sensory loss, cranial nerve involvement, other movement disorders, intellectual disability, dementia, epilepsy, optic atrophy, and, in the case of SCA7, retinal macular degeneration. Peripheral neuropathy commonly occurs in SCAs, most frequently of mixed type with axonal and demyelinating characteristics, but with differences among genetic subtypes.2
We investigated a 5-generation Belgian family with dominantly inherited late-onset cerebellar ataxia and axonal peripheral neuropathy. We could exclude all known SCAs by direct testing or by linkage analysis. By combining linkage data and whole exome sequencing (WES), we identified a mutation in the MME gene as the causative of the disease. Of interest, recessive MME mutations have been recently found in participants with a very similar peripheral neuropathy but without cerebellar involvement.3
METHODS
We collected DNA samples of 28 family members, including 7 living affected individuals. We reviewed the clinical records of the 7 living affected individuals. Experienced neurologists at university hospitals in Brussels or Liège had collected personal and family history and performed general clinical and neurologic examinations. Results of EMG and nerve conduction studies were available for all 7 patients. Brain MRI was performed in 2 participants, and a sural nerve biopsy was obtained from 1 individual.
Linkage analysis.
We performed linkage analyses on DNA from the 7 affected and 14 unaffected family members. First, we performed a whole genome scan with a set of 400 highly polymorphic microsatellite markers spaced at an average distance of 8.7 cM throughout the human genome (Marshfield set 10). Next, linkage was retested by using an array of single nucleotide polymorphisms (SNPs). The programs LINKAGE and GENEHUNTER were used for linkage analysis.
Whole exome sequencing.
DNA was extracted from the lymphocytes according to standard procedures. WES was first performed on an affected individual (III-4 figure 1) at the Beijing Genomics Institute (BGI). Exonic sequences were enriched by hybridization on an Agilent SureSelect All Exon v1 capture kit, amplified, and 90 bp paired-end sequenced using an Illumina HiSeq2000 sequencer (Illumina, San Diego, CA). WES was then performed on another affected individual (IV-19, figure 1) at AROS Applied Biotechnology A/S (Aarhus, Denmark), where enrichment was accomplished by using Illumina TruSeq Exome Enrichment Kit, followed by 100 bp paired-end Illumina HiSeq2000 sequencing. Sequence reads were filtered to eliminate reads with more than 6 undetermined nucleotides or 40 identical bases in tandem, then aligned to the human genome GRCh36 assembly using the SOAPaligner 2.20 software, and the genotypes were called using the SOAPsnp program.4 We used the Burrows–Wheeler Alignment Tool and the Genome Analysis Toolkit to detect insertions and deletions (indels). Single nucleotide variants and indels were annotated using the Ensembl V54 database. We then filtered variants for being heterozygous, shared by the 2 sequenced individuals, unique in our local exome database, predicted to be of moderate or high effect by the SnpEff toolbox (http://snpeff.sourceforge.net), and mapping to nonexcluded genomic regions, i.e., with Lod scores >−2 by multipoint linkage analysis.
Figure 1. Family pedigree.

Extended pedigree of the Belgian spinocerebellar ataxia family. The proband, individual III-4, is indicated with an arrowhead. MME genotypes are indicated as WT (C143) or M (Y143).
Follow-up DNA analysis.
To confirm the candidate mutation by Sanger sequencing, we designed oligonucleotide primers to amplify a 250 bp fragment of the MME gene where the identified variant is located (forward primer: 5′-TCAAGAACCCAAAACTGAAGA-3′, reverse primer: 5′-TTGCTACTGGCCACCCATA-3′). The amplified fragments were sequenced using the Big Dye Terminator Cycle Sequencing Kit v2 (Applied Biosystems, Foster City, CA) and analyzed on a 3130 Genetic Analyzer sequencing machine (Applied Biosystems). All available family members (24/28), 7 of whom affected, were tested for the candidate mutation to confirm cosegregation with the disease. The mutation was also searched by Sanger sequencing in 96 unrelated individuals with an SCA of unidentified genetic cause.
Standard protocol approvals, registrations, and patient consents.
The Ethics Committee of the Hôpital Erasme, Brussels, Belgium, approved this study. All family members provided written informed consent for genetic analysis.
RESULTS
Clinical description.
The proband, a woman aged 69 years at the time of examination, presented with gait and balance problems and pain in the distal lower limbs from around age 58. Clinical neurologic examination showed bilateral pes cavus, mild distal lower limb atrophy, bilaterally weak knee tendon reflexes and absent Achilles tendon reflexes, ataxic gait with difficulties in turning, mild upper and lower limb ataxia, mild upper limb cogwheel rigidity, bilaterally positive palmomental reflex, hypometric saccades, and mild dysarthria. Sensory examination was normal, and there was no orthostatic hypotension. Her medical history included hypercholesterolemia and appendectomy. She did not smoke, and there was no history of excessive alcohol consumption. EMG performed at ages 69 and 70 demonstrated progressive, severe motor neuropathy in the lower limbs with significantly increased F-response latency but preserved sensory responses. Nerve conduction velocities were normal. Lumbar CT and MRI, as well as lumbar puncture, were normal. Serum antiganglioside and anti-MAG antibodies were negative. Brain MRI showed moderate cerebellar vermis atrophy (figure 2).
Figure 2. Brain MRI scan of the proband.
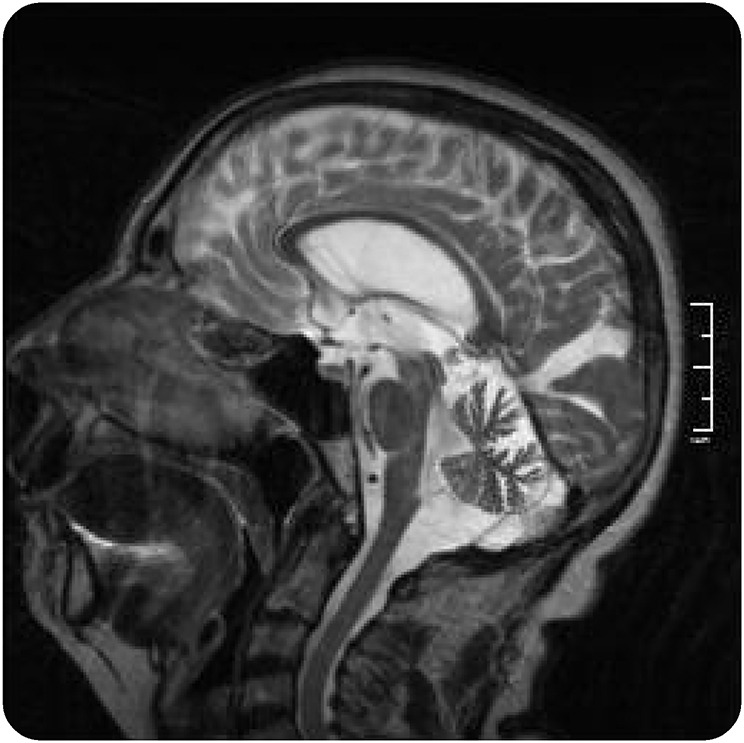
Sagittal T2-weighted MRI image from individual III-4 showing moderate atrophy of the cerebellar vermis.
Seven other family members, of whom 1 was deceased when the molecular genetic study was started, presented with late-onset (42–68 years old) balance problems. Clinical examination performed in the remaining 6 patients when they were 47–72 years old showed signs of polyneuropathy, associated with a cerebellar syndrome in 5 (table). None of them reported cognitive problems, but formal cognitive assessment was not performed. Two participants had an MRI showing cerebellar vermis atrophy. One individual (III-14) underwent sural nerve biopsy, revealing axonal pathology consistent with a Charcot–Marie–Tooth type 2 (CMT2) polyneuropathy. The pedigree was consistent with autosomal dominant inheritance (figure 1).
Table.
Demographics and clinical features of affected individuals

Genetic analyses.
SCA1, 2, 3, 6, and 7 were excluded from the routine diagnostic testing in the proband (III-4, figure 1). The first genome scan allowed us to exclude all the known SCA loci at the time it was performed (2004), as well as most of the genome, with 4 remaining unexcluded regions on chromosomes 3p, 3q, 7p, 8q, and 22p. A subsequent analysis with an SNP array allowed us to exclude the 7p region and to refine the other candidate linked regions. The highest LOD score (Z = 2.47) was obtained on chromosome 3q23-q26.31, defining a 31.3 Mb candidate region between the markers D3S3694 and rs1488547.
Four variants identified by WES fulfilled our filtering criteria. They affected the MME and GOLIM genes on chromosome 3q, the FER1L6 gene on chromosome 8q, and the PPIL2 gene on chromosome 22p. Variants in the GOLIM, FER1L6, and PPIL2 genes were listed in the SNP database (dbSNP, http://www.ncbi.nlm.nih.gov/SNP/index.html) and/or in the exome variant server (EVS, http://evs.gs.washington.edu/EVS/), making them very unlikely candidates for a rare dominant disease with a clear neurologic phenotype. The shared heterozygous variant in the MME gene, a G>A transition on chromosome 3q25.2 at position 156317031, was not present in dbSNP and EVS. It was also absent from the exAC database (http://exac.broadinstitute.org/), spanning 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies. It results in a Cys>Tyr substitution (p.C143Y) predicted to be damaging by SIFT (score 0, median information 2.66), PolyPhen-2 (score 1.000), and PROVEAN (score −10.185). Furthermore, the mutated residue is in a highly conserved region and is invariant in all species carrying a homolog of the MME gene. Segregation of the mutation with the phenotype was confirmed by Sanger sequencing in the 2 participants who had WES and in 22 additional family members, including all affected individuals (figure 2). Sequencing in 96 individuals presented with dominant ataxia of unknown cause did not identify any other carrier of this mutation.
DISCUSSION
Affected individuals in this family developed a late-onset, motor-predominant axonal polyneuropathy associated with cerebellar signs in all cases but one, presenting with ataxia with mixed afferent and cerebellar features. Pectus carinatum, although not omnipresent, seems to be a distinctive clinical feature. Genetic analyses, including WES performed in 2 affected individuals, identified a variant in the MME gene, p.C143Y, fulfilling several criteria for pathogenicity, including absence in control databases, cosegregation with the phenotype, and predicted damaging effect on the encoded protein.5 Although we could not find a second SCA family with the same mutation among the 96 we screened, the recent identification of recessive MME mutations in 10 unrelated individuals from Japan with an essentially identical polyneuropathy, but no cerebral or cerebellar atrophy, supports the causality of the MME mutation in our study family, despite the dominant mode of transmission and the presence of cerebellar involvement.3 In this regard, it is well recognized that mutations in the same gene may lead to different phenotypes and modes of transmission, and such observations are becoming common as next-generation sequencing allows solving a much larger number of cases of genetic disorders, including ataxias.6 Experimental functional studies on the p.C143Y MME variant are needed to understand the reason for its dominant effect. While all prediction algorithms we used (SIFT, PolyPhen-2, and PROVEAN) indicated a strong deleterious impact of the p.C143Y variant, the identification of recessive mutations, some leading to complete loss of the MME expression,3 makes haploinsufficiency an unlikely mechanism.
MME encodes neprilysin (NEP), a zinc-dependent metalloprotease that cleaves peptides at the amino side of the hydrophobic residues. NEP is expressed in most tissues, including the central and peripheral nervous systems (CNS and PNS). In the CNS, the expression is almost exclusively neuronal, particularly in axons and synaptic terminals,7 while in the PNS, the expression is mostly in Schwann cells,8 but NEP has also been shown to be transported along sciatic nerve axons.9 Together with the endothelin-converting enzymes (ECE-1 and ECE-2), the erythrocyte surface antigen KELL, and the PEX gene product, NEP belongs to the M13 subfamily of mammalian neutral endopeptidases.10 These are cell membrane proteins composed of a short N-terminal cytoplasmic domain, followed by a single transmembrane helix, and a large C-terminal extracellular portion composed of 2 major alpha-helical (peptidase M13) domains connected by a linker region. There are 12 cysteine residues in the NEP extracellular portion, all involved in disulfide bridges, 10 of which are also conserved in ECE, KELL, and PEX. Cysteine 143, mutated in our study family, is one of these 10 cysteines. It is localized in the N-terminal peptidase M13 domain and forms a disulfide bridge with cysteine 411 in the linker portion10 (figure 3).
Figure 3. Position of the mutated residue.

Schematic of neprilysin showing the position of the mutated residue (Cys143) and the disulfide bond with Cys411 that is disrupted by the mutation.
NEP acts on many substrates, including neuropetides such as glucagon, enkephalins, cholecystokinin, neuropeptide Y, substance P, somatostatin, neurotensin, oxytocin, prodynorphin and bradykinin, as well as the amyloid beta (Aβ) peptides accumulated in Alzheimer disease (AD). Because of its activity on Aβ and its higher expression in entorhinal, hippocampal, and cortical areas, NEP has been intensively scrutinized for a possible pathogenic role and as a potential therapeutic target in AD.11 However, although Mme knockout mice and mice exposed to NEP inhibitors have increased brain Aβ levels,12 they do not develop AD-like pathology. While some studies reported amyloid deposition and neurodegenerative changes in their hippocampus,13 a more recent study surprisingly found cognitive improvement in these animals as they age and no amyloid plaques in their brains.14 No MME mutations are known to cause familial AD, while genetic association studies have provided only limited evidence of association between MME polymorphisms and risk of AD.15 Furthermore, none of the Japanese CMT2 patients with MME mutations were found to be cognitively impaired after detailed neuropsychological testing, and amyloid PET imaging was negative in the single case in which it was performed.3 Cognitive impairment was also not a feature in our study family. Taken together, these findings indicate that although NEP expression has been reported to decrease with age and in AD, NEP deficiency does not lead to the development of AD, possibly because other enzyme systems degrade excess Aβ and prevent amyloid plaque formation.
The pathogenesis of peripheral neuropathy due to MME mutations is still unclear. It has been hypothesized that NEP has a role in axon–Schwann cell interaction,3 but experimental studies are needed to explore its physiologic role and the consequences of its impaired or loss of function. Equally unknown is the role of NEP in the cerebellum and the reason why recessive loss-of-function mutations do not affect this structure, while the p.C143Y mutation causes cerebellar dysfunction and atrophy. A possible lead comes from the fact that prodynorphin, the precursor of the opioid neuropetides α-neoendorphin and dynorphins A and B (Dyn A and B), is an NEP substrate, and mutations affecting the prodynorphin (PDYN) gene cause SCA23.16 Of interest, some SCA23 mutations alter the equilibrium between Dyn A and B and raise dynorphin peptide levels, which could be mimicked by altered NEP function.17 The p.C143Y mutation may have some specific effect on NEP activity on dynorphin peptides, possibly competing with wild-type NEP for substrate affinity and catalytic activity, which eventually triggers cerebellar degeneration. Conversely, as dynorphin is a potent NEP inhibitor,18 impaired NEP activity might be implicated in SCA23 pathogenesis. Clearly, further research is needed to address these questions. Existing Mme knockout mice should in principle model human recessive polyneuropathy cases, but interestingly they do not develop a severe axonal neuropathy, suggesting important interspecific differences. For this reason, there is a high risk that a mouse model carrying the p.C143Y mutation, obtained by classical knock-in approaches or by genome editing, would also not recapitulate the human phenotype associated with this mutation.
By combining linkage analysis and WES, we found a variant in the MME gene, p.C143Y, that responds to several criteria for pathogenicity, i.e., rarity, cosegregation with the phenotype, predicted deleterious effect, absence of other identified potential pathogenic variants by exome sequencing, and involvement of a gene proven to be mutated in families with a closely related phenotype. While functional studies will be needed to study pathogenic mechanisms, we consider that current evidence suffices to propose this mutation as disease causing in our study family, identifying a novel SCA genotype (SCA43) as well as a novel phenotype associated with a specific MME mutation.
GLOSSARY
- AD
Alzheimer disease
- BGI
Beijing Genomics Institute
- CMT2
Charcot–Marie–Tooth type 2
- ECE
endothelin-converting enzyme
- EVS
exome variant server
- NEP
neprilysin
- PNS
peripheral nervous system
- SNP
single nucleotide polymorphism
- SCA
spinocerebellar ataxia
- WES
whole exome sequencing
AUTHOR CONTRIBUTIONS
Chantal Depondt: design or conceptualization of the study, clinical evaluation, analysis and interpretation of the data, revising the manuscript for intellectual content. Simona Donatello and Myriam Rai: analysis and interpretation of the data, revising the manuscript for intellectual content. François Charles Wang, Mario Manto, and Nicolas Simonis: analysis and interpretation of the data. Massimo Pandolfo: design or conceptualization of the study, interpretation of the data, and writing the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
Chantal Depondt has served on the editorial board of BMC Neurology; and has received research support from UCB and EU Fp7 grant no. 279062. Simona Donatello, Myriam Rai, and François Charles Wang report no disclosures. Mario Manto has served on the scientific advisory boards/editorial boards of The Cerebellum, Cerebellum and Ataxias, and the Journal of NeuroEngineering and Rehabilitation; has received publishing royalties from Cambridge University Press, Sauramps Médical, Springer France, and Elsevier; has been a consultant for Guerbet France; and has received research support from FNRS Belgium (salary). Nicolas Simonis has received research support from Belgian Fonds National de la Recherche Scientifique. Massimo Pandolfo has served on the scientific advisory boards of Apopharma and Voyager Therapeutics; has served on the editorial boards of Acta Neurologica Belgica, the Orphanet Journal of Rare Diseases, and Neurology® Genetics; holds a patent for Direct molecular diagnosis of Friedreich's ataxia; has been a consultant for Biomarin; has been a Scientific Advisory Board member for Voyager Therapeutics; has received research support from Biomarin, Fonds National de la Recherche Scientifique (Belgium), Offrez-moi-la-lune, Friedreich's Ataxia Research Alliance, and Association Belge contre les Maladies neuro-Musculaires; and has received royalty payments from Athena Diagnostics. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Pandolfo M, Manto M. Cerebellar and afferent ataxias. Continuum (Minneap Minn) 2013;19:1312–1343. [DOI] [PubMed] [Google Scholar]
- 2.Linnemann C, Tezenas du Montcel S, Rakowicz M, et al. . Peripheral neuropathy in spinocerebellar ataxia type 1, 2, 3, and 6. Cerebellum 2016;15:165–173. [DOI] [PubMed] [Google Scholar]
- 3.Higuchi Y, Hashiguchi A, Yuan J, et al. . Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol 2016;79:659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu S, Fang L, Xu X. Using SOAPaligner for short reads alignment. Curr Protoc Bioinformatics 2013;44:11.11.1–11.11.17. [DOI] [PubMed] [Google Scholar]
- 5.MacArthur DG, Manolio TA, Dimmock DP, et al. . Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyle A, Smertenko T, Bargiela D, et al. . Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015;138:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukami S, Watanabe K, Iwata N, et al. . Abeta-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci Res 2002;43:39–56. [DOI] [PubMed] [Google Scholar]
- 8.Kioussi C, Matsas R. Endopeptidase-24.11, a cell-surface peptidase of central nervous system neurons, is expressed by Schwann cells in the pig peripheral nervous system. J Neurochem 1991;57:431–440. [DOI] [PubMed] [Google Scholar]
- 9.Ohkushi G, Suzuki N, Kobayashi S, Chikuma T. Axonal transport of neprilysin in rat sciatic nerves. J Mol Neurosci 2014;53:96–102. [DOI] [PubMed] [Google Scholar]
- 10.Oefner C, D'Arcy A, Hennig M, Winkler FK, Dale GE. Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. J Mol Biol 2000;296:341–349. [DOI] [PubMed] [Google Scholar]
- 11.Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-beta peptide and Alzheimer's disease. Pharmacol Ther 2005;108:129–148. [DOI] [PubMed] [Google Scholar]
- 12.Iwata N, Tsubuki S, Takaki Y, et al. . Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 2000;6:143–150. [DOI] [PubMed] [Google Scholar]
- 13.Madani R, Poirier R, Wolfer DP, et al. . Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. J Neurosci Res 2006;84:1871–1878. [DOI] [PubMed] [Google Scholar]
- 14.Walther T, Albrecht D, Becker M, et al. . Improved learning and memory in aged mice deficient in amyloid beta-degrading neutral endopeptidase. PLoS One 2009;4:e4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarimon J, Muñoz FJ, Boada M, et al. . Possible increased risk for Alzheimer's disease associated with neprilysin gene. J Neural Transm (Vienna) 2003;110:651–657. [DOI] [PubMed] [Google Scholar]
- 16.Bakalkin G, Watanabe H, Jezierska J, et al. . Prodynorphin mutations cause the neurodegenerative disorder spinocerebellar ataxia type 23. Am J Hum Genet 2010;87:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smeets CJLM, Jezierska J, Watanabe H, et al. . Elevated mutant dynorphin A causes Purkinje cell loss and motor dysfunction in spinocerebellar ataxia type 23. Brain 2015;138:2537–2552. [DOI] [PubMed] [Google Scholar]
- 18.Martin-Kleiner I, Balog T, Marotti T. Dynorphin inhibits NEP activity in R1.1 mouse thymoma cells. Cell Biochem Funct 2008;26:916–919. [DOI] [PubMed] [Google Scholar]