

Abstract
The histone deacetylase inhibitor trichostatin A (TSA) has been demonstrated to alleviate certain symptoms associated with osteoarthritis (OA). However, the exact mechanisms underlying this protective effect remain to be elucidated. The present study therefore examined the effects of TSA on the expression levels of interleukin-1β (IL-1β)-induced matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases-1 (TIMP-1) in vitro and in vivo. In vitro, reverse transcription-quantitative polymerase chain reaction was performed to investigate alterations in mRNA expression levels in TSA-treated chondrocytes in the presence or absence of IL-1β; in addition, protein expression and acetylation levels were assessed by western blotting. In vivo, TSA was administered to rats by intra-articular injection, following which the mRNA and protein expression levels were analyzed. In addition, macroscopic and histological observations were conducted. Chondrocytes treated with IL-1β demonstrated increased mRNA and protein expression levels of MMP-1, MMP-3 and MMP-13, and decreased expression levels of TIMP-1 mRNA and protein; these alterations were significantly attenuated by TSA treatment. In addition, increased MMPs and decreased TIMP-1 expression levels were observed in vivo in the OA rat model. TSA treatment demonstrated in vivo efficacy through the attenuation of various OA-associated molecular and physiological changes. Taken together, the results of the present study suggest that TSA has potential therapeutic value for the treatment of OA.
Keywords: osteoarthritis, trichostatin A, matrix metalloproteinases, tissue inhibitors of metalloproteinases-1, acetylation
Introduction
Osteoarthritis (OA), the predominant form of arthritis (1), is characterized by slow, progressive joint degeneration. It is a primary cause of continuous pain and chronic disability that restricts the normal daily lives of many elderly individuals (2–4). Although there are numerous risk factors for OA, including obesity, aging and trauma, the mechanisms underlying these associations remain to be elucidated (5–7). Previous studies have demonstrated that synthesis and catabolism disorders in chondrocytes are involved in the onset of OA (8,9). It is generally accepted that the loss of articular cartilage matrix components, including collagen type II and aggrecan, resulting from the activation of proteolytic enzymes, such as matrix metalloproteinases (MMPs), is a frequent characteristic of OA (10–14). MMPs are a family of endopeptidases that contribute to numerous physiological processes via the degradation of extracellular matrix components. MMP-1 and MMP-13, also referred to as collagenases, degrade all types of collagen, particularly those that supply mechanical strength to tissues, such as collagen type II in articular cartilage (14,15). MMP-3, also referred to as pro-MMP, is a type of stromelysin that activates other MMPs, including MMP-1 and MMP-13 (16). The biological activities of MMPs are antagonized by tissue inhibitors of metalloproteinases (TIMPs), such as TIMP-1 (17,18). Thus, an imbalance in the activities of MMPs and TIMPs is considered to be critical in OA progression (19,20).
Inflammatory cytokines, including interleukin (IL)-1β, tumor necrosis factor-α and IL-6, are crucial for chondrocyte apoptosis and degradation of the extracellular matrix (21,22), and are thus involved in the pathogenesis and development of OA. A previous study has confirmed that IL-1β induces upregulation of MMPs and downregulation of TIMPs, resulting in degradation of proteoglycans and collagen (21).
Trichostatin A (TSA), originally developed as an anti-fungal agent (23), is a non-selective histone deacetylase inhibitor (HDACi) (24,25). The acetylation of lysine residues on histones H3 and H4 opens the chromatin structure to promote active gene transcription (26–29). The regulation of inflammatory genes by TSA has attracted considerable attention. Previous studies have demonstrated that TSA exerts anti-OA and anti-rheumatoid arthritis effects via suppression of the expression of MMPs, cyclooxygenase-2, inducible nitric oxide synthase and a disintegrin and MMP with thrombospondin motifs (4,30). Furthermore, it has been revealed that the underlying anti-inflammatory mechanism may involve inhibition of the mitogen-activated protein kinase and nuclear factor-κB (NF-κB) signaling pathways, and subsequent upreg-ulation of sirtuin 1 expression (31). However, the underlying mechanism by which TSA ameliorates the OA inflammatory response remains to be fully elucidated, and there is currently not enough evidence to determine whether the anti-OA effect of TSA involves altering the level of acetylation in chondrocytes. Therefore, the present study investigated the association between the anti-OA effects of TSA and the levels of acetylation in chondrocytes.
Materials and methods
Reagents
TSA and IL-1β were purchased from Sig ma-Aldrich (St. Louis, MO, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and collagenase II were obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Isolation and culture of rat chondrocytes
Rat cartilage was collected from the knee and hip joints of six 2-week-old female Sprague Dawley rats (weight, 35–45 g) from the Zhejiang Academy of Medical Sciences (Hangzhou, China). Rats were housed under pathogen-free conditions at room temperature (22–25°C) and 50% humidity, and has a 12-h light/dark cycle. Food and water were offered ad libitum. To isolate chondrocytes, rat cartilage was digested with 0.2% collagenase II (Sigma-Aldrich) in DMEM for 4 h at 37°C. Rat cartilage was subsequently filtered through a 150 µm mesh filter and centrifuged at 300 × g for 5 min at 4°C, and chondrocytes were then collected and cultured in DMEM with 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a 5% CO2 atmosphere at 37°C. Chondrocytes at passage 3 were used for all analyses, according to a previous study (32).
Determination of TSA concentration
To select the optimal TSA concentration for the in vitro and in vivo experiments, cell viability in the presence of TSA was evaluated using the Cell Counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) assay. Cells were seeded in 96-well plates at 6×103 cells/well and allowed to adhere overnight. Cells were then cultured in various concentrations of TSA in diluted in serum-free DMEM for 24 h. The medium was then removed, and 10 µl CCK-8 solution in 90 µl fresh medium was added to each well. Following incubation for 3 h at 37°C the absorbance was measured at 450 nm using a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Treatment of rat chondrocytes
Chondrocytes were seeded in 6-well plates at 2×105 cells/well. At 80% confluency, they were serum-starved for 12 h. Cells were then pre-treated with various concentrations of TSA for 2 h, and stimulated with IL-1β (5 ng/ml) for 12 h or left unstimulated. Cells were harvested for mRNA expression analysis of MMPs and TIMP-1. For determination of total protein and acetylation levels of MMPs and TIMP-1, IL-1β stimulation was maintained for 24 h.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Briefly, total RNA was extracted from cell and cartilage samples with TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.). RNA (0.5 µg) was reversed-transcribed to cDNA using the PrimeScript-RT reagent kit (Takara Bio, Inc., Otsu, Japan). cDNA samples were amplified and quantified by qPCR (ABI Prism 7500; Applied Biosystems; Thermo Fisher Scientific, Inc.) using the SYBR Premix Ex Taq (Takara Bio, Inc.). qPCR protocol cycling conditions were as follows: Holding stage, 95°C for 30 sec; cycling stage, 95°C for 5 sec, then 60°C for 30 sec, for a total of 40 cycles. Stage Primers for MMP-1, MMP-3, MMP-13, TIMP-1, and 18S ribosomal RNA (18S rRNA) genes were obtained from Sangon Biotech Co., Ltd. (Shanghai, China); the sequences are listed in Table I. 18S rRNA served as an internal control. Relative expression was calculated using the 2−ΔΔCq method (33).
Table I.
Primer sequences.
| Gene | Forward | Reverse |
|---|---|---|
| 18S rRNA | 5′-TTGACGGAAGGGCACCA-3′ | 5′-CAGACAAATCGCTCCACCAA-3′ |
| MMP-1 | 5′-GGAACAGATACGAAGAGGAAACA-3′ | 5′-TGTGGGAATCAGAGGTAGAAGA-3′ |
| MMP-3 | 5′-GCATTGGCTGAGTGAAAGAGAC-3′ | 5′-ATGATGAACGATGGACAGATGA-3′ |
| MMP-13 | 5′-TGAGAGTCATGCCAACAAATTC-3′ | 5′-CAGCCACGCATAGTCATGTAGA-3′ |
| TIMP-1 | 5′-GGTTCCCTGGCATAATCTGAG-3′ | 5′-ATCGCTCTGGTAGCCCTTCT-3′ |
rRNA, ribosomal RNA; MMP, matrix metalloproteinase; TIMP-1, tissue inhibitor of metalloproteinases-1.
Western blot analysis
Cell and cartilage samples were washed twice with ice-cold phosphate-buffered saline (PBS). Proteins were then extracted using radioimmunoprecipitation assay lysis buffer (Wuhan Boster Biological Technology, Ltd., Wuhan, China) for 30 min. Protein levels were quantified with the Pierce Bicinchoninic Acid Protein assay (Thermo Fisher Scientific, Inc.). Proteins (5 µg) were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 110 V for 90 min, and transferred to polyvinylidene difluoride membranes. Following incubation in blocking buffer (5% non-fat milk) for 1 h at room temperature, the membranes were sectioned based on molecular weights, and probed with the following primary antibodies: Rabbit-anti β-actin (catalog no., DW130656; Hang Zhou Dawen Biotec Co., Ltd., Hangzhou, China), rabbit-anti MMP-1 (catalog no., BS1229; Bioworld Technology, Inc., Louis Park, MN, USA), rabbit-anti MMP-3 (catalog no., 17,873-1-AP; ProteinTech Group, Inc., Chicago, IL, USA), rabbit-anti MMP-13 (catalog no., BS6668; Bioworld Technology, Inc.), rabbit-anti TIMP-1 (catalog no., sc-5538; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), and mouse anti-H3 (96C10, catalog no., #3638), mouse anti-H4 (L64C1, catalog no., #2935), rabbit anti-acetyl-H3 (Lys27, catalog no., #8173), and rabbit anti-acetyl-H4 (Lys8, catalog No., #2594; all Cell Signaling Technology, Inc., Danvers, MA, USA). All primary antibodies were diluted 1:1,000. The membranes were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary goat anti-rabbit IgG antibody (catalog no., sc-2004; Santa Cruz Biotechnology, Inc.; 1:5,000) or HRP-conjugated secondary goat anti-mouse IgG antibody (catalog no., sc-2005; Santa Cruz Biotechnology, Inc.; 1:2,000) for 1 h at room temperature. Following washing, signals were detected using Enhanced Chemiluminescence (ECL; Thermo Fisher Scientific, Inc.) and densitometry was performed using the Quantity One version 4.6.2 software (Bio-Rad Laboratories Inc., Munich, Germany).
Animal study
A total of 30 4-week-old male Sprague Dawley rats (weight, 120–140 g), from the Animal Center at Zhejiang University (Hangzhou, China), were used in the present study. Experiments were conducted with the approval of the Zhejiang University Animal Care and Use Committee (Hangzhou, China). Rats were housed under pathogen-free conditions at room temperature (22–25°C) and 50% humidity, and had a 12-h light/dark cycle. Food and water were offered ad libitum. Rats were randomly divided into three groups: i) Control, ii) OA and iii) TSA-treated. Following anesthesia with 2% pentobarbital sodium (0.1 ml/100 g; Sigma-Aldrich) (34), rats in the OA and TSA-treated groups received an intra-articular injection of monoiodoacetate (MIA; 1 mg in 50 µl PBS; Sigma-Aldrich) into the knee joint (35). Rats in the control group were injected with 50 µl PBS. Three days later, rats in the TSA-treated group were bilaterally intra-articularly injected into the knees with 50 µl TSA (0.2 µM), while the other groups received 50 µl PBS injected intra-articularly. These injections were repeated once a week for four weeks. All of the rats were sacrificed seven days following the final injection. Mice were anaesthetized with 2% pentobarbital sodium (0.1 ml/100 g; Sigma-Aldrich) and cervical vertebrae were dislocated. Then femoral condyles were collected for gene and protein expression analysis and histological assessment.
Histological analysis
Specimens were fixed in 4% paraformaldehyde in 0.1 M PBS for 72 h (31) and decalcified with 10% buffered formic acid at room temperature for two months. Specimens were embedded in paraffin wax and cut into 5 mm sections. The sections were stained with Safranin-O and fast green (36). The Mankin scoring system was used for blinded histological assessment (36).
Statistical analysis
Data are expressed as the mean ± standard deviation. Comparisons were made using an unpaired Student's t-test and one-way analysis of variance, followed by Dunnett's analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
Assessment of TSA concentrations
To select the optimal concentrations of TSA for in vitro and in vivo experiments, a CCK-8 assay was performed to evaluate various TSA concentrations. As presented in Fig. 1, TSA concentrations <0.4 µM exerted no significant effect on chondrocyte viability at 24 h (0.8 vs. 0 µM TSA; P=0.03), and therefore were selected for use in subsequent experiments.
Figure 1.
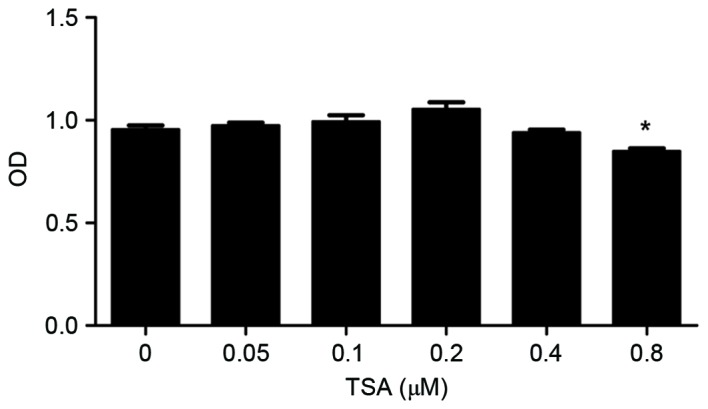
Determination of optimal TSA concentrations for in vitro and in vivo experiments. Chondrocytes were treated with increasing concentrations of TSA (0.05–0.8 µM) for 24 h. Cell viability was determined using a cell counting kit-8 assay. *P<0.05 vs. 0 µM TSA. Data are presented as the mean ± standard deviation. TSA, trichostatin A; OD, optical density.
TSA decreases MMP and increases TIMP-1 expression levels in chondrocytes
RT-qPCR revealed that treatment of chondrocytes with IL-1β decreased mRNA expression levels of MMP-1, MMP-3 and MMP-13 (Fig. 2A). By contrast, mRNA expression levels of TIMP-1 increased following IL-1β administration. Notably, IL-1β-mediated regulation of each of these genes was significantly attenuated by increasing concentrations of TSA. While mRNA expression levels of MMP-1, MMP-3 and MMP-13 were not affected by treatment with TSA alone, TIMP-1 was significantly upregulated in cells incubated with TSA alone (0.05, 0.1, 0.2 vs. 0 µM TSA; P=0.034, <0.001, <0.001, respectively; Fig. 2B). Examinations of protein expression levels of MMPs and TIMP-1 were consistent with the RT-qPCR data. That is, TSA significantly attenuated IL-1β-mediated regulation of MMP-1, MMP-3, MMP-13 and TIMP-1 protein expression levels (MMP-1, MMP-2, MMP-13 and TIMP-1 incubated with 0.2 vs. 0 µM TSA; P<0.001, P=0.004, 0.006 and 0.004, respectively; Fig. 2C). Treatment of chondrocytes with TSA alone increased TIMP-1 protein expression levels, which was consistent with the transcriptional analysis (P<0.05; Fig. 2D).
Figure 2.

Effect of TSA on MMP-1, MMP-3, MMP-13, and TIMP-1 mRNA and protein expression levels in chondrocytes. (A) Chondrocytes were pretreated with increasing concentrations of TSA for 2 h, followed by stimulation with 5 ng/ml IL-1β for 12 h. *P<0.05 vs. IL-1β alone. (B) Chondrocytes were treated with increasing concentrations of TSA for 12 h. *P<0.05 vs. untreated. Total RNA was isolated and reverse transcription-quantitative polymerase chain reaction was performed to determine relative mRNA expression levels. Levels were calculated using the 2−ΔΔCq method and expressed as the mean ± SD. (C) Chondrocytes were pretreated with increasing concentrations of TSA for 2 h, followed by stimulation with 5 ng/ml IL-1β for 24 h. (D) Chondrocytes were treated with increasing concentrations of TSA for 24 h. Proteins were harvested for western blot, and bands were quantified using densitometry and quantity one software. Data are expressed as the mean ± SD. TSA, trichostatin A; MMP, matrix metalloproteinase; TIMP-1, tissue inhibitor of metalloproteinases-1; IL-1β, interleukin-1β; SD, standard deviation.
TSA treatment delays knee joint degeneration in the rat OA model and is accompanied by decreased MMP and increased TIMP-1 mRNA and protein expression levels in cartilage
The gross appearance of femoral condyles collected from the various treatment groups were examined (Fig. 3A). The condyle surface in the control group was intact and glossy. By contrast, the condyle surface of rats in the OA group revealed gross morphological alterations, including defects, osteophytes and tissue adhesions. Notably, a marked reversion to normal morphology was observed in the TSA-treated group. Morphological analysis was accompanied by Safranin-O/fast green staining of cartilage (Fig. 3B). Compared with the control group, Safranin-O staining was largely absent in the OA group (demonstrated by the absence of red staining in the OA group image). TSA administration markedly restored Safranin-O staining, indicative of cartilage retention, although not to the level of the control group. TSA treatment markedly attenuates numerous changes associated with OA progression, as measured by the Mankin scoring system for histological analyses (Table II). Structural and cellular changes were analyzed and assigned histological scores. The sum of the scores was significantly increased in the OA group (7.47±1.07) compared with the control (1.02±0.18; P=0.005). Notably, and consistent with our earlier findings (4,30), TSA treatment promoted reversion back to normal, with a sum of 3.01±0.41 (P=0.003 vs. OA group).
Figure 3.

Effect of TSA on the rat OA model and on MMP-1, MMP-3, MMP-13 and TIMP-1 mRNA and protein expression levels in cartilage. An in vivo study was performed using Sprague Dawley rats, which were randomly divided into three groups (n=10/group): Control, OA and OA with TSA treatment. (A) Representative images of macroscopic observations. (B) Safranin-O/fast green staining (magnification, ×100). (C) Total RNA was isolated from cartilage, and reverse transcription-quantitative polymerase chain reaction was performed to determine relative mRNA expression levels. Levels were calculated using the 2−ΔΔCq method and expressed as the mean ± SD. *P<0.05 vs. OA. (D) Proteins were extracted from cartilage for western blot analysis, and bands were quantified using densitometry and quantity one software. Data are expressed as the mean ± SD. TSA, trichostatin A; OA, osteoarthritis; MMP, matrix metalloproteinase; TIMP-1, tissue inhibitor of metalloproteinases-1; SD, standard deviation.
Table II.
Histological scoring of articular cartilage.
| Parameter | Control group | OA group | TSA-treated group |
|---|---|---|---|
| Structural changes | 0.15±0.04 | 2.60±0.16a | 0.83±0.09b |
| Cellular changes | 0.19±0.03 | 2.04±0.26a | 0.86±0.15b |
| Safranin-O staining | 0.54±0.09 | 2.68±0.72a | 1.18±0.16b |
| Tide mark | 0.14±0.04 | 0.15±0.04 | 0.14±0.03 |
| Sum of score | 1.02±0.18 | 7.47±1.07a | 3.01±0.41b |
Mankin scores were positively correlated with OA severity. Using Mankin's histological scoring principles, a total score of 13 is possible. Structural changes, scored from normal to severely damaged (0–3); Cellular changes, scored from normal to severe hyperplasia (0–3); Safranin-O staining, scored from normal to non-staining (0–4); Tide mark, scored from normal to fuzzy (0–3). Data are expressed as the mean ± standard deviation.
P<0.05 vs. control
P<0.05 vs. OA. OA, osteoarthritis; TSA, trichostatin A.
Molecular analyses of in vivo samples were performed. Results from RT-qPCR revealed that mRNA expression levels of MMP-1, MMP-3 and MMP-13 were significantly upregulated, and TIMP-1 was downregulated, in cartilage from OA rats compared with control rats (Fig. 3C). Consistent with the in vitro observations, TSA treatment significantly attenuated the OA-associated alterations in expression levels of each of these genes (P<0.05). Alterations in protein expression levels of MMPs and TIMP-1 were consistent with the RT-qPCR data (P<0.05; Fig. 3D).
TSA increases acetylation levels in chondrocytes and cartilage
Data from the in vitro and in vivo experiments indicate that TSA may be beneficial in the treatment of OA. As TSA is an HDACi, alterations in levels of acetylation in cells (Fig. 4A and B) and in the OA animal model (Fig. 4C) were examined. Levels of acetylation at H3K27 and H4K8 were not significantly altered in cells treated with IL-1β or in cartilage from the OA group, indicating that acetylation at these specific residues is not involved in OA progression. Notably, TSA administration significantly increased acetylation at these residues, providing evidence of functional TSA treatment (P<0.05).
Figure 4.

Effect of TSA on acetylation levels in chondrocytes and cartilage. (A) Chondrocytes were pretreated with increasing concentrations of TSA for 2 h, followed by stimulation with 5 ng/ml IL-1β for 24 h. (B) Chondrocytes were treated with increasing concentrations of TSA for 24 h. (C) Proteins were extracted from the cartilage of control, OA, and OA with TSA treatment rats. Western blot protein bands were quantified using densitometry and quantity one software. Data are expressed as the mean ± standard deviation. TSA, trichostatin A; IL-1β, interleukin-1β; OA, osteoarthritis; H, histone.
Discussion
The results of the present study revealed that TSA significantly attenuated the IL-1β-induced upregulation of MMP-1, MMP-3 and MMP-13, and the downregulation of TIMP-1. In addition, the level of acetylation increased in a TSA dose-dependent manner, independent of IL-1β treatment. Consistent with these observations in vitro, increased expression levels of MMP-1, MMP-3 and MMP-13, and decreased TIMP-1 expression levels, were detected in the OA rat model compared with control rats. These findings suggest that the altered MMP/TIMP-1 ratio is an important contributing factor in OA progression. In addition, TSA treatment demonstrated a beneficial effect in vivo, possibly via an increase in TIMP-1 expression levels. Furthermore, levels of acetylated histones, H3 and H4 were increased in the TSA-treated group compared with the OA and control groups. These observations in vitro and in vivo suggest that TSA treatment alters the MMP/TIMP-1 ratio to protect against OA. These conclusions are supported by the function of TIMP-1 as an important inhibitor of MMP activity during the process of cartilage degeneration (37). MMPs degrade various extracellular matrix components, including collagen type II, which is a key pathological feature of OA (38).
In the present study, the role of histone acetylation in OA progression and following TSA treatment was examined. Analyses were limited to acetylation of H3K27 and H4K8. Acetylation of these residues was not identified to be significantly altered in vitro by IL-1β treatment or in vivo in the OA rat model. These findings suggest that altered histone acetylation of these specific residues is not involved in OA development and progression. Alternatively, alterations in acetylation of these residues may occur only at key genes involved in OA and, as such, may not be observable at the global level when probed by western blot analysis. However, increased acetylation of the two residues following TSA treatment in vitro and in vivo was observed. In addition, while TSA treatment increased TIMP-1 expression levels, it had no direct effect on expression levels of MMP-1, MMP-3 or MMP-13. This suggests that TSA attenuates the OA-associated increase in MMPs via upregulating TIMP-1, rather than by directly affecting MMP transcription. Previous studies have demonstrated that histone hyperacetylation is associated with anti-inflammatory properties (39) via altered gene expression (40). A similar mechanism of action may underlie the anti-OA effect of TSA that was observed in in the present study. However, this hypothesis requires additional investigation. In future experiments, analysis of a larger panel of histone acetylation modifications and chromatin immunoprecipitation would be performed to determine which genes are marked by histone modifications. This would facilitate the determination of the precise underlying mechanism of action of TSA in the protection against OA.
The results of the present study revealed that TSA treatment does not significantly alter transcript levels of MMP-1, MMP-3 and MMP-13. These findings suggest that MMP transcription in this setting is not directly linked to histone acetylation. Consistent with this, a previous study demonstrated that the IL-1β-mediated production of MMPs primarily occurs via the NF-κB signaling pathway (41). However, in our previous study on the effect of TSA on the NF-κB signaling pathway in chondrocytes treated with IL-1β, no significant alterations in NF-κB activity were observed (32). Additional experiments are therefore required to determine the exact underlying mechanism by which the anti-OA effects of TSA are exerted. In conclusion, the results of the present study demonstrated that TSA protects against OA formation in chondrocytes in vitro and in an in vivo OA rat model. It is proposed that TSA functions via the upregulation of TIMP-1 and subsequent inhibition of MMP activity. The findings of the present study suggest that TSA may be a potential therapy for the treatment of OA, and that histone acetylation may be an important contributing factor in this disease.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (grant nos. 81201429 and 81371996).
References
- 1.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felson DT, Chaisson CE, Hill CL, Totterman SM, Gale ME, Skinner KM, Kazis L, Gale DR. The association of bone marrow lesions with pain in knee osteoarthritis. Ann Intern Med. 2001;134:541–549. doi: 10.7326/0003-4819-134-7-200104030-00007. [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 4.Chen WP, Bao JP, Hu PF, Feng J, Wu LD. Alleviation of osteoarthritis by Trichostatin A, a histone deacetylase inhibitor, in experimental osteoarthritis. Mol Biol Rep. 2010;37:3967–3972. doi: 10.1007/s11033-010-0055-9. [DOI] [PubMed] [Google Scholar]
- 5.Neogi T, Zhang Y. Osteoarthritis prevention. Curr Opin Rheumatol. 2011;23:185–191. doi: 10.1097/BOR.0b013e32834307eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee R, Kean WF. Obesity and knee osteoarthritis. Inflammopharmacology. 2012;20:53–58. doi: 10.1007/s10787-011-0118-0. [DOI] [PubMed] [Google Scholar]
- 7.Loeser RF. Aging and osteoarthritis. Curr Opin Rheumatol. 2011;23:492–496. doi: 10.1097/BOR.0b013e3283494005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aigner T, McKenna L. Molecular pathology and pathobiology of osteoarthritic cartilage. Cell Mol Life Sci. 2002;59:5–18. doi: 10.1007/s00018-002-8400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu PF, Chen WP, Tang JL, Bao JP, Wu LD. Protective effects of berberine in an experimental rat osteoarthritis model. Phytother Res. 2011;25:878–885. doi: 10.1002/ptr.3359. [DOI] [PubMed] [Google Scholar]
- 10.Loeuille D, Chary-Valckenaere I, Champigneulle J, Rat AC, Toussaint F, Pinzano-Watrin A, Goebel JC, Mainard D, Blum A, Pourel J, et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: Correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005;52:3492–3501. doi: 10.1002/art.21373. [DOI] [PubMed] [Google Scholar]
- 11.Aigner T, Sachse A, Gebhard PM, Roach HI. Osteoarthritis: Pathobiology-targets and ways for therapeutic intervention. Adv Drug Deliv Rev. 2006;58:128–149. doi: 10.1016/j.addr.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 12.Attur MG, Dave M, Cipolletta C, Kang P, Goldring MB, Patel IR, Abramson SB, Amin AR. Reversal of autocrine and paracrine effects of interleukin 1 (IL-1) in human arthritis by type II IL-1 decoy receptor. Potential for pharmacological intervention. J Biol Chem. 2000;275:40307–40315. doi: 10.1074/jbc.M002721200. [DOI] [PubMed] [Google Scholar]
- 13.Moos V, Fickert S, Müller B, Weber U, Sieper J. Immunohistological analysis of cytokine expression in human osteoarthritic and healthy cartilage. J Rheumatol. 1999;26:870–879. [PubMed] [Google Scholar]
- 14.Bramono DS, Richmond JC, Weitzel PP, Kaplan DL, Altman GH. Matrix metalloproteinases and their clinical applications in orthopaedics. Clin Orthop Relat Res. 2004:272–285. doi: 10.1097/01.blo.0000144166.66737.3a. [DOI] [PubMed] [Google Scholar]
- 15.Mandal M, Mandal A, Das S, Chakraborti T, Sajal C. Clinical implications of matrix metalloproteinases. Mol Cell Biochem. 2003;252:305–329. doi: 10.1023/A:1025526424637. [DOI] [PubMed] [Google Scholar]
- 16.Poole AR. Biochemical/immunochemical biomarkers of osteoarthritis: Utility for prediction of incident or progressive osteoarthritis. Rheum Dis Clin North Am. 2003;29:803–818. doi: 10.1016/S0889-857X(03)00056-5. [DOI] [PubMed] [Google Scholar]
- 17.Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 18.Johnston P, Larson D, Clark IM, Chojnowski AJ. Metalloproteinase gene expression correlates with clinical outcome in Dupuytren's disease. J Hand Surg Am. 2008;33:1160–1167. doi: 10.1016/j.jhsa.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Hayakawa T. Multiple functions of tissue inhibitors of metalloproteinases (TIMPs): A new aspect involving osteoclastic bone resorption. J Bone Miner Metab. 2002;20:1–13. doi: 10.1007/s774-002-8440-0. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez-Barrantes S, Bernardo M, Toth M, Fridman R. Regulation of membrane type-matrix metalloproteinases. Semin Cancer Biol. 2002;12:131–138. doi: 10.1006/scbi.2001.0421. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi M, Squires GR, Mousa A, Tanzer M, Zukor DJ, Antoniou J, Feige U, Poole AR. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005;52:128–135. doi: 10.1002/art.20776. [DOI] [PubMed] [Google Scholar]
- 22.Aida Y, Maeno M, Suzuki N, Shiratsuchi H, Motohashi M, Matsumura H. The effect of IL-1beta on the expression of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in human chondrocytes. Life Sci. 2005;77:3210–3221. doi: 10.1016/j.lfs.2005.05.052. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 24.Ghosh AK, Mori Y, Dowling E, Varga J. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: Involvement of Sp1. Biochem Biophys Res Commun. 2007;354:420–426. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- 25.Piekarz R, Bates S. A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr Pharm Des. 2004;10:2289–2298. doi: 10.2174/1381612043383980. [DOI] [PubMed] [Google Scholar]
- 26.Monneret C. Histone deacetylase inhibitors. Eur J Med Chem. 2005;40:1–13. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: A dynamic perspective. Mol Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 30.Chen WP, Bao JP, Tang JL, Hu PF, Wu LD. Trichostatin A inhibits expression of cathepsins in experimental osteoarthritis. Rheumatol Int. 2011;31:1325–1331. doi: 10.1007/s00296-010-1481-7. [DOI] [PubMed] [Google Scholar]
- 31.Matsushita T, Sasaki H, Takayama K, Ishida K, Matsumoto T, Kubo S, Matsuzaki T, Nishida K, Kurosaka M, Kuroda R. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1β in human chondrocytes. J Orthop Res. 2013;31:531–537. doi: 10.1002/jor.22268. [DOI] [PubMed] [Google Scholar]
- 32.Zhong HM, Ding QH, Chen WP, Luo RB. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-kB nuclear translocation. Int Immunopharmacol. 2013;17:329–335. doi: 10.1016/j.intimp.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Zhou XD, Yang KH, Fan TD, Chen WP, Jiang LF, Bao JP, Wu LD, Xiong Y. Hinokitiol reduces matrix metalloproteinase expression by inhibiting Wnt/β-Catenin signaling in vitro and in vivo. Int Immunopharmacol. 2014;23:85–91. doi: 10.1016/j.intimp.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Lee SW, Song YS, Shin SH, Kim KT, Park YC, Park BS, Yun I, Kim K, Lee SY, Chung WT, et al. Cilostazol protects rat chondrocytes against nitric oxide-induced apoptosis in vitro and prevents cartilage destruction in a rat model of osteoarthritis. Arthritis Rheum. 2008;58:790–800. doi: 10.1002/art.23220. [DOI] [PubMed] [Google Scholar]
- 36.Bulstra SK, Buurman WA, Walenkamp GH, Van der Linden AJ. Metabolic characteristics of in vitro cultured human chondrocytes in relation to the histopathologic grade of osteoarthritis. Clin Orthop Relat Res. 1989:294–302. [PubMed] [Google Scholar]
- 37.Schurigt U, Stopfel N, Hückel M, Pfirschke C, Wiederanders B, Bräuer R. Local expression of matrix metalloproteinases, cathepsins, and their inhibitors during the development of murine antigen-induced arthritis. Arthritis Res Ther. 2005;7:R174–R188. doi: 10.1186/ar1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, Shah M, Thompson EW. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. 2007;150:829–831. doi: 10.1038/sj.bjp.0707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 41.Lauder SN, Carty SM, Carpenter CE, Hill RJ, Talamas F, Bondeson J, Brennan P, Williams AS. Interleukin-1beta induced activation of nuclear factor-kappab can be inhibited by novel pharmacological agents in osteoarthritis. Rheumatology (Oxford) 2007;46:752–758. doi: 10.1093/rheumatology/kel419. [DOI] [PubMed] [Google Scholar]