

Summary
Invariant Natural Killer T (iNKT) cells are evolutionarily conserved innate T cells that influence inflammatory responses. We have shown that iNKT cells, previously thought to be rare in huamns, were highly enriched in human and murine adipose tissue, and that as adipose tissue expands in obesity, iNKT cells were depleted, correlating with proinflammatory macrophage infiltration. iNKT cell numbers were restored in mice and humans after weight loss. Mice lacking iNKT cells had enhanced weight gain, larger adipocytes, fatty livers and insulin resistance on high fat diet. Adoptive transfer of iNKT cells into obese mice or in vivo activation of iNKT cells with their lipid ligand alpha-galactocylceramide decreased body fat, triglycerides, leptin, fatty liver, and improved insulin sensitivity through Th2 cell-type cytokine production by adipose-derived iNKT cells. This finding highlights the potential of iNKT cell-targeted therapies, previously proven to be safe in humans, in the management of obesity and its consequences.
Introduction
The discovery that TNFα was elevated in obesity and correlated with insulin resistance was a seminal finding that kick-started the field of inflammation and immunometabolism in 1993 (Hotamisligil et al., 1993). It is now accepted that inflammation, particularly within adipose tissue itself (Hotamisligil, 2006; Hotamisligil et al., 1993; Nishimura et al., 2008; Shoelson et al., 2006), is critically linked to obesity and its accompanying metabolic disorders, including impaired glucose tolerance, insulin resistance, hepatic steatosis, dyslipidemia and eventually Type 2 diabetes (Reaven, 1988). Adipose tissue is immunologically dynamic, with resident CD4+ (Winer et al., 2009) and CD8+ T cells (Nishimura et al., 2009), T regulatory (Treg) cells (Feuerer et al., 2009), B cells (Winer et al., 2011) and macrophages (Lumeng et al., 2007; Weisberg et al., 2003; Wentworth et al., 2010), each of which have been shown to play positive or negative roles in metabolic dysregulation and the development of obesity (Feuerer et al., 2009; Lumeng et al., 2007; Nishimura et al., 2009; Wentworth et al., 2010; Winer et al., 2011; Winer et al., 2009).
Chronic low grade inflammation in obese fat may activate resident innate immune cells, leading to inappropriate immune responses and development of insulin resistance (Xu et al., 2003). Likewise, inflammation associated with adiposity is partly generated by fat-resident immune cells, the most widely studied being macrophages, which infiltrate fat during obesity and undergo phenotypic switching, leading to the development of insulin resistance (Lumeng et al., 2007; Weisberg et al., 2003). Macrophage activation is modified by T cells. IFNγ production by T cells enhances pro-inflammatory M1 macrophage differentiation, while T cells secreting anti-inflammatory cytokines such as IL-4, IL-13 and IL-10 promote anti-inflammatory M2 macrophage development (Tiemessen et al., 2007)(Kim et al., 2008). Classical CD11c expressing M1 macrophages (Fujisaka et al., 2009; Lumeng et al., 2007) are often found aggregated around necrotic adipocytes and produce excess pro-inflammatory cytokines such as IL-6 and TNF-α, characteristic of obese adipose tissue in humans (Wentworth et al., 2010) and mice (Lumeng et al., 2007). Alternatively-activated M2 macrophages (F4/80+CD11c−CD206+) found in lean adipose tissue generate high amounts of anti-inflammatory cytokines such as IL-10 but are decreased in obesity (Fujisaka et al., 2009; Lumeng et al., 2007). While the importance of macrophages and their interaction with adipocytes and other immune cells is now accepted as key in the development of adipocyte and metabolic dysfunction, the mechanisms regulating these interactions are not well understood.
We have explored a role for the innate T lymphocyte population of invariant natural killer T (iNKT) cells in obesity and have previously shown that iNKT cells are enriched in human adipose tissue, but are decreased in human obesity (Lynch et al., 2009). iNKT cells comprise a unique innate T cell population which are highly conserved and express an invariant T cell receptor (TCR), Vα24Jα18, paired with Vβ11 in humans and Vα14Jα18 coupled with TCR Vβ7, Vβ8.2 or Vβ2 in mice. This specific TCR recognizes glycolipid ligands presented by the MHC-like molecule CD1d (Brigl and Brenner, 2004; Gumperz, 2006; Matsuda et al., 2000). The most studied lipid antigen is alpha-galactosylceramide (αGC)(Matsuda et al., 2000), as physiological lipid ligand(s) have yet to be fully defined (Fox et al., 2009; Speak et al., 2007). A striking feature of iNKT cells is their rapid production of both Th1 and Th2 cell cytokines upon activation with αGC (Bendelac et al., 2007; Berzins et al.; Matsuda et al., 2008), likely accounting for their immunoregulatory potential. Indeed, iNKT cell cytotoxicity and rapid production of IFNγ have been shown to be protective against tumor development, while iNKT cells also play immunoregulatory roles in Type 1 diabetes, multiple sclerosis and rheumatoid arthritis (Berzins et al.; Swann et al., 2007; Wu and Van Kaer, 2009). Our previous study reported that iNKT cells are enriched in human fat at higher numbers than elsewhere in the body (Lynch et al., 2009). Here we have shown that, like humans, murine adipose tissue was enriched with iNKT cells. Under physiological conditions of clonal abundance, iNKT cells likely exceed the number of a given antigen-specific T cells by up to 10,000-fold, and thus are likely to play an important immune regulatory role in adipose tissue. Furthermore, we showed that iNKT cells resident in fat represent a unique subset with distinct Th2 cell cytokine production, compared to iNKT in liver and spleen. Given the potent influence of other fat-resident immune cells on the development of obesity and metabolic outcomes, the conservation of iNKT cells in fat in mammals, and the unique anti-inflammatory profile of fat-resident iNKT cells, we hypothesized that local fat-resident iNKT cell populations would play a physiological role in adipose tissue regulation. Our data confirm this hypothesis and show that iNKT cells confer protection against the development of metabolic syndrome and inflammation following a high fat diet challenge.
Results
iNKT cells are depleted in fat and liver during the development of obesity
We have previously shown that iNKT cells are enriched in human fat but are depleted in obesity (Lynch et al., 2009). As it was not possible to obtain adipose tissue from patients at multiple time points post-bariatric surgery, we looked in human peripheral blood and found that iNKT cells were also reduced in the circulation of obese patients compared to lean healthy age-matched controls (Fig. 1a). Cross-sectional analysis found that circulating iNKT cell numbers were increased in obese patients who had lost weight after Roux-en-Y gastric bypass (RYGB) surgery, compared to obese patients pre-RYGB surgery, although iNKT cells were still reduced compared to lean controls (Fig. 1a). We then followed a group of patients (n=7) longitudinally pre- and post-RYGB surgery, whose body mass index (BMI) decreased from Grade III obesity (mean BMI>50 kg/m2) to Grade II obesity (mean BMI 35–40 kg/m2) 18 months post-surgery (Fig. 1a). Peripheral iNKT cell numbers increased in each patient after weight loss. (Fig. 1a).
Figure 1. iNKT cells are depleted in obesity, but are restored following weight loss in mice and humans.

(a) iNKT cells (expressed as a % of T cells) in peripheral blood of obese patients before bariatric surgery (pre-op) (BMI>50, n=26) compared to lean age-matched controls (BMI= 20–25, n=22) and unmatched patients 18 months after surgery (postop) (n=18, p=0.0002; Mann Whitney test). 7 patients were analyzed both pre- and post-bariatric surgery. Middle: BMI of patients before and 18 months post-op.. Right: Peripheral iNKT cell levels in each patient (n=7, paired t-test). (b) Left: iNKT (αGC loaded tetramer+) as % of T cells (top) and unloaded CD1d tetramer negative control (bottom) in a representative sample of matched spleen, liver and fat. Right: iNKT cells (expressed as a % of T cells) in each individual wt spleen, liver and fat from 6–10 week old mice (n=24). (c) Left: Representative flow cytometric plots showing percentage of iNKT cells producing each cytokine intracellularly following αGC injection (representative of n=4), Right: Concentrations of intracellular cytokines production by iNKT cells, mean + s.d. (n=4). (d) iNKT cytokine production after in vitro stimulation with αGC-loaded CD1d transfected C1R cells (3 independent experiments performed in duplicate). (e) Representative dot plots of iNKT cell (αGC-loaded tetramer+) numbers in matched fat, liver and spleen from 10–14 week old mice on a SFD or HFD (representative of 11 experiments). (f) iNKT cells in fat, liver and spleen from mice on HFD for 8 weeks (n=11) or ob/ob mice (n=3) compared to age-matched mice on SFD (n=11). (g) iNKT cell numbers measured each week on HFD in matched fat, liver and spleen (n=4 per week). See also Fig. S2. (h) Mice were removed from HFD after 6 weeks (n=4) and after 10 weeks (n=4). Graphs show overall weight (total) and fat pad weight during HFD (green bars) and after removal from HFD (orange bars) after 6 weeks (p=0.0007) or 10 weeks (p=0.001). iNKT numbers in fat, liver and spleen after removal from HFD. Graph show mean+ s.d. n=4. *p<0.05, **, p<0.01, ***p<0.0001
To further explore the relationship between obesity and iNKT cells, we turned to murine models. We first determined whether iNKT cells were also present in murine fat. As in humans, iNKT cells are enriched in murine adipose tissue (Fig. 1b) at numbers equivalent to those found in murine liver. Fat-derived iNKT cells produced significantly less IFN-γ, and more IL-4 and IL-10 compared to iNKT cells from liver and spleen following αGC activation in vivo (Figure 1c) and in vitro (Figure 1d). We also compared other lymphoid populations from adipose tissue to those from liver and spleen. Similar to liver, fat had high levels of NK and NKR+ T cells, and high numbers of CD4−CD8− double negative (DN) T cells, reflecting the increased numbers of DN iNKT cells in liver and fat (Fig. S1a). Furthermore, of the iNKT cells, there were less CD4+ iNKT and more DN iNKT cells in fat compared to liver and spleen (Fig. S1b). Adipose tissue contained a small, but significant population of Treg (FoxP3+CD25+CD4+) cells (6%) similar to spleen (7% of T cells), in agreement with numbers previously reported (Feuerer et al., 2009) (Fig. S1a & S1c). iNKT cells were previously shown to express FoxP3 in certain circumstances (Monteiro et al., 2010). Given the high levels of iNKT cells and the substantial popuation of Treg cells in fat, it was necessary to determine whether there was any overlap between both populations. Fig. S1c shows that iNKT cells are distinct from FoxP3+ Treg cells. Thus, the immune repertoire in fat is unique, but with similarities to both liver in NK, NKR+T and iNKT levels, as well as to spleen in Treg cell levels.
We next investigated the effects of obesity on iNKT cells using two murine models of obesity; diet-induced obesity (DIO) or obesity due to leptin deficiency (ob/ob). Mice fed a high fat diet (HFD) for 6 weeks had markedly reduced numbers of iNKT cells in adipose tissue and liver (Fig. 1e,f). iNKT cell depletion was even more pronounced in ob/ob mice (Fig. 1f), which were heavier and had higher fasting blood glucose than DIO mice. We next analyzed iNKT cell numbers during the development of obesity. iNKT numbers in fat were reduced as early as week 2 of HFD and steadily declined each week during the course of the HFD (Fig. 1g). iNKT levels also declined in the liver upon HFD challenge, with significant differences seen from week 6 onwards. Splenic iNKT cell levels fluctuated, with a trend towards depletion at week 10 (Fig. 1g). We also calculated absolute numbers of iNKT cells per organ and per gram of fat (Table S1). We estimate that lean adipose tissue contains 7.3 × 104 iNKT cells, and in obesity, the numbers were reduced to 2.2 × 104 iNKT cell per 2 epididymal fat pads. This compares with 7.3 × 104 iNKT in liver from lean mice and 3 × 104 iNKT in liver from obese mice. We observed no changes in activation markers CD69 and CD25 on iNKT cells in obesity (not shown). We also looked at fluctuations in other T cells in obese fat. CD8+ T cells increased in obese fat, there were no significant obesity-induced changes in other T cells in liver, and there was a decrease in CD4+ T cells and an increase in CD8+ T cells in the spleens of obese mice (Fig S1d).
HFD was replaced by standard fat diet (SFD) for a week after 6 weeks or 10 weeks, which caused only a slight drop in total weight, but a dramatic reduction in fat pad weight (Fig. 1h). After switching to SFD after 6 weeks of HFD, there was a significant increase in iNKT cells in fat and liver; iNKT levels also began to increase after switching to SFD after 10 weeks of HFD (Fig. 1h). These findings indicate that murine and human iNKT response to obesity and weight loss is similar.
Contribution of iNKT cells to the development of obesity
We next explored the hypothesis that iNKT cells may protect against obesity and related metabolic consequences. Jα18-deficient mice, lacking iNKT cells, but having an otherwise normal immune system, were fed HFD alongside age-matched wt mice on HFD or SFD. Jα18-deficient mice were significantly larger before HFD challenge. They also gained significantly more weight than wt mice on HFD, and had significantly larger fat pads, while lean mass was unchanged (Fig. 2a) Food intake was similar between Jα18-deficient and wt mice (Fig. 2b). Adipocytes were larger in Jα18-deficient mice compared to wt on HFD (Fig. 2c,d). Furthermore, Jα18-deficient mice on HFD had higher fat deposition in liver (Fig. 2e), an elevated fasting blood glucose, impaired GTT, and increased insulin resistance compared to wt on HFD (Fig. 2f). Serum leptin concentrations were equivalent in wt and Jα18-deficient mice on HFD compared to SFD (Fig. 2g).
Figure 2. Impact of iNKT cell deficiency on weight gain, glucose tolerance, adipocyte size and number and fat accumulation in liver.

(a) Weight of Jα18−/− and wt mice on commencement of and during 8 weeks of HFD compared to wt on SFD (n=4 per group per week). Overall weight gain afer 8 weeks of HFD. Lean mass and epididymal fat pad weight of wt and Jα18−/− mice on HFD, wt mice on SFD are shown for comparison. (b) HFD food intake of wt and Jα18-deficient mice. (c) Adipocyte diameter was measured on osmium-fixed adipocytes with a particle counter. Adipocyte size from Jα18−/− and wt mice on HFD (4 samples per mouse, 4 mice per group). (d) Histology of adipocytes from epididymal fat. Adipocyte size from wt mice on SFD, wt mice and HFD and Jα18−/− mice on HFD. ob/ob mice also shown for comparison. Scale bars, 100μm. (e) Histology of fat infiltration in liver of wt and Jα18−/− mice on HFD (Representative of 4 individual experiments). (f) Fasting glucose (left), glucose tolerance (middle) and insulin resistance (right) in wt on SFD, wt on HFD, and Jα18-deficient mice on HFD for 6 weeks (n=4 per group, t tests, and 2 way ANOVA with Tukey for glucose tolerance tests). Insulin resistance as measured by HOMA-IR (t test). (g) Serum leptin levels in wt and Jα18-deficient mice on HFD compared to wt on SFD (n=4 per group, ANOVA). Graphs show mean+ s.d. *p<0.05, **, p<0.01, ***p<0.0001
We examined DIO in another mouse model lacking iNKT cells; Cd1d1−/− mice, which lack the restriction element for iNKT cells and therefore also selectively lack iNKT cells. Cd1d1−/− mice gained significantly more weight than wt mice on a HFD (Fig. S2a). Cd1d1−/− mice on HFD had higher fasting blood glucose and worse glucose handling than wt on HFD. Fasting insulin and insulin resistance were not significantly higher, possibly due to higher variability inherent in these insulin assays (Fig. S2b,c). Cd1d1−/− mice also had larger adipocytes, as measured by immunohistochemistry (Fig. S2d).
The above experiments were all performed on males. As there have been reported gender differences in some of the metabolic complications of obesity (Fox et al., 2007; Medrikova et al., 2011), we also investigated metabolic outcome in female Jα18-deficient versus female wt mice on HFD (Fig. S3). Like males, female Jα18-deficient mice gained significantly more weight in the first 4 weeks of HFD challenge; thereafter, weight gain was increased but not signifcantly different, compared to wt (Fig. S3a). This correlated with eating behaviour; at 4 weeks female Jα18-deficient mice had reduced food intake compared to wt females, unlike males which had almost identical food intake patterns in wt and Jα18-deficient mice (Fig. S3b). Lean mass was similar, but both overall fat mass and fat pad weight were significantly higher in Jα18-deficient females (Fig. S3a). Adipocytes were significantly larger and fewer in number (Fig. S3c) and the degree of fatty liver was greater in Jα18-deficient females compared to wt on HFD (Fig. S3d). However in contrast to males, fasting glucose was unchanged and GTT was not impaired in Jα18-deficient females compared to wt females on HFD (Fig. S3e). Our findings agree with other obesity studies (Fox et al., 2007; Medrikova et al., 2011) illustrating that female mice have larger fat pads and adipocytes but are less susceptible to HFD-induced glucose impairment and insulin resistance than male mice
iNKT cell numbers correlate inversely with macrophage infiltration in adipose tissue
iNKT cells can recruit and regulate other immune cells (Cerundolo et al., 2009). We investigated the influence of adipose-derived iNKT cells on macrophage infiltration and activation. As expected, pro-inflammatory macrophages (F4/80+CD11c+) increased in adipose tissue during the development of obesity, with significant increases detectable as early as 1 week into HFD. After cessation of HFD for 1 week, proinflammatory macrophages were significantly decreased in fat (Fig. 3a). We found a strong inverse correlation between iNKT cell levels in fat and proinflammatory macrophages (Fig. 3a).
Figure 3. The relationship between iNKT cells and macrophages.

(a) Left, Proinflammatory macrophages (Pro-inflam Macs; F4/80+ CD11c+ MMR+) levels in fat (as % stromovascular fraction (SVF) of wt mice fed HFD (red) or SFD (blue) for 10 weeks. Dashed line shows when HFD was replaced with SFD (black). Right, Correlation between iNKT cell levels and macrophage number in fat, Pearson r= −0.9612, p=0.0001. (b) Left: Representative dot plots of % F4/80+ total macrophages per fat pad (top), and the % macrophages that are CD11c+ (proinflammatory) (middle) and the % macrophages that are CD206+ (anti-inflammatory) (bottom) in wt SFD, wt HFD and Jα18-deficient on HFD. Right: Levels and phenotypes of macrophages from individual mice groups (n=4 mice per group), including number of CD68+ macrophages as measured by immunohistochemistry (n=20 low power field (LPF) per group). (c) Immunohistochemical staining of F4/80+ macrophages in fat from wt on SFD, wt on HFD and Jα18−/− mice on HFD, and ob/ob mice on SFD (Representative of 4 mice per group). (d) Immunohistochemical staining of CD68+ M1 macrophages in fat from wt on SFD, and wt and Jα18-deficient on HFD (representative of 4 mice per group). Scale bars, 100 μm. *p<0.05, **, p<0.01, ***p<0.0001
To determine whether iNKT cells play a causal role in the infiltration and phenotype of macrophages, we investigated macrophage numbers in Jα18-deficient mice in obesity. In the absence of iNKT cells, total macrophage numbers were higher in adipose tissue (Fig. 3b,c,d). Adipose tissue macrophages displayed a trend towards increased proinflammatory (CD11c) and decreased anti-inflammatory (CD206) phenotype in Jα18-deficient mice, compared to wt on HFD (Fig. 3b). Cd1d1−/− mice had similarly high numbers of F4/80 macrophages in adipose tissue as wt mice on HFD (Fig. S2e) but had significantly more proinflammatory macrophages than wt mice on HFD (Fig. S2f).
Mice lacking iNKT cells show metabolic disorder on SFD
Both Jα18-deficient and Cd1d1−/− mice have overtly normal immune systems with no pathological susceptibilities, unless challenged with certain pathogens or tumors. We investigated if there was any evidence of metabolic syndrome in Jα18-deficient and Cd1d1−/− mice fed ad lib for 4–5 months on SFD. Both Jα18-deficient mice and Cd1d1−/− mice consistently weighed more (Fig. 4a) and had larger adipocytes on SFD compared to aged-matched wt mice (Fig. 4b). Adipose tissue macrophages were markedly increased in both iNKT-deficient, but had similar proinflammatory (not shown) and anti-inflammatory phenotypes in each group on SFD (Fig. 4c). Consistent with these findings, both Jα18-deficient and Cd1d1−/− mice had greatly increased serum triglycerides, TNFα concentrations, and somewhat elevated IL-6 concentrations (Fig. 4d). Jα18-deficient mice and Cd1d1−/− mice on SFD had elevated fasting glucose, and GTT was slightly impaired in Cd1d1−/− mice although these differences were not significant (Fig. 4e).
Figure 4. iNKT null mice have more pro-inflammatory cytokines and macrophages on a SFD.

(a) Weight of wt, Jα18−/−, and CD1d1−/− mice fed SFD ad lib until 20 weeks of age (n=3 per group, one-way ANOVA with post-hoc Tukey). (b) Adipocyte size in wt, Jα18-deficient and CD1d1−/− mice on SFD (representative of 3 mice per group). (c) Macrophage level and phenotype in the 3 mice groups on SFD. Left top, total macrophages, and bottom, M2 (CD206+) macrophages, CD206 hi (top gate), CD206lo (middle gate) and CD206− (bottom gate). (d) Fasting serum triglyceride (TGL) concentration in the 3 groups (n=3, all one-way ANOVA with post-hoc Tukey). Serum TNFα and IL-6 concentration in wt and Jα18−/− mice on SFD (n=3, t test), CD1d1−/− mice not tested. (e) Fasting glucose and glucose tolerance of 20 week old wt, Jα18−/−, and CD1d1−/− mice on SFD. Graphs show mean+ s.d. *p<0.05, **, p<0.01, ***p<0.0001.
Transfer of iNKT cells in obese Jα18-deficient mice improves glucose handling
To directly test the hypothesis that iNKT cells play a protective role against the development of obesity-induced metabolic syndrome, we adoptively transfered 5×105 wt hepatic iNKT cells into obese mice (>8 weeks on HFD), who were continued on HFD for 4 days. Adoptive transfer of 5×105 NKT-depleted T cells or PBS were used as controls. Adoptive transfer of iNKT cells resulted in significant weight loss and a dramatic reduction in adipocyte size, compared to transfer of T cells or PBS (Fig. 5a and b). Mice that received NKT cells had similar numbers of macrophages as mice that received T cells but had less pro-inflammatory marker CD11c expression (Fig. S4). Food intake was similar between mice receiving iNKT transfer or PBS alone (not shown). Mice that received iNKT cells had significantly smaller fat pads than PBS controls, while fat pad size in the T cell recipients was intermediate between PBS and iNKT cells (Fig. 5c). Importantly, lean control mice that recieved the same number of iNKT cells did not lose weight (Fig. 5d) or have hypoglycemia (Fig. 5e) compared to PBS alone. Obese mice that received iNKT cells had significantly lower fasting glucose (Fig. 5f) and improved GTT (Fig. 5g) compared to mice receiving PBS, while T cell recipients were again intermediate between PBS and iNKT recipients. Insulin sensitivity improved after iNKT cell transfer, but not T cell transfer (Fig. 5h). Next, we cultured fat pads overnight from mice that received NKT cells or PBS in vivo and performed an array analysis on the supernatant from the cultured fat pads. Mice that received NKT cells had similar amounts of the adipokine resistin, but had signifcantly less leptin and significantly more adiponectin. Furthermore, angiopoietin-like 3 (ANGPTL3) production was greatly reduced, and IL-10 production was increased, likely produced by iNKT cells (Fig. 5i).
Figure 5. Adoptive transfer of iNKT cells protect from weight gain and adipocyte hypertrophy and reverse obesity-associated metabolic disorder.
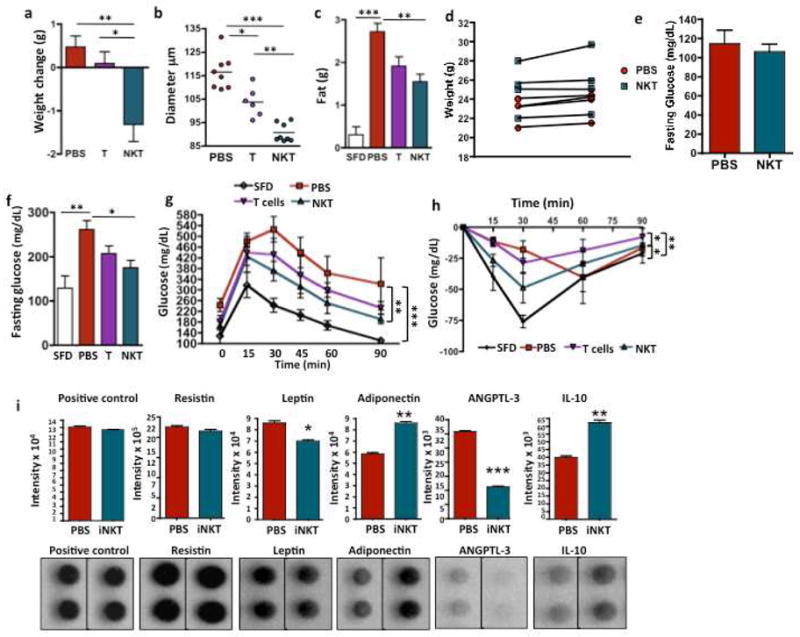
Wt iNKT cells (>95% pure), iNKT cell-depleted T cells (T), or no treatment (PBS) control were injected ip into 18–20 wk old diet-induced obese mice on HFD for 12 weeks which continued on HFD for 4 days. (a) Difference in weight pre-and 4 days post-adoptive transfer of mice that received iNKT cells (n=10) or PBS (n=6) or T cells (n=6). (b) Adipocyte diameter from obese mice that received iNKT cells vs T cells or PBS control, 2 samples per mouse, n=4 for PBS and NKT, n=3 for T cells, ANOVA with Tukey). (c) Epididymal fat weight after iNKT transfer (n=10) compared to PBS (n=6) or T cell transfer (n=6) and wt on SFD as comparison (n=4). (d) Weight change and (e) fasting glucose in lean wt mice that relieved NKT cell transfer. (f) Fasting glucose (ANOVA) and (g) glucose tolerance following iNKT (n=8) transfer compared to PBS (n=4) or T cell transfer (n=4) with wt on SFD for comparison (n=4, 2 way ANOVA) (h) Insulin tolerance test in obese mice was measured 4 days post transfer of iNKT cells (n=4), PBS (n=4), T cells or wt on SFD (n=3) (2 way ANOVA with Tukey post-hoc). (i) Following adoptive transfer of iNKT cells for 3 days, adpose tissue was harvested and cultured. Production of resistin, leptin, adiponectin, ANGPTL3 and IL-10 were measured by protein array (n=2 of 2 pooled mice each), Graphs show mean+ s.d. *p<0.05, **, p<0.01, ***p<0.0001.
αGC treatment causes weight loss, adipocyte hypertrophy and improves fatty metabolic disorder
We investigated whether αGC, the prototypical ligand for iNKT cells, could activate residual iNKT cells in obesity and subsequently improve metabolic outcome. Following one injection of αGC into wt obese mice (HFD >8 weeks), and continued HFD for 4 days, mice lost a significant amount of weight; their percentage body fat also decreased significantly, although lean mass did not change (Fig. 6a,b). αGC also caused a rapid reduction in adipocyte size (Fig. 6c). Food intake was similar between treatment and control. αGC caused a marked reduction in fasting blood glucose compared to vehicle control (Fig. 6d). Obese Jα18-deficient mice lacking iNKT cells did not lose weight after αGC treatment (Fig. 6a) or have improved glucose concentrations, which were extremely high (Fig. 6d). αGC returned GTT almost to normal in wt HFD, unlike in obese Jα18-deficient mice. αGC treatment did not significantly affect fasting glucose or GTT in normal weight mice on SFD (Fig. 6d). αGC significantly improved insulin resistance as measured by Homeostasis model of assesment- insulin resistance (HOMA-IR), although the ITT was not significantly different due to variability in baseline fasting insulin (Fig. 6e). αGC lowered serum triglycerides, circulating leptin and IL-6 in obese wt mice (Fig. 6f). Surprisingly, TNFα concentrations were increased (Fig. 6f). IL-4 was not significantly increased after αGC and serum IL-10 was below the detection range (not shown). αGC treatment improved fatty liver; fat droplets were smaller and less frequent, compared to mice that received vehicle (Fig. 6g).
Figure 6. αGC treatment reverses obesity-associated metabolic disorders.

(a,b) Effect of αGC treatment on weight gain and adipocytes. (a) Difference in weights before and 4 days after αGC or vehicle (VEH) treatment. (n=7 per group, t test). (b) Lean mass and % body fat measured by DEXA following αGC treatment (n=5, t test). (c) Adipocyte size following αGC treatment (2 samples per mouse, n=4 mice, t test). (d) Oil Red O staining in liver samples from wt obese mice 4 days post αGC injection. 2 representative images per treatment (representing 5 mice per group). (e) Fasting glucose and GTT of wt obese mice 4 days post αGC injection (n=5, fasting glucose: t test; GTT: 2 way ANOVA with post hoc shown; Area under curve ***p=0.0007). (f) Glucose tolerance test following αGC treatment into lean wt mice on SFD (n=4 per group). (g) Insulin resistance as measured by HOMA-IR and (h) insulin tolerance test following aGC treatment (n=4). (i) Circulating triglyceride (TGL), leptin IL-6, TNF-a, and IL-4 following aGC treatment (n=5 each, all t tests. Graphs show mean+ s.d. *p<0.05, **, p<0.01, ***p<0.0001.
We examined iNKT cells 4 days post-treatment with αGC, at the time of metabolic analysis. αGC caused dramatic expansion in numbers of iNKT cells in fat, as well as a smaller but significant expansion in spleen and liver (Fig. 7a). αGC activated iNKT cells to produce anti-inflammatory cytokines (Fig. 7a), likely leading to the improvement in metabolism despite high fat diet feeding (Fig 6 a–e), although iNKT cells also produced some IFNγ following αGC. Furthermore, iNKT cells in adipose tissue were still producing cytokines 4-days post-injection, unlike those in spleen and liver (Fig. 7a). We next investigated whether αGC could mediate metabolic protection if IL-4 and IL-10 were neutralized before αGC injection. When both IL-4 and IL-10 were neutralized immediately prior to αGC treatment, mice still lost weight (Fig. 7b), however fasting glucose concentrations were elevated compared to αGC treatment alone (Fig. 7c). Furthermore neutralizating these cytokines resulted in increased glucose concentrations during the GTT at 60 and 90 min, like vehicle control, and unlike αGC alone which significantly improved GTT at each time point. Blocking these cytokines resulted in increased insulin resistance at 15 and 30 min timepoints compared to αGC treatment alone (Figure 6e). We next blocked these cytokines individually. Neutralization of IL-4 alone prior to αGC partly prevented the improvement in fasting glucose seen with αGC (Fig. S6a& S6c). It was possible that blocking IL-4 or IL-10 without αGC could also be beneficial through another pathway such as macrophages, however, neutralizing antibody treatment alone without αGC did not improve or worsen GTT compared to with PBS (Fig. S5d).
Figure 7. αGC treatment expands adipose iNKT cells and activates their IL-4 and IL-10 production resulting in improved metabolic health.

(a) Left: iNKT cell levels in adipose tissue, liver and spleen at 4 days post-αGC injection. Right: IFNγ, IL-4 and IL-10 production by iNKT cells from each organ post-activation. Quadrant percentages represent cytokine production by iNKT cells after subtraction of control (control for fat shown on bottom row). (b) Obese mice received neutralizing IL-4 and IL-10 (n=7) antibodies before αGC treatment. Weight loss post treatment with or without neutralizing antibodies. (c) Glucose tolerance test and insulin resistance test in obese mice where IL-10 and IL-4 were neutralized prior to αGC (n=7, 2-way ANOVA and t test). *p<0.05, **, p<0.01, ***p<0.0001.
Discussion
We have identified a role for iNKT cells in the regulation of body weight and metabolic state. Our results indicate that iNKT cells may act through regulation of inflammation in adipose tissue, although whether this effect is direct or indirect is not fully clear. Previous data from human and murine studies suggest that adiposity due to diet (and negative energy expenditure) is the trigger for increased adipose tissue inflammation, which subsequently leads to insulin resistance and metabolic disorder. In obesity, excess lipid leads to larger, stressed adipocytes that produce pro-inflammatory adipokines and cytokines, leading to downstream negative effects on insulin sensitivity. Influencing the function of adipocytes has potentially significant health benefits. We and others have shown that increasing adipocyte size correlates with insulin resistance, triglyceride levels and degree of hepatic steatosis (Lonn et al., 2009; O’Connell et al., 2010). One hypothesis is that adipocytes are dysfunctional when filled to capacity (~3μg lipid/cell) (Danforth, 2000), causing ‘overflow’ of lipids to muscle and liver (Gregor and Hotamisligil, 2007), leading to over-production of glucose and high fatty acid levels, which can activate the kinases JNK and IKK leading to insulin resistance at these sites (reviewed by Guilherme et al., 2008; Hotamisligil and Erbay, 2008). iNKT cell-deficient mice had larger adipocytes, even on SFD, and when iNKT cells were reintroduced or activated, adipocytes rapidly reduced in size. Importantly, iNKT cell transfer did not cause weight loss or hypoglycemia in lean control mice. Furthermore mice that received iNKT cells had significantly higher levels of adipose tissue adiponectin, an insulin-sensitizing adipokine produced largely by adipocytes. Moreover, fat from iNKT cell transfers had significantly less leptin and ANGPTL2 production, which are key mediators of chronic adipose tissue inflammation and insulin resistance. The production of anti-inflammatory cytokines by iNKT cells may have direct affects on adipocytes. IL-10, which was increased in adipose tissue following iNKT cell transfer, can protect adipocytes from the physiological insulin desensitizing effects of TNF-α seen in obesity (Hong et al., 2009; Lumeng et al., 2007), and likely leads to ‘healthier’ adipocytes, reflected in lower levels of circulating pro-inflammatory cytokines and leptin.
iNKT cells may also indirectly effect adipocytes through their effects on macrophage function. The production of IL-10 was unique to iNKT cells in adipose tissue and IL-10 and IL-4 are potent inhibitors of pro-inflammatory cytokines (Williams et al., 2004). IL-10 promotes a phenotypic switch in macrophage activation towards an anti-inflammatory phenotype (Williams et al., 2004). Adipocyte hypertrophy can lead to cell death, triggering an inflammatory response from macrophages, which form crown-like structures surrounding dead adipocytes. This causes a phenotypic switch in macrophages from M2 to M1, which also contributes to adipose tissue inflammation (Dalmas et al., 2011). iNKT numbers were strongly inversely correlated with pro-inflammatory macrophage numbers in fat, and reintroduction or activation of iNKT cells caused a marked decrease in adipose tissue-associated macrophages. These findings, and another recent study (Winer et al., 2009), show that adoptive transfer of conventional T cells offered some relief in the metabolic syndrome, compared to PBS, although the effect was not as potent as iNKT cell transfer. The somewhat protective effect of T cell transfer into obese mice is not surprising given that CD4+ T cell transfer into obese mice reversed weight gain and insulin resistance (Winer et al., 2009), an improvement that was found to be due to both FoxP3 positive and negative CD4 T cells. Another recent study found that Tregs alone did not significantly restore all metabolic function in obesity (Feuerer et al., 2009), although this could be due to difficulties in complete gain or loss experiments involving Treg cells. Our conventional T cell transfer also included Treg cells, which likely contributed to the benefits in metabolism. Conversely, it is possible that iNKT cells were likely a contaminant in previous studies that used adoptive transfer of CD4+ T cells in obesity, given that iNKT cells are a sub-population of CD4+ T cells in fat, liver and spleen. From these combined data, we could draw the conclusion that iNKT cells are sufficient, and possibly more effective at improving glucose handling, but are not the only immune cell type in adipose tissue that are involved in metabolic regulation.
Our results from two iNKT cell–deficient models show that mice lacking iNKT cells are more susceptible to HFD, and even on a SFD, gained more weight with age than controls. In this study, we did not use littermate controls in our iNKT-deficient models due to lack of availability. Therefore there is a possibility that weight gain or metabolic changes seen could be due wholly or partly due to different weaning or early changes, which have been shown to be very important in metabolism during the life of animals. In this case, we believe this is unlikely, since iNKT cells are thought to arise postnatally at day 5 in the thymus and at 1–2 weeks in the periphery (Pellicci et al., 2002). We found that wt and mutant mice had similar sized litters with similar birth weight; additionally, it was only with age (>4 months) that differences were seen on a SFD. Our results also suggest that the severe obesity in leptin deficient ob/ob mice causes iNKT cell depletion. The presence of leptin itself does not appear to be essential for iNKT cell survival, as leptin levels are elevated in obesity, while iNKT cells are depleted. Previous studies show that ob/ob mice have aberrant hematopoiesis and immune defects (Fantuzzi and Faggioni, 2000; Macia et al., 2006) including decreased iNKT cells in livers from ob/ob mice (Yang et al., 2007). As iNKT cells are thought to be absent at birth (Kronenberg and Gapin, 2002; Pellicci et al., 2002), and ob/ob mice are already significantly larger than their wt littermates very early after birth, excess adiposity cannot be ruled out.
The role of iNKT cells in regulation of metabolism is just emerging, although it has previously been described that iNKT cells are reduced in liver of obese mice (Ma et al., 2008; Yang et al., 2007) leading to hepatic insulin resistance and that increasing iNKT cells in obese liver improved hepatic steatosis and glucose tolerance (Ma et al., 2008), (Elinav et al., 2006). During the review of our manuscript, Ji et al, also reported that iNKT cells are decreased in human and murine obesity, confirming our previous (Lynch et al., 2009) and current findings. Furthermore, both our groups found that activation of iNKT cells with αGC led to macrophage polarization to an M2 phenotype and improved glucose sensitivity through anti-inflammatory cytokine signaling (Ji et al., 2012). Similarly, Kotas et al. found that iNKT cells were selectively depleted in obese livers and that Cd1d1−/− mice had slight, but significantly increased steatosis and impaired hepatic glucose tolerance (Kotas et al., 2011). Their findings, however, were not as dramatic as our results in obese iNKT-deficient mice. Furthermore, the protective role of iNKT cells was further questioned when Ohmura et al. reported that mice lacking iNKT cells were protected against obesity. However, this study used β2-microglobulin-deficient mice, which lack both CD8+ T cells and iNKT cells and CD8 T cells are reported to be pathogenic in obesity (Nishimura et al., 2009). To address this, Mantell et al. used CD1d−/− mice and in contrast to ours and other studies, found no difference between CD1d−/− and wt mice on a HFD (Mantell et al., 2011), although the conclusion was drawn that the deletion of NKT cells was insufficient to protect against the development of the metabolic abnormalities of obesity. Another study (Satoh et al., 2012) described that CD1d−/− mice gained less weight than wt mice with less steatohepatitis, implying that NKT cells are pathogenic in obesity in contrast to previously published studies (Elinav et al., 2006; Ji et al., 2012; Kotas et al., 2011; Ma et al., 2008; Yang et al., 2007). These discrepancies between iNKT-deficient studies are difficult to resolve, although there are some key differences in the experimental approach between studies. Most importantly, Mantell et al use NK1.1+ T cells as a marker of iNKT cells when not all invariant NKT cells express NK1.1, and in fat, iNKT cells express less NK1.1 than elsewhere, and lastly not all NK1.1+ T cells are CD1d-restricted. However, this study found that iNKT cells were depleted in obesity, similar to ours and others that describe a loss of iNKT cells in obesity, leading to worsening metabolic outcome. The discrepancies in results with iNKT-deficient mice between different labs may not yet be explained, but the adoptive transfer of iNKT cells into obese mice and specific activation of iNKT cells with αGC provide a more conclusive readout of the positive benefits of iNKT cells against obesity related disorder. Furthermore, our murine data parallel our human results on the effects of weight gain and loss on iNKT cells.
In humans, iNKT cell levels correlate negatively with weight gain (Lynch et al., 2009). Following return to SFD in mice, or weight loss in humans, iNKT levels are increased, demonstrating the reversibility of iNKT cell defects. Likewise, despite severe numerical defects, the reduced iNKT cell pool in murine obesity is still capable of marked expansion and production of anti-inflammatory cytokines, leading to dramatically positive effects on adipocytes and metabolism. Whether treatments that increase or activate iNKT cells will have beneficial effects in humans with metabolic syndrome remains to be determined. The use of αGC to activate iNKT cells in humans has been used in multiple cancer settings. It has proven to be safe, with no reports of hepatotoxicity, unlike in mice where repeated higher doses in older animals can cause hepatotoxicity (Exley et al., 2011). The observation that targeting the iNKT cell system with αGC in mice did not cause hypocglycaemia in the diabetic or euglycaemic model suggests a restoration of physiological balance. This is potentially very attractive from a therapeutic perspective.
Immuno-metabolic interaction in obesity is now established as a key factor in adipose tissue inflammation and subsequent development of type 2 diabetes. It also holds exciting possiblities in the development of a new treatment paradigm for this disorder which is currently at epidemic proportions. Our study supports the emerging view that T cells and macrophages play important roles in adipose tissue function, and identifies iNKT cells as a major regulatory T cell population in fat. Further studies exploring the therapeutic potential of iNKT cells in obesity and metabolic syndrome are warranted.
Experimental Procedures
Mice
Male (and where indicated, female) wt C57BL/6 and ob/ob mice were purchased from Jackson Laboratories (Bar Harbor, ME). Jα18-deficient mice and C57BL/6J CD1d1−/− mice have been described(Exley et al., 2003). Jα18-deficient (F9) and CD1d1−/− (F12) were backcrossed on the C57BL/6 background. Mice were house adjacently from birth and fed the same chow (SFD or HFD). In general, experiments began with six-wk-old male mice. For metabolic studies, the mice received either SFD or HFD (Research Diets, 60 kcal% fat for the HFD), from 6 weeks of age for 6 weeks, or 12 weeks where indicated. Mice were housed under specific pathogen-free conditions. Animal experiments were performed in accordance with protocols approved by Institutional Animal Care and Use Committee.
Subjects
10 mls of peripheral blood were obtained from 26 consecutive obese subjects who were referred to our hospital-based weight-management clinic (mean age 47, range 24–60 years; mean BMI 48), and 18 patients attending the weight management clinic 18 months after bariatric surgery (mean age 46, range 36–54 years; mean BMI 38) and 22 lean healthy controls (mean age 39, range 23–54 years; mean BMI 24). All blood samples were obtained with written informed consent. The ethics committee at St. Vincent’s University Hospital, Dublin granted approval for this study.
Reagents
αGC analogue PBS-57-loaded or empty CD1d tetramers were provided by the NIH tetramer facility (Emory Vaccine Center, Atlanta, GA). αGC (KRN 7000) was kindly provided by Kirin Ltd, Japan. Immune cells were cultured in RPMI-1640, adipose tissue-derived cells in Dulbecco’s Modified Eagle Media (DMEM), supplemented with penicillin, streptomycin (Mediatech, Manassas, VA), and 5% FBS (Hyclone, Logan, UT).
Diet and metabolic studies
Wt, Jα18-deficient and CD1d1−/− were weighed weekly and food intake was monitored on HFD. Body fat content was measured by an X-ray emitting DEXA scan performed after mice were sacrificed. Whole abdominal adipose fat pads were weighed after dissecting out the testes and lymph nodes. After 6 weeks on HFD, fasting blood glucose (OneTouch Ultra) and insulin concentrations (Crystal Chem ELISA) were measured. For glucose tolerance tests, fasted (10 h) mice received 1 g glucose per kg body weight intraperitoneally (i.p) and glucose levels were measured every 15mins for 90mins. For insulin resistance, homeostasis model of assessment for insulin resistance (HOMA-IR) calculation (Matthews et al., 1985) was used: fasting blood glucose x fasting insulin/22.5. Two samples of 5mm liver were collected and fixed in formalin overnight, prior to paraffin mounting and preparation of H&E or OIL Red O stained slides for measurement of fatty liver. For H&E and Oil Red O staining, biopsies were viewed using the 20x objective. Degree of fatty liver was measured by Oil Red O staining intensity around 5 portal tract areas per slide.
Adipocyte size
Adipocyte size and number were measured by osmium and immunohistochemistry. Two samples of 20–30 mg of adipose tissue per mouse were immediately fixed in osmium tetroxide (3% solution in collidine 0.05M), minced into 1mm pieces and incubated in the dark at room temperature for 48 hrs. Adipose cell size and number were determined by a Beckman Coulter Multisizer III with a 400μm aperture. Adipose tissue was also fixed in formalin overnight, prior to paraffin mounting and preparation of H&E slides. Adipocyte number was counted per field of view, in ten fields per sample and related back to the original weight of each fat pad.
Spleen, liver and adipose tissue, and human blood preparations
Isoflurane-anesthetized mice were systemically perfused with PBS. Single cell suspensions from spleens were prepared by standard techniques. Liver MNC were isolated as previously described without collagenase digestion (Nowak et al., 2009). Briefly, livers were perfused with PBS, minced and iNKT cells were enriched by centrifugation in a two-step Percoll gradient. Enriched populations typically contained 20–30% iNKT cells. Epididymal/inguinal adipose tissue was dissected carefully avoiding lymph nodes, minced with opposing scalpels and digested with collagenase (Sigma, 0.2 mg ml−1 in DMEM for 45 min at 37 °C on a rotary shaker). The digests were filtered through 40 μm cell strainers and pelleted to enrich fat-associated lymphocytes in the stromovascular fraction (SVF). Cell yields and viability were measured with trypan blue staining. Ten milliliters of venous blood was collected in heparinized tubes for measurement of iNKT cell levels. Peripheral blood mononuclear cells were prepared by standard density gradient centrifugation over Lymphoprep (Nycomed) at 400 g for 25 min. Cells were then washed twice with HBSS supplemented with HEPES buffer solution (Invitrogen Life Technologies) and antibiotics. Cell pellets were re-suspended in 1 ml of RPMI 1640 medium, and cell yields and viability were assessed by ethidium bromide/acridine orange staining. The cell suspension was adjusted to 1×106 cells/ml in RPMI for staining (100μl/tube).
Flow cytometry
Single cell suspensions of splenocytes, LMNCs and adipose SVF were blocked with anti-CD16/32 mAb and stained for 30 min at 4°C in the dark with PBS-57-loaded or empty CD1d tetramer-PE (NIH tetramer facility) and CD3 (1:150 dulution, eBiosciences). Macrophages were labeled with phycoerythrin-conjugated antibody to F4/80 (1 in 100) and CD11c (1in 200) and CD206 (1 in 200) to differentiate M1 from M2 macrophages in the SVF as previously described (Fujisaka et al., 2009). For human peripheral blood, mouse anti-human CD3 combined with the iNKT TCR (6B11) and isotype-matched controls were used (BD Biosciences). iNKT cells were also stained with Vα24 and Vβ11 TCR chains from Coulter Immunotech (Marseilles, France). Cells were washed and fixed in 1% PFA and acquired on an LSR II flow cytometer (BD Bioscience) and with FlowJo and Kaluza software.
iNKT cell isolation and adoptive transfer
Hepatic mononuclear cells were stained with CD1d tetramer-PE and sorted to >95% purity using a FacsAriaII (Becton Dickinson, CA). Purified iNKT cells (5×105) were injected i.p. into Jα18-deficient mice which had been on HFD for 8 weeks. Metabolic parameters were analyzed after 4 days, mice were sacrificed, adipose tissue was weighed, and adipocytes were measured by osmium and immunohistochemistry.
In vivo stimulation of iNKT cells and intracellular cytokine staining
Mice were injected i.p. with 2 μg of αGG or vehicle alone and mice were sacrificed after 5 hours or 4 days, at the time of metabolic analysis. Single cell suspension of splenocytes, liver mononuclear cells and adipose tissue stromovascular fractions (SVF) were obtained as before, but with the inclusion of Brefeldin A in all media. Firstly, Single cell suspensions of splenocytes or liver mononuclear cells were stained with cell surface labeling anti-CD3 mAb and αGC-loaded CD1d tetramer. Cells were then fixed, permeabilized, and stained intracellularly for IL-4, IL-10 and IFN-γ were performed using Cytofix/cytoperm (BD Biosciences), according to manufacturers instructions. To neutralize cytokines prior to αGC treatment, anti-IL-4 (11B11) or anti-IL-10 (JES5-2A5) were injected i.p. prior to injection of αGC.
Statistical analyses
Error bars represent the standard error of the mean. The statistical significance of differences between two groups was determined using Mann-Whitney or Student’s t-tests where appropriate following determination of Gaussian distribution of the data. Differences among mice groups (>2) were evaluated using one-way or two-way ANOVA followed by post hoc Tukey tests. Values of p < 0.05 were considered significant.
Supplementary Material
Highlights.
iNKT cells are enriched in mamalian adipose tissue.
iNKT cells in adipose tissue are unique IL-10 producers
Without iNKT cells, mice are fatter and more insulin resistant
Restoring iNKT cells in obesity reverses type 2 diabetes.
Acknowledgments
We are thankful to Prof. B. Kahn, Dr. Odile Peroni and the Metabolic Physiology Core, Boston for assistance with DEXA imaging and adipocyte measurement. We thank Prof. Gokhan Hotamisligil of Harvard School of Public Health, Prof. Ulrich von Andrian of Harvard Medical School for fruitful discussions and Prof. Michael Brenner for kind assistance and discussion of this maunscript. We gratefully acknowledge Dr. F. Scheuplein and Ms. S. Jordan for mouse care. We also thank the BIDMC Flow Cytometry Core, especially John Tigges, and BIDMC Histology Core, especially Ms S. White and Dr. LH. Ang. This study was supported by NIH R01 DK066917, U19 AI066313 (MAE), US DOD W81XWH-09-1-0156 (SPB), UNESCO-L’Oreal Fellowship (LL), European Comission Marie Curie Fellowship (LL), Science Foundation Ireland (CO’F), and the Health Research Board, Ireland (LL, AEH, DOS). Drs. Balk and Exley have consulting relationships with NKT Therapeutics Inc.
Footnotes
The other authors have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol. 11:131–142. doi: 10.1038/nri2904. [DOI] [PubMed] [Google Scholar]
- Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009;9:28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- Dalmas E, Clement K, Guerre-Millo M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol. 2011 doi: 10.1016/j.it.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- Elinav E, Pappo O, Sklair-Levy M, Margalit M, Shibolet O, Gomori M, Alper R, Thalenfeld B, Engelhardt D, Rabbani E, Ilan Y. Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J Pathol. 2006;209:121–128. doi: 10.1002/path.1950. [DOI] [PubMed] [Google Scholar]
- Exley MA, Bigley NJ, Cheng O, Shaulov A, Tahir SM, Carter QL, Garcia J, Wang C, Patten K, Stills HF, et al. Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology. 2003;110:519–526. doi: 10.1111/j.1365-2567.2003.01779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley MA, Lynch L, Varghese B, Nowak M, Alatrakchi N, Balk SP. Developing understanding of the roles of CD1d-restricted T cell subsets in cancer: Reversing tumor-induced defects. Clin Immunol. 2011 doi: 10.1016/j.clim.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. 2000;68:437–446. [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, Trott DL, Ndonye RM, Veerapen N, Besra GS, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 2009;7:e1000228. doi: 10.1371/journal.pbio.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, Tsuneyama K, Nagai Y, Takatsu K, Urakaze M, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58:2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48:1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumperz JE. The ins and outs of CD1 molecules: bringing lipids under immunological surveillance. Traffic. 2006;7:2–13. doi: 10.1111/j.1600-0854.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes. 2009;58:2525–2535. doi: 10.2337/db08-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, Qi L. Activation of Natural Killer T Cells Promotes M2 Macrophage Polarization in Adipose Tissue and Improves Systemic Glucose Tolerance via Interleukin-4 (IL-4)/STAT6 Protein Signaling Axis in Obesity. J Biol Chem. 2012;287:13561–13571. doi: 10.1074/jbc.M112.350066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–640. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotas ME, Lee HY, Gillum MP, Annicelli C, Guigni BA, Shulman GI, Medzhitov R. Impact of CD1d deficiency on metabolism. PLoS One. 2011;6:e25478. doi: 10.1371/journal.pone.0025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002;2:557–568. doi: 10.1038/nri854. [DOI] [PubMed] [Google Scholar]
- Lonn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 24:326–331. doi: 10.1096/fj.09-133058. [DOI] [PubMed] [Google Scholar]
- Lonn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2009;24:326–331. doi: 10.1096/fj.09-133058. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L, O’Shea D, Winter DC, Geoghegan J, Doherty DG, O’Farrelly C. Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol. 2009;39:1893–1901. doi: 10.1002/eji.200939349. [DOI] [PubMed] [Google Scholar]
- Ma X, Hua J, Li Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol. 2008;49:821–830. doi: 10.1016/j.jhep.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia L, Delacre M, Abboud G, Ouk TS, Delanoye A, Verwaerde C, Saule P, Wolowczuk I. Impairment of dendritic cell functionality and steady-state number in obese mice. J Immunol. 2006;177:5997–6006. doi: 10.4049/jimmunol.177.9.5997. [DOI] [PubMed] [Google Scholar]
- Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O’Doherty RM. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS One. 2011;6:e19831. doi: 10.1371/journal.pone.0019831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-restricted iNKT cells, the ‘Swiss-Army knife’ of the immune system. Curr Opin Immunol. 2008;20:358–368. doi: 10.1016/j.coi.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, Koezuka Y, Kronenberg M. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med. 2000;192:741–754. doi: 10.1084/jem.192.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Medrikova D, Jilkova ZM, Bardova K, Janovska P, Rossmeisl M, Kopecky J. Sex differences during the course of diet-induced obesity in mice: adipose tissue expandability and glycemic control. Int J Obes (Lond) 2011 doi: 10.1038/ijo.2011.87. [DOI] [PubMed] [Google Scholar]
- Monteiro M, Almeida CF, Caridade M, Ribot JC, Duarte J, Agua-Doce A, Wollenberg I, Silva-Santos B, Graca L. Identification of regulatory Foxp3+ invariant NKT cells induced by TGF-beta. J Immunol. 2010;185:2157–2163. doi: 10.4049/jimmunol.1000359. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Seo K, Yamashita H, Hosoya Y, Ohsugi M, Tobe K, Kadowaki T, Nagai R, Sugiura S. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest. 2008;118:710–721. doi: 10.1172/JCI33328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M, Lynch L, Yue S, Ohta A, Sitkovsky M, Balk SP, Exley MA. The A2aR adenosine receptor controls cytokine production in iNKT cells. Eur J Immunol. 2009 doi: 10.1002/eji.200939897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, Lynch L, Cawood TJ, Kwasnik A, Nolan N, Geoghegan J, McCormick A, O’Farrelly C, O’Shea D. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One. 5:e9997. doi: 10.1371/journal.pone.0009997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, Lynch L, Cawood TJ, Kwasnik A, Nolan N, Geoghegan J, McCormick A, O’Farrelly C, O’Shea D. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One. 2010;5:e9997. doi: 10.1371/journal.pone.0009997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicci DG, Hammond KJ, Uldrich AP, Baxter AG, Smyth MJ, Godfrey DI. A natural killer T (NKT) cell developmental pathway iInvolving a thymus-dependent NK1.1(−)CD4(+) CD1d-dependent precursor stage. J Exp Med. 2002;195:835–844. doi: 10.1084/jem.20011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Satoh M, Andoh Y, Clingan A, Ogura H, Fujii S, Eshima K, Nakayama T, Taniguchi M, Hirata N, Ishimori N, et al. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatis and insulin resistance. PLos ONE. 2012;7(2):e30568. doi: 10.1371/journal.pone.0030568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speak AO, Salio M, Neville DC, Fontaine J, Priestman DA, Platt N, Heare T, Butters TD, Dwek RA, Trottein F, et al. Implications for invariant natural killer T cell ligands due to the restricted presence of isoglobotrihexosylceramide in mammals. Proc Natl Acad Sci U S A. 2007;104:5971–5976. doi: 10.1073/pnas.0607285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JB, Coquet JM, Smyth MJ, Godfrey DI. CD1-restricted T cells and tumor immunity. Curr Top Microbiol Immunol. 2007;314:293–323. doi: 10.1007/978-3-540-69511-0_12. [DOI] [PubMed] [Google Scholar]
- Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE, Harrison LC. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59:1648–1656. doi: 10.2337/db09-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LM, Ricchetti G, Sarma U, Smallie T, Foxwell BM. Interleukin-10 suppression of myeloid cell activation--a continuing puzzle. Immunology. 2004;113:281–292. doi: 10.1111/j.1365-2567.2004.01988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Van Kaer L. Natural killer T cells and autoimmune disease. Curr Mol Med. 2009;9:4–14. doi: 10.2174/156652409787314534. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Jhaveri R, Huang J, Qi Y, Diehl AM. Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab Invest. 2007;87:927–937. doi: 10.1038/labinvest.3700603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.