

Abstract
The organic electrochemical transistor (OECT), capable of transducing small ionic fluxes into electronic signals in an aqueous environment, is an ideal device to utilize in bioelectronic applications. Currently, most OECTs are fabricated with commercially available conducting poly(3,4-ethylenedioxythiophene) (PEDOT)-based suspensions and are therefore operated in depletion mode. Here, we present a series of semiconducting polymers designed to elucidate important structure–property guidelines required for accumulation mode OECT operation. We discuss key aspects relating to OECT performance such as ion and hole transport, electrochromic properties, operational voltage, and stability. The demonstration of our molecular design strategy is the fabrication of accumulation mode OECTs that clearly outperform state-of-the-art PEDOT-based devices, and show stability under aqueous operation without the need for formulation additives and cross-linkers.
Introduction
Semiconducting materials have long played a pivotal role in the development and advancement of organic electronic applications such as organic light-emitting diodes, organic field-effect transistors, and organic solar cells.1−4 More recently, semiconducting polymers have made their entry into the new field of organic bioelectronics, which broadly encompasses any application that couples a relevant function of organic electronic materials with a targeted biological event.5,6 In this context, recent endeavors have seen organic electronic materials utilized for example in biologically relevant ion sensing7,8 and ion pumps,9,10 and as transducers of neural activity.11,12
Organic electronic materials provide the ideal platform for transduction of biological signals. In contrast to inorganic materials, organics can easily be modified for biocompatibility, while there is already a comprehensive understanding of structure–property relations from the well-established organic field-effect transistor (OFET) community.13−15 Despite this large body of work relating to the transduction of electronic signals in organic semiconductors, high-performing bioelectronic devices such as organic electrochemical transistors (OECTs) are predominantly fabricated with the commercially available blend of poly(3,4-ethylenedioxythiophene) and polystyrenesulfonate (PEDOT:PSS).16,17 From a synthetic point of view, it is very challenging to modify PEDOT:PSS, which makes it difficult to control important device parameters such as operational voltage. The intrinsically conducting nature of the PEDOT:PSS blend furthermore means that the OECT must be operated in depletion mode rather than accumulation mode, giving rise to a device that is on at no external bias and shows inferior on/off ratios at low bias, which is often desired in biological applications. Therefore, we have recently focused on developing new accumulation mode OECT materials with an underlying motivation of addressing the current lack of understanding relating to the chemical design of semiconducting OECT materials. Our aim is to help establish molecular design criteria and to elucidate important structure–property relations for the synthesis of high-performing semiconducting polymers for OECT applications. In this context, we present here a series of five thiophene- and benzodithiophene-based polymers and show how our rational molecular design strategy affords a top-performing polymer that, in an accumulation mode OECT configuration, for the first time, outperforms state-of-the-art PEDOT:PSS-based depletion mode devices with peak transconductances above 20 mS, peak currents in the mA regime, on/off ratios above 105, and excellent switching times below 1 ms.
Since OECTs are mainly operating as transducers, the figure of merit is the transconductance (gm) defined as dID/dVG; transconductance therefore describes the peak performance of an OECT in transducing a small change in effective gate voltage (VG) into a large modulation in current running through the transistor channel (drain current, ID). In the saturation regime, the transconductance is furthermore proportional to the charge carrier mobility (μ), the capacitance per unit volume (C*), and the active channel area defined by the channel width (W), length (L), and thickness (d) as displayed in eq 1, where VT is the threshold voltage.
| 1 |
Therefore, to optimize OECT performance from a molecular design point of view, charge and ion transport must be optimized in a balanced fashion to maximize the product μC*. To address this, we have investigated a series of thiophene-based polymers with ethylene glycol side chains as further outlined below. Whereas one can simplistically associate the polythiophene backbone with the charge transport and the ethylene glycol side chains with the ion transport, there is ample literature evidencing that the nature, positioning, and density of the solubilizing side chains play a huge role in the charge transport properties of the semiconducting material. With this caveat in mind, we have aimed to explore synthetic design criteria in a systematic fashion in order to optimize both the charge transport and the ion transport and more importantly the product of the two (μC*).
The polymers chosen for this study are displayed in Chart 1. For efficient ion transport in the solid state, triethylene glycol (TEG) chains were grafted onto benzo[1,2-b:4,5-b′]dithiophene (BDT) and 2,2′-bithiophene (2T), and the resultant monomers were either homopolymerized, or copolymerized with thiophene or bithiophene. These comonomers were chosen due to their electron-rich conjugated systems, and the fact that they could facilitate high degrees of backbone coplanarity, thus achieving low operational voltages and good charge transport properties.
Chart 1. Chemical Structures of Polymers.

The polymers are designed with the aim of investigating a number of important structure–property relations including the following: (1) Backbone conformation is modulated as illustrated for example by the polymers gBDT, gBDT-T, and gBDT-2T. While BDT ensures a linear connectivity along the polymer backbone, the thiophene and bithiophene comonomers introduce various degrees of backbone curvature as illustrated in Figure 1. This is likely to influence important parameters such as side chain orientation, polymer solubility, packing, and, consequently, charge transport. (2) TEG chain density is modulated along the polymer backbone as illustrated for example by the polymer series discussed above or by gBDT-2T versus gBDT-g2T. This will again have an effect on polymer solubility and packing, but also importantly on the polymers’ ionization potentials (because the oxygen is directly grafted onto the thiophene ring) and their abilities to promote ion penetration/transport in the solid state. (3) Structure of TEG-functionalized monomer through comparison of gBDT-T and g2T-T to investigate the role of the π-conjugated backbone and its charge transport properties without making significant changes to the backbone conformation or the side chain density.
Figure 1.
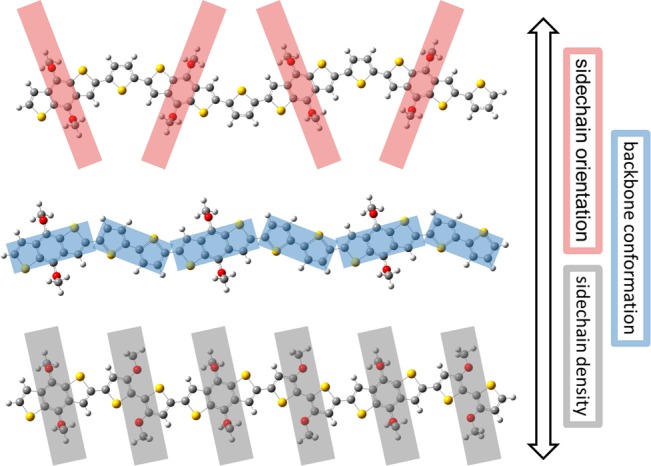
Energy-minimized structures for gBDT-T (top), gBDT-2T (middle), and gBDT-g2T (bottom) illustrating important molecular design criteria employed in this study.
Results and Discussion
Benzo[1,2-b:4,5-b′]dithiophene (BDT) and 2,2′-bithiophene (2T) were functionalized with methoxy-terminated triethylene glycol (TEG) chains according to literature protocols to afford the two TEG-functionalized comonomers.8 Utilizing conventional Stille-type cross-coupling polymerization reactions, these building blocks allowed for the synthesis of the BDT homopolymer (gBDT) and the two thiophene copolymers (gBDT-T and g2T-T), while the TEG-functionalized BDT monomer could furthermore be copolymerized with either unsubstituted 2T or TEG-functionalized 2T to afford gBDT-2T and gBDT-g2T, respectively. Full synthetic details are included in the SI. Molecular weight analysis was carried out by size exclusion chromatography in N,N-dimethylformamide or chlorobenzene as summarized in Table S1. Good degrees of polymerization were observed for gBDT-g2T and g2T-T, with number-average molecular weights of 22 and 63 kDa, respectively, while the very limited solubility of the three other polymers resulted in inconclusive molecular weight analysis.
UV–vis spectroscopy on thin films of the polymers (Figure 2, Table S1) revealed a gradual red-shift in absorption maximum when going from gBDT (533 nm) to gBDT-T (549 nm) and gBDT-2T (562 nm) with all three polymers showing strong absorption features from both the 0–0 and the 0–1 optical transitions. The absorption profiles of gBDT-g2T and g2T-T were further red-shifted with λmax values of 585 and 650 nm, respectively, while these spectra were less resolved in terms of vibronic features. Optical band gaps were determined from the onsets of absorption and ranged from 2.18 eV for gBDT to 1.73 eV for g2T-T as further detailed in Table S1. The observed red-shift and narrowing of band gap when increasing the thiophene content relative to benzodithiophene in the polymer is likely attributed to the stronger quinoidal contribution from the thiophene-rich polymers resulting in a higher degree of backbone coplanarity. Additional backbone planarization of the g2T-based polymers can also be envisioned due to attractive intramolecular S–O interactions. We furthermore note that density functional theory (DFT) calculations support the observed trend in optical properties (SI, Table S2).
Figure 2.
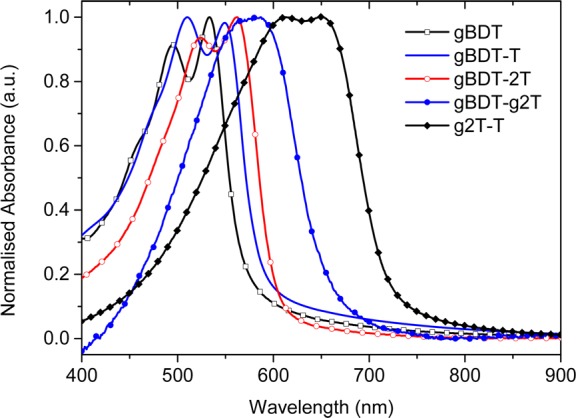
UV–vis spectra of thin spin-cast films of the polymers.
Oxidative electrochemical properties were investigated by cyclic voltammetry (CV) on thin polymer films on indium tin oxide (ITO) coated glass substrates using a conventional three-electrode setup in acetonitrile solution. The onsets of oxidation were determined for all polymers, while all polymers except gBDT also showed well-defined first half-wave potentials as summarized in Table 1 (voltammograms in SI, Figure S3). Ionization potentials (IPs) were estimated from the onsets of oxidation; gBDT, gBDT-T, and gBDT-2T all have IPs around 4.8–4.9 eV (slightly higher than what was measured for poly(3-hexylthiophene) (4.78 eV) under identical conditions), whereas the electron-rich all-thiophene polymer g2T-T and the fully TEGylated gBDT-g2T have significantly lower IPs around 4.4 eV as expected (Figure 3). DFT calculations of the highest occupied molecular orbital (HOMO) energy levels are in good agreement with the experimental trend discussed above (Table S2). From the CV data it is evident that our molecular design strategy provides good control over the onset of oxidation which can easily be shifted nearly 600 mV within this series of polymers. Importantly, we also note qualitatively that the current density and thus the capacitance are significantly higher for g2T-T than for the four other polymers (Figure S4).
Table 1. Oxidative Electrochemical Properties of the Polymers.
| polymer | Eonset (V)a | E1/2 (V)a | IP (eV)a | Eonset (V)b |
|---|---|---|---|---|
| gBDT | 0.48 | 4.90 | 0.39 | |
| gBDT-T | 0.41 | 0.57 | 4.83 | 0.32 |
| gBDT-2T | 0.50 | 0.67 | 4.92 | 0.47 |
| gBDT-g2T | –0.01 | 0.15 | 4.43 | –0.15 |
| g2T-T | –0.06 | 0 | 4.38 | –0.14 |
Cyclic voltammetry of polymer thin films on ITO coated glass substrates in acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte.
Water with 0.1 M sodium chloride as the supporting electrolyte.
Figure 3.
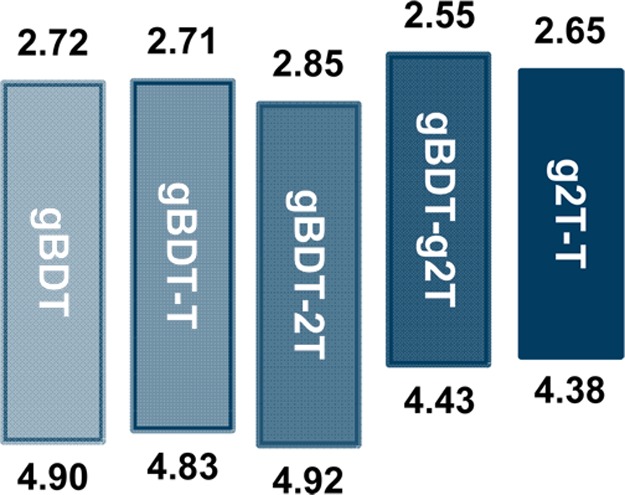
Ionization potentials (bottom) and electron affinities (top, estimated from ionization potential and optical band gap) for the polymers.
To investigate the electrochemical properties under more OECT relevant conditions, the oxidative properties were also measured in aqueous solution. The resulting cyclic voltammograms are depicted in Figure 4 and in the SI (Figure S5). Generally, the same trends were observed within the polymer series when comparing the onsets of oxidation in acetonitrile and water although the onsets in water were 30–140 mV lower than those in acetonitrile, which is likely due to the smaller anion (chloride vs hexafluorophosphate) and better solvated TEG side chains in water than in acetonitrile and hence easier ion penetration. This hypothesis is supported by the fact that the difference in onset is largest for gBDT-g2T, which has the highest TEG side chain density. Thus, it appears that, for this system, the CV characteristics are a convolution of not only the energetics of the polymer, but also the energetics associated with ion penetration (injection) and transport. In conjunction with the aqueous CV experiment, the electrochromic properties of the polymers were also studied by means of spectroelectrochemistry. The data for the three polymers included in Figure 4 illustrate the different electrochromic properties observed for the polymer series. Barely any electrochromic response was observed for the homopolymer gBDT (Figure 4, top panel) when sweeping the potential from −0.2 to 0.6 V versus Ag/Ag+, whereas gBDT-T and gBDT-2T (Figure 4, middle panel, and Figure S5, respectively) both showed partial extinction of the π–π* absorption band with the simultaneous gradual appearance of a very broad absorption feature around 750 nm. The new absorption feature is likely from the oxidized polymer (polaron) and appears stable when subjected to consecutive cycling within the potential range −0.2–0.7 V (Figure 4, right panel). The more electron-rich polymers gBDT-g2T and g2T-T (Figure S5 and Figure 4, bottom panel, respectively) both show a more complete oxidation of the bulk polymer film evident from the disappearance of the π–π* absorption band with a concurrent appearance of a broad polaron absorption band around 850 nm. Again, the two polymers are stable to several oxidative CV cycles with no sign of significant current or absorbance loss as evident from Figure 4 (right panel). From a structural point of view, it is clear that the two low-IP polymers containing the TEGylated bithiophene unit show superior electrochromic properties (judged from the complete bleaching of the π–π* transition and the stable cycling between colored and bleached state) compared to the three other TEGylated BDT polymers. The applied potentials are kept low deliberately since these materials are investigated for OECTs that should be operational in aqueous media and hence a narrow electrochemical potential window. Moreover, with application of higher potentials, gBDT, gBDT-T, and gBDT-2T degrade rapidly. Thus, the spectroelectrochemical properties reported here represent the highest degree of doping that can be obtained under conditions relevant for OECT operation and without polymer degradation taking place; in other words, increasing the bias further does not result in further doping but merely in degradation of the active material.
Figure 4.

Cyclic voltammograms in water with 0.1 M sodium chloride as the supporting electrolyte (left panel) and corresponding UV–vis absorption spectra (middle panel) with color coding indicating the applied potential during recording of the UV–vis spectrum. Right panel shows the switching behaviors with the two wavelengths used for monitoring the optical switching indicated in the corresponding UV–vis spectra by the green and red lines.
Grazing incidence wide-angle X-ray scattering (GIWAXS) was employed to study the structural organization of thin spin-cast films of the polymers on Si substrates (Table 2, Figure 5, and Figure S10). Polymers gBDT and gBDT-T were found to orient preferably in a face-on fashion with π-stacking distances of 3.72 Å, whereas gBDT-2T showed a predominant edge-on orientation with nearly identical π-stacking distances of 3.71 Å. The lamellar stacking distances gradually decrease from roughly 20 to 14 Å when increasing the unsubstituted thiophene content within this series due to the decrease in side chain density. This observation could be explained by a less extended and more disordered side chain orientation. The variation of structural order and orientation on a substrate with varying comonomer (gBDT vs gBDT-T vs gBDT-2T) is well-known from the literature.18 For the polymers discussed herein, the explanation most likely relates to π-stacked lamellar platelets forming in solution, thereby favoring an edge-on orientation (platelets orient face-on to the substrate, with the polymer backbones correspondingly oriented edge-on) as seen for the least soluble polymer, gBDT-2T.19 Going from gBDT-2T to gBDT-g2T, the side chain density is increased significantly along the polymer backbone, which not only brings the lamellar stacking distance back to approximately 20 Å as seen for gBDT, but also increases the π-stacking distance to 3.84 Å. The increased side chain density furthermore causes a shift in preferential orientation from edge-on to face-on for gBDT-g2T in agreement with the trend noted above. Polymer g2T-T compares well with gBDT-T in having a lamellar repeat distance around 17 Å, whereas the π-stacking distance is markedly decreased to 3.59 Å. In resemblance with gBDT-2T, g2T-T orients predominantly edge-on relative to the substrate, which should be favorable for horizontal charge transport as required in a conventional OECT configuration.20,21 Upon further comparison, it is evident that the high side chain density of gBDT-g2T, with TEG chains on each comonomer unit, is detrimental to effective π-stacking whereas g2T-T has much stronger π–π interactions indicative of good interchain charge transport.
Table 2. Solid State Packing Parameters for the Polymers.
| lamellar
stacking |
π-stacking |
||||
|---|---|---|---|---|---|
| polymer | q (Å–1) | d (Å) | q (Å–1) | d (Å) | dominant texture |
| gBDT | 0.315 | 19.9 | 1.690 | 3.72 | face-on |
| gBDT-T | 0.364 | 17.3 | 1.688 | 3.72 | face-on |
| gBDT-2T | 0.444 | 14.2 | 1.694 | 3.71 | edge-on |
| gBDT-g2T | 0.307 | 20.5 | 1.635 | 3.84 | face-on |
| g2T-T | 0.359 | 17.5 | 1.749 | 3.59 | edge-on |
Figure 5.
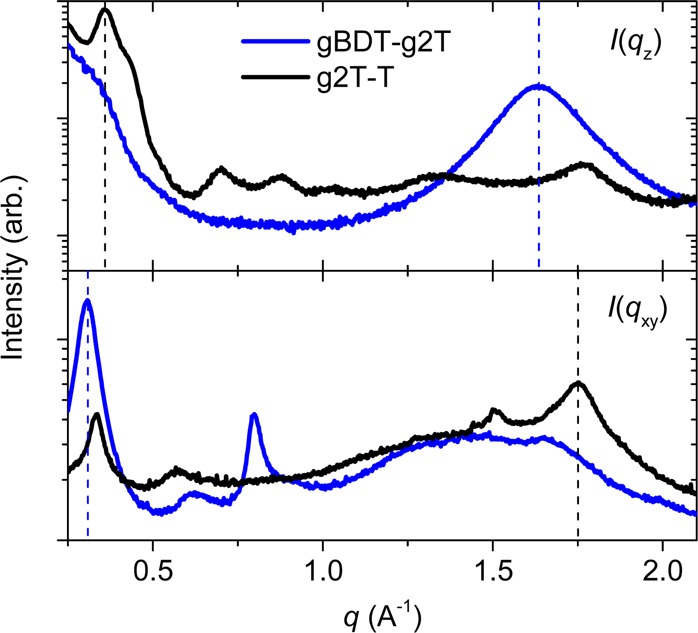
Vertical (top) and horizontal (bottom) line-cuts of the two-dimensional GIWAXS spectra for gBDT-g2T and g2T-T; qz and qxy are the perpendicular and parallel wave vector transfers with respect to substrate surface.
Organic field-effect transistors (OFETs) were successfully fabricated for the high-IP polymers as further evidenced in the SI (Figure S8) with extracted hole mobilities of 1 × 10–3 cm2/(V s) for both gBDT and gBDT-T and 9 × 10–3 cm2/(V s) for gBDT-2T. The nearly 1 order of magnitude higher mobility of the latter is in good agreement with the preferential edge-on orientation relative to the substrate, which should favor in-plane charge transport as noted above.20,21 Difficulties with fabricating and testing stable OFET devices with gBDT-g2T and g2T-T, due to unintentional doping, were circumvented by extracting charge carrier mobilities from the OECTs affording hole mobilities of 0.01 and 0.28 cm2/(V s), respectively.22,23 Here, we note that the significantly tighter π-stacking of g2T-T compared to gBDT-g2T, as well as the edge-on packing observed, results in a considerably higher charge carrier mobility, as expected. Furthermore, from a structural point of view, it appears that the higher backbone flexibility of the thiophene-based polymer, g2T-T, improves film formation and morphology, which also aid charge transport compared to the more rigid BDT-containing polymer. Because the hole mobilities of gBDT, gBDT-T, and gBDT-2T are extracted differently from those of gBDT-g2T and g2T-T, we are not able to compare directly the charge transport properties between the two sets of materials.
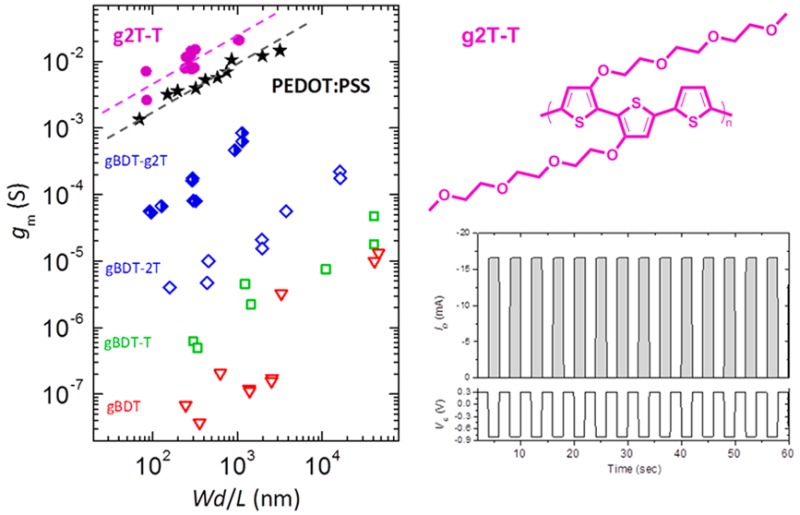
Organic electrochemical transistors were initially fabricated with thin spin-cast active layers with comparable thicknesses of 82–110 nm in order to ensure a fair comparison of the five polymers since device performance scales with Wd/L, where W and L are the channel width and length, respectively, and d is the active layer thickness.12 Channel dimensions (W × L, in μm) of 100 × 10, 10 × 10, and 50 × 50 were tested. Full details on OECT fabrication and operation can be found in the SI. As detailed in Table 3, gBDT and gBDT-T show the poorest performances with transconductances around 0.1–0.5 μS, which is corroborated nicely by the poor electrochromic activity and the low charge carrier mobilities. Polymer gBDT-2T shows a transconductance around 0.06 mS and a similar 100-fold improvement in peak current when compared to gBDT and gBDT-T. This improvement is supported by the improved electrochromic properties as well as the better hole mobility of gBDT-2T, which can be ascribed to its edge-on orientation. It should be noted that these polymers do not show a peak transconductance, as is commonly observed for PEDOT:PSS, within the stable operation window. For this reason, we quote the highest attainable reproducible transconductance value which occurs at a gate bias of −0.6 V. This suggests that these materials could perform better if they were able to operate in a stable manner at higher gate biases. A further and quite significant improvement in device performance of 1 and 2 orders of magnitude, respectively, is seen for gBDT-g2T and g2T-T. We note again that a good correlation is observed with the much improved electrochromic properties compared to the other polymers, while it also appears that the strong π–π interactions and the edge-on orientation of g2T-T result in superior OECT performance with a peak transconductance (gmpeak) of nearly 8 mS at a gate voltage (VG) of −0.6 V, a peak drain current (IDpeak) of −3.3 mA (at VG = −0.7 V), and a good on/off ratio above 105. The nearly 20-fold increase in peak transconductance when going from gBDT-g2T to g2T-T at identical device dimensions is in good agreement with the 28-fold improvement in charge carrier mobility as discussed above. We show below (Figure 6), when considering multiple devices and dimensions, that the improvement in device performance of g2T-T compared to gBDT-g2T is actually closer to 100-fold, which indicates that not only hole transport but also ion transport are greatly improved.
Table 3. OECT Parameters for the Polymersa.
| polymer | d (nm) | gmpeak (mS) | IDpeak (mA) | ION/OFF |
|---|---|---|---|---|
| gBDT | 85 | 1.2 × 10–4 | –2.9 × 10–5 | 2.7 × 101 |
| gBDT-T | 82 | 5.5 × 10–4 | –3.3 × 10–5 | 1.9 × 101 |
| gBDT-2T | 110 | 6.0 × 10–2 | –3.4 × 10–3 | 4.1 × 103 |
| gBDT-g2T | 110 | 0.47 | –4.9 × 10–2 | 9.8 × 103 |
| g2T-T | 103 | 7.9 | –3.3 | 1.7 × 105 |
Accumulation mode p-type OECT operation; all devices are spin-cast devices with active areas of 100 μm × 10 μm (W × L), and all transfer characteristics and transconductances are measured at a drain voltage of −0.4 V.
Figure 6.
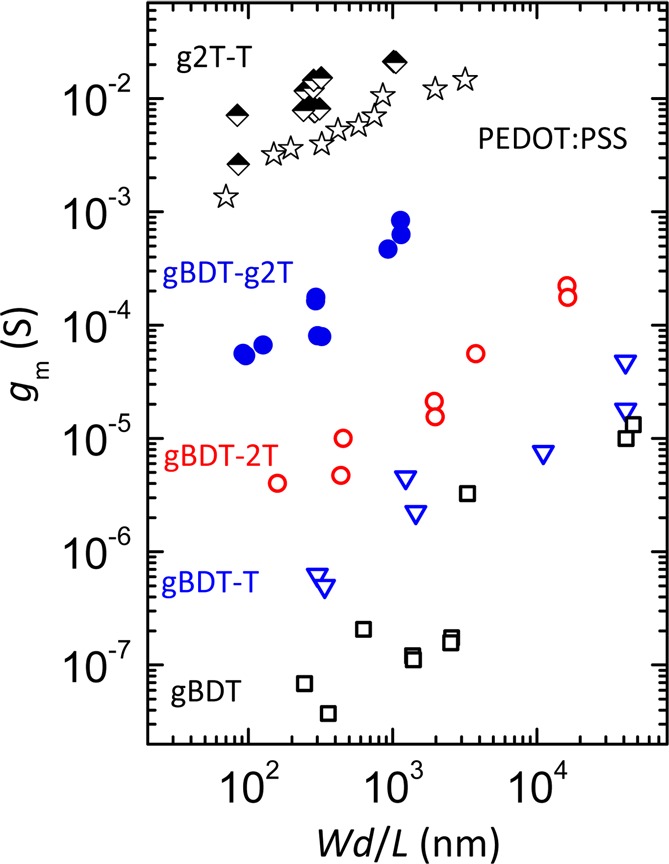
Peak transconductances as a function of active layer dimensionality (Wd/L) for all polymers and including PEDOT:PSS-based devices with ethylene glycol dispersion additive (data from ref (12)).
Focusing on the top-performing material, g2T-T, we subsequently turned our attention to OECTs with drop-cast active layers in order to fabricate thicker devices. A drastic improvement in device performance was observed when compared to spin-cast devices, already achieving a high peak transconductance of 15 mS, a peak current of −6.6 mA, and an on/off ratio of 4 × 105 for a drop-cast device of similar thickness (110 nm) to that of the spin-cast device. With a 160 nm active layer thickness, the peak transconductance had increased to 21 mS with a peak current of −7.7 mA and an on/off ratio of 2.6 × 105. To date, this is the highest reported peak transconductance for an OECT in accumulation mode,24,25 while it also outperforms the archetypical PEDOT-based depletion mode devices reported in the literature thus far.25,26 Given the expected scaling of gm with channel thickness, the ∼40% increase in transconductance from the 110 to 160 nm device is well-explained.12
To unequivocally demonstrate the superior performance of g2T-T in OECTs relative to that of PEDOT-based devices, a thorough study of the relationship between peak transconductance and dimensionality (in the form of Wd/L) was carried out as summarized in Figure 6. From this, it is clear that g2T-T-based devices display higher transconductances than PEDOT:PSS-based devices at all tested active layer dimensions. We also note that the trend outlined in Table 3 and discussed above comparing the five polymers in this study is valid across all tested dimensions. It is moreover evident from Figure 6 that the peak transconductance for each of these materials can be varied 1–2 orders of magnitude by varying the active layer dimensions, as previously reported.12
With the change in device preparation from spin- to drop-casting and the significantly increased active layer thicknesses, we found it relevant to reinvestigate the structural order in the solid state. Specular X-ray diffraction (XRD) data on thick drop-cast films was generally in good agreement with the GIWAXS data on thinner spin-cast films as further detailed in the SI (Figure S9).
Output and transfer characteristics along with the related peak transconductances for optimized OECTs with gBDT-g2T and g2T-T are displayed in Figure 7. The peak current is obtained at slightly less negative potential for g2T-T relative to gBDT-g2T, while a more significant shift is seen for the peak transconductance, which is recorded at a gate potential around −0.6 V for gBDT-g2T and −0.2 V for g2T-T. The lower turn-on voltage of g2T-T-based devices relative to gBDT-g2T is observed for all tested device dimensions as illustrated in Figure 8. An excellent turn-on voltage around 0 V is found for g2T-T independent of Wd/L, while gBDT-g2T devices turn on around −0.3 V and the higher IP polymers all turn on from −0.5 to −0.7 V as one would expect from the differences in oxidation potentials (Table 1) measured both in organic and aqueous media. All the polymers show minimal variations in turn-on voltage over 2–3 orders of magnitude of Wd/L, emphasizing how this device dimensionality parameter can be used to control the peak transconductance without adversely affecting the turn-on voltages.
Figure 7.

Output characteristics (left), transfer characteristics and related transconductances (middle), and transient characteristics (right) for optimized drop-cast OECTs using gBDT-g2T (top, 400 nm thick device) and g2T-T (bottom, 162 nm thick device) measured in 0.1 M NaCl aqueous solution; devices have active areas of 100 μm × 10 μm (W × L), and transfer characteristics and transconductances are measured at a drain voltage of −0.4 V.
Figure 8.
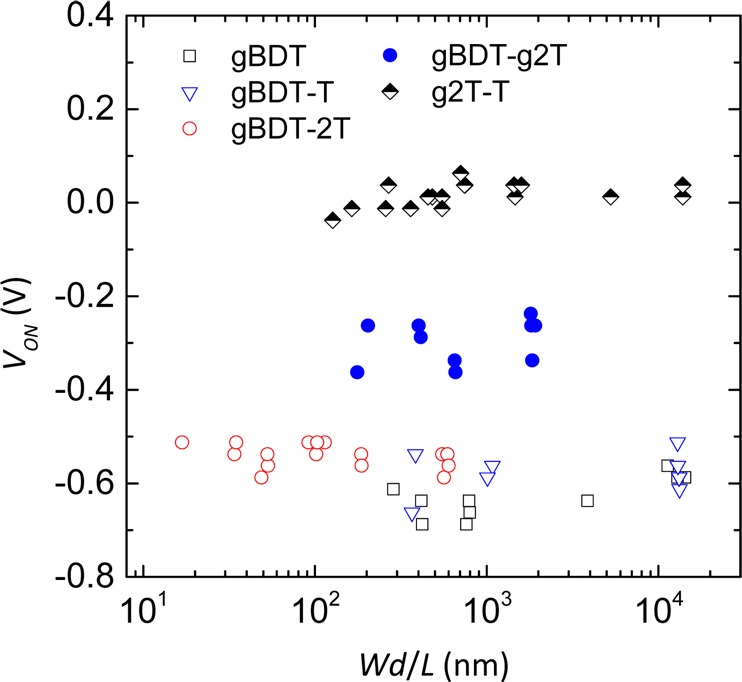
OECT turn-on voltages as a function of active layer dimensionality (Wd/L) for all polymers.
Another important aspect of OECTs and their application in biological environments is the transient behavior. As depicted in Figure 7 (right panel), we have investigated this aspect by repeated cycling of the OECTs between their on and off states using a square-wave applied gate potential. gBDT-g2T is switched from 0.2 to −0.4 V with excellent stability as evident from the constant peak current of roughly −2.5 μA obtained for each cycle. Similarly, g2T-T also shows excellent stability when switched from 0.3 to −0.2 V with a peak current around −1 mA; a faster rise in peak current is observed upon switching from 0.3 to −0.8 V (Figure S7). We find no discernible variation in the transient stability with device dimensions as evidenced for several g2T-T-based OECTs in the SI (Figure S7). Notably, these excellent switching properties are achieved with 2 s pulses of on and off gate voltages. The time constant (τ) for the temporal response of a spin-coated g2T-T-based device (d = 103 nm) was 0.64 ms for a 10 μm × 10 μm active area, while a larger active area of 100 μm × 10 μm as expected afforded a slightly slower response of 1.4 ms. In comparison with the τ-values of the solid state accumulation (40–192 ms) and depletion (300–360 ms) mode devices having the highest peak transconductances previously reported in the literature,25 the τ-values for g2T-T-based devices are 2–3 orders of magnitude lower. The lowest τ-values for accumulation mode devices have been reported by Inal and colleagues, who showed, with a polyelectrolyte-based OECT, that τ could be decreased from 38 to 0.4 ms by active layer casting with ethylene glycol cosolvent.24 Our results clearly indicate that equally fast temporal responses can be achieved through judicious synthetic design, optimizing not only the conjugated polymer backbone for efficient charge transport but also the solubilizing side chains for efficient swellability and ion transport. This approach circumvents the need for solvent additives and thus allows for a simpler device processing protocol.
In conclusion, we have prepared a series of semiconducting thiophene-based polymers with triethylene glycol side chains with the aim of simultaneously optimizing both charge and ion transport in the bulk polymer film in order to improve accumulation mode OECT performance. Our molecular design strategy entailed the copolymerization of two different TEGylated monomers with each other or with unsubstituted thiophene and bithiophene. This allowed us to access a series of polymers with various degrees of TEG side chain density and orientation, various backbone curvatures, and ionization potentials ranging from 4.4 to 4.9 eV. Cyclic voltammetry and in particular spectroelectrochemistry clearly indicated that polymers containing the TEGylated bithiophene unit were more electrochemically stable than polymers with the TEGylated benzodithiophene unit. The copolymer containing both units, gBDT-g2T, showed better stability than the other BDT-based polymers indicating that the intrinsic stability issues with the BDT unit could be reduced by incorporating the TEGylated bithiophene. This very electron-rich unit causes a significant lowering of the ionization potential of approximately 0.4–0.5 eV (both when it replaces TEGylated BDT and unsubstituted bithiophene), which is likely an underlying reason for the improved electrochemical stability. Moreover, this shift brings the oxidative process into an electrochemical potential window compatible with stable aqueous operation.
Structural characterization by XRD and GIWAXS confirmed the semicrystalline properties of these polymers, while it was also evident that the choice of comonomer could be used to control the polymer backbone orientation relative to the substrate. This was emphasized by the fact that both the TEGylated and the unsubstituted bithiophene unit promoted a favorable edge-on orientation. A high degree of side chain density was furthermore found to unfavorably increase the π-stacking distance in the solid state. The latter point nicely illustrates the balance one must strike when optimizing side chain density for swellability and ion transport, while simultaneously maintaining a high degree of structural order in the solid state for efficient charge transport. The conclusions from the structural characterization were corroborated by the charge carrier mobilities of the polymers; the edge-on oriented materials generally showed higher hole mobilities, and stronger π–π interactions were likewise found to promote better charge carrier mobilities as expected. OECT fabrication and analysis confirmed that the polymers with the better electrochromic properties, better structural organization, and higher hole mobilities also showed significantly higher device performance. After device optimization, the highest reported peak transconductances of above 20 mS were achieved for g2T-T with excellent turn-on voltages around 0 V, high on/off ratios around 105, and fast temporal responses in the millisecond regime with high operational stability at peak transconductance.
This molecular design strategy has elucidated a number of important structure–property relationships for the continued development of high-performing accumulation mode organic electrochemical transistors, and it is evident that the TEGylated bithiophene unit (g2T) is a very promising building block in this context. It is furthermore evident from this study that cyclic voltammetry and in particular spectroelectrochemistry are excellent screening tools for the development of new OECT materials.
Acknowledgments
This work was carried out with financial support from EC FP7 Project SC2 (610115), EC FP7 Project ArtESun (604397), EC FP7 Project PolyMed (612538), and EPSRC Project EP/G037515/1. E.B. thanks the Spanish Ministry of Economy and Competitiveness for his predoctoral contract. M.S. acknowledges support from the I edition of the BBVA Foundation Grants for Researchers and Cultural Creators.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b05280.
Details on polymer synthesis and characterization, DFT data and molecular orbital visualizations, UV–vis spectra, cyclic voltammograms and spectroelectrochemistry data; details on OECT preparation and OECT parameters for alternative active layer dimensions; OFET, GIWAXS, and XRD data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Friend R. H.; Gymer R. W.; Holmes A. B.; Burroughes J. H.; Marks R. N.; Taliani C.; Bradley D. D. C.; Santos D. A. D.; Bredas J. L.; Logdlund M.; Salaneck W. R. Nature 1999, 397, 121. 10.1038/16393. [DOI] [Google Scholar]
- Ashraf R. S.; Meager I.; Nikolka M.; Kirkus M.; Planells M.; Schroeder B. C.; Holliday S.; Hurhangee M.; Nielsen C. B.; Sirringhaus H.; McCulloch I. J. Am. Chem. Soc. 2015, 137, 1314. 10.1021/ja511984q. [DOI] [PubMed] [Google Scholar]
- Nielsen C. B.; Ashraf R. S.; Treat N. D.; Schroeder B. C.; Donaghey J. E.; White A. J. P.; Stingelin N.; McCulloch I. Adv. Mater. 2015, 27, 948. 10.1002/adma.201404858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Li Y.; Yang G.; Jiang K.; Lin H.; Ade H.; Ma W.; Yan H. Nat. Energy 2016, 1, 15027. 10.1038/nenergy.2015.27. [DOI] [Google Scholar]
- Berggren M.; Richter-Dahlfors A. Adv. Mater. 2007, 19, 3201. 10.1002/adma.200700419. [DOI] [Google Scholar]
- Rivnay J.; Owens R. M.; Malliaras G. G. Chem. Mater. 2014, 26, 679. 10.1021/cm4022003. [DOI] [Google Scholar]
- Jobe K.; Brennan C. H.; Motevalli M.; Goldup S. M.; Watkinson M. Chem. Commun. 2011, 47, 6036. 10.1039/c1cc11213a. [DOI] [PubMed] [Google Scholar]
- Giovannitti A.; Nielsen C. B.; Rivnay J.; Kirkus M.; Harkin D. J.; White A. J. P.; Sirringhaus H.; Malliaras G. G.; McCulloch I. Adv. Funct. Mater. 2016, 26, 514. 10.1002/adfm.201503791. [DOI] [Google Scholar]
- Simon D. T.; Kurup S.; Larsson K. C.; Hori R.; Tybrandt K.; Goiny M.; Jager E. W. H.; Berggren M.; Canlon B.; Richter-Dahlfors A. Nat. Mater. 2009, 8, 742. 10.1038/nmat2494. [DOI] [PubMed] [Google Scholar]
- Jonsson A.; Song Z.; Nilsson D.; Meyerson B. A.; Simon D. T.; Linderoth B.; Berggren M. Sci. Adv. 2015, 1, e1500039. 10.1126/sciadv.1500039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodagholy D.; Doublet T.; Quilichini P.; Gurfinkel M.; Leleux P.; Ghestem A.; Ismailova E.; Hervé T.; Sanaur S.; Bernard C.; Malliaras G. G. Nat. Commun. 2013, 4, 1575. 10.1038/ncomms2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivnay J.; Leleux P.; Ferro M.; Sessolo M.; Williamson A.; Koutsouras D. A.; Khodagholy D.; Ramuz M.; Strakosas X.; Owens R. M.; Benar C.; Badier J.-M.; Bernard C.; Malliaras G. G. Sci. Adv. 2015, 1, e1400251. 10.1126/sciadv.1400251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch I.; Heeney M.; Chabinyc M. L.; DeLongchamp D.; Kline R. J.; Cölle M.; Duffy W.; Fischer D.; Gundlach D.; Hamadani B.; Hamilton R.; Richter L.; Salleo A.; Shkunov M.; Sparrowe D.; Tierney S.; Zhang W. Adv. Mater. 2009, 21, 1091. 10.1002/adma.200801650. [DOI] [Google Scholar]
- Nielsen C. B.; Turbiez M.; McCulloch I. Adv. Mater. 2013, 25, 1859. 10.1002/adma.201201795. [DOI] [PubMed] [Google Scholar]
- Sirringhaus H. Adv. Mater. 2014, 26, 1319. 10.1002/adma.201304346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White H. S.; Kittlesen G. P.; Wrighton M. S. J. Am. Chem. Soc. 1984, 106, 5375. 10.1021/ja00330a070. [DOI] [Google Scholar]
- Owens R. M.; Malliaras G. G. MRS Bull. 2010, 35, 449. 10.1557/mrs2010.583. [DOI] [Google Scholar]
- Zhang X.-R.; Richter L. J.; DeLongchamp D. M.; Kline R. J.; Hammond M. R.; McCulloch I.; Heeney M.; Ashraf R. S.; Smith J. N.; Anthopoulos T. D.; Schroeder B.; Geerts Y. H.; Fischer D. A.; Toney M. F. J. Am. Chem. Soc. 2011, 133, 15073. 10.1021/ja204515s. [DOI] [PubMed] [Google Scholar]
- Ho P. K. H.; Chua L. L.; Dipankar M.; Gao X. Y.; Qi D. C.; Wee A. T. S.; Chang J. F.; Friend R. H. Adv. Mater. 2007, 19, 215. 10.1002/adma.200601285. [DOI] [Google Scholar]
- Sirringhaus H.; Brown P. J.; Friend R. H.; Nielsen M. M.; Bechgaard K.; Langeveld-Voss B. M. W.; Spiering A. J. H.; Janssen R. A. J.; Meijer E. W.; Herwig P.; de Leeuw D. M. Nature 1999, 401, 685. 10.1038/44359. [DOI] [Google Scholar]
- Nielsen C. B.; McCulloch I. Prog. Polym. Sci. 2013, 38, 2053. 10.1016/j.progpolymsci.2013.05.003. [DOI] [Google Scholar]
- Bernards D. A.; Malliaras G. G. Adv. Funct. Mater. 2007, 17, 3538. 10.1002/adfm.200601239. [DOI] [Google Scholar]
- Rivnay J.; Ramuz M.; Leleux P.; Hama A.; Huerta M.; Owens R. M. Appl. Phys. Lett. 2015, 106, 043301. 10.1063/1.4906872. [DOI] [Google Scholar]
- Inal S.; Rivnay J.; Leleux P.; Ferro M.; Ramuz M.; Brendel J. C.; Schmidt M. M.; Thelakkat M.; Malliaras G. G. Adv. Mater. 2014, 26, 7450. 10.1002/adma.201403150. [DOI] [PubMed] [Google Scholar]
- Zeglio E.; Vagin M.; Musumeci C.; Ajjan F. N.; Gabrielsson R.; Trinh X. T.; Son N. T.; Maziz A.; Solin N.; Inganäs O. Chem. Mater. 2015, 27, 6385. 10.1021/acs.chemmater.5b02501. [DOI] [Google Scholar]
- Inal S.; Rivnay J.; Hofmann A. I.; Uguz I.; Mumtaz M.; Katsigiannopoulos D.; Brochon C.; Cloutet E.; Hadziioannou G.; Malliaras G. G. J. Polym. Sci., Part B: Polym. Phys. 2016, 54, 147. 10.1002/polb.23938. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.