

Abstract
The one-pot, three-component, coupling reaction of indoles/pyrroles, dimethyl malonate, and acetic acid was performed using Mn(III) acetate as an oxidant. In the presence of Mn(OAc)3, indole-2, and indole-3-carbonyl compounds were alkylated at the 3- and 2- positions, respectively, with subsequent oxidation and nucleophilic capture occurring at the newly formed benzylic carbon. In contrast, oxidation of 2- and 3-indole carboxylic acids afforded the corresponding 2-oxindol-3-ylidenes and 3-oxindol-2-ylidenes. The reaction conditions, scope, and mechanism are discussed herein.
Graphical abstract

1. Introduction
The indole aromatic scaffold is ubiquitous in both bioactive natural products and pharmaceuticals,1 and tremendous effort has been expended to develop methods for the robust and efficient functionalization of the heteroaromatic skeleton. In the same vein, reliable investigation of the bioactivity of the natural frameworks has often relied upon de novo synthesis to supply the necessary quantities for study. Indole alkaloids functionalized at both the 2- and 3- carbons of the aromatic scaffold, such as undulifoline,2 actinophyllic acid,3 vinblastine and vincristine,4 piqued our interest into radical indole alkylation as they pose both strategic and methodological challenges (Figure 1).
Figure 1.

Stuctures of indole alkaloid natural products with C2 quaternary centers
We initially proposed that the congested C2 substituent in many of these natural products could be derived from the coupling of indole with versatile malonate derivatives using Mn(III). Manganese(III) acetate is a single-electron oxidant commonly used in radical-mediated annulations, cyclizations, as well as in the coupling of 1,3-dicarbonyl compounds to alkenes and aromatic systems.5 Oxidative radical cyclizations mediated by Mn(III) are well established and have been employed as key steps in the synthesis of natural products such as mersicarpine6 and phloroglucinol.7 Additionally, in the efforts towards the synthesis of tronocarpine, Kerr and coworkers were able to achieve oxidative cyclization of a wide variety of N-tethered dimethyl malonates of indoles, pyrroles, and indolines.8a,8b-c Conversely, there exist a limited number of reports involving intermolecular arene functionalization utilizing the radical chemistry of Mn(III), most notable being the seminal publications of Citterio and Santi, and Chuang.9 We herein expand the scope of Mn(OAc)3-mediated arene functionalization to include the successful coupling of unprotected indoles and pyrroles to dimethyl malonate.
2. Results and discussion
Initial efforts focused on accessing oxidatively coupled product 3 via the reaction of dimethyl malonate with indole-3-carboxaldehyde using Mn(OAc)3 (Scheme 1). Interestingly, over oxidized, alkylated indole 4a, derived from nucleophilic capture of the azafulvene indolidenium ion by acetic acid, was isolated in 38% yield with no detectable traces of 3. Initial reports of malonate radical coupling with arenes describe the identification of over oxidized products, although the application to indole scaffolds has been relatively unexplored.10 Previous oxidative, and Lewis acid mediated generation of similar indolidenium intermediates have proven valuable in the synthesis of a number of indole alkaloids.3f,4l,11
Scheme 1.

Attempted alkylation of indole-3-carboxaldehyde 1a
We attempted to further optimize the oxidative coupling reaction by investigating the effects of solvent, temperature, concentration, order of substrate addition, as well as the addition of co-oxidants; however, all attempts failed to significantly improve the overall yield. The best results were obtained from slow and simultaneous addition of substrates 1a and 2a over a period of 4–6 hours to a slurry of Mn(OAc)3 in AcOH at 85 °C.12 Full conversion of the indole starting material is observed after an additional 12 hours of stirring; however, undesirable by-products such as malonate dimers are also formed during the extended reaction time.
Exploring the scope of indole substitution, the optimized conditions were applied to a number of 4- and 5-substituted indole-3-carboxaldehyde compounds (Table 1, entries 1–3). In general, substitution lowered the yield and resulted in the formation of multiple unidentified side products. The scope of the dicarbonyl compound was also explored by reacting 1a with diethyl methyl malonate 2b (entry 4) providing the 2-quaternary-substituted indole 4ab in 28% yield. We were pleased to observe a significantly improved yield of 80% when 3-carbomethoxy substituted indole 1d was used (Table 1, entry 5). Again, substitution on the indole benzene unit was tolerated for compounds 1e–f but did not provide better overall results (entries 6–7). When ester-substituted indole 1d was reacted with tertiary malonate 2b there was a decrease in yield similar to that observed previously with the aldehyde-substituted substrate 1a (entry 8) as the oxidative coupling adduct (4db) was isolated in a modest 18% yield. It is interesting to note that nitrogen protection of indoles 1d–f with a pivaloyl group did not increase the conversion to oxidatively coupled products and instead resulted in 80% recovery of deprotected starting material. Finally, the reaction of 3-acetylindole also gave good yields of the oxidatively coupled product, (4g, Table 1, entry 9).
Table 1.
Scope of Mn(III)-mediated oxidative coupling with 3-indole (1)a

| |||
|---|---|---|---|
| Entry | 1 (R1, R2) | 2 (R3, R4) | Product (% Yield)b |
| 1 | 1a (H, H) | 2a (H, Me) |

|
| 2 | 1b (5-Br, H) | 2a (H, Me) |

|
| 3 | 1c (4-CO2Me, H) | 2a (H, Me) |
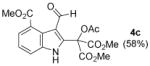
|
| 4 | 1a (H, H) | 2b (Me, Et) |

|
| 5 | 1d (H, OMe) | 2a (H, Me) |

|
| 6 | 1e (5-Br, OMe) | 2a (H, Me) |

|
| 7 | 1f (5-OMe, OMe) | 2a (H, Me) |

|
| 8 | 1d (H, OMe) | 2b (Me, Et) |

|
| 9 | 1g (H, Me) | 2a (H, Me) |

|
Reactions were performed by tandem slow addition of indoles 1(1 equiv.) and malonates 2 (2 equiv.) over 5 h to a stirred slurry of Mn(OAc)3•2H2O (6 equiv.) in AcOH at 85 °C.
Isolated yield.
A proposed mechanism for the formation of indole coupled products 4 is illustrated in Scheme 2. First, the Mn(III) enolate formed with the 1,3-dicarbonyl compound undergoes a one-electron oxidation to generate a doubly stabilized radical (I). Addition occurs preferentially to the least sterically hindered, non-substituted carbon of the indole 2,3-unsaturation. Subsequent oxidation by Mn(III) and acetate-mediated proton transfer rearomatizes the indole scaffold. The dialkyl arylmalonate product can again undergo rapid Mn(III)-mediated enolization and oxidation affording an indolidenium intermediate (V) that is ultimately trapped by acetate, furnishing the observed products, 4.
Scheme 2.

Proposed mechanism for the formation of 4
Overall, the ease of product formation seems to correlate with the ability of each substrate to stabilize the benzylic radical intermediate (II, Scheme 2). This is evidenced by the difference in yield that occurs when the 3-substituent on the indole decreases in electron-withdrawing ability, proceeding from the 3-formyl- (4a, 38%), to the 3-acetyl- (4g, 62%), to the 3-methylcarboxy- (4d, 80%) substituted indole. Substitution on the indole carbocycle seems to have a lesser effect, with no clear trend progressing from electron deficient to electron rich indole substrates.
During the course of the reaction, as outlined in Scheme 2, over oxidation occurs at the benzylic carbon, which is subsequently trapped by an external acetate nucleophile. Given this sequence of events, it was postulated that oxidative cyclization may compete with acetoxylation on a substrate such as indole-3-carboxylic acid (5a), which contains a pendant oxygen nucleophile able to form lactone 6 (Scheme 3). Surprisingly, upon the reaction of carboxylic acid 5a and malonate 2a in the presence of Mn(OAc)3, no traces of cyclized product were detected and instead 3-oxindole-2-ylidene 7a was obtained in 40% yield. Gribble and co-workers recently reported a similar transformation in which Mn(OAc)3-mediated oxidative coupling of 2-nitroindole and dimethyl malonate led to the formation of the corresponding 2-oxoindolin-3-ylidene (7b) via an in situ Nef reaction.13 Interestingly, the authors report failing to observe the 3-oxoindolin-2-ylidene (7a) when 3-nitroindole was used as substrate, rationalizing this result as the lack of a captodative stabilizing effect at C3 of the radical intermediate of 3-nitroindole. Testing the developed oxidative Mn(OAc)3 coupling conditions with indole-2-carboxylic acid (5b) we observed the formation of the same 2-oxindolin-3-ylidene 7b reported by Gribble and coworkers in 38% yield.
Scheme 3.

Oxidative cyclization of 5 to oxindolinylidenes 7.
The postulated mechanism occurs similarly to that suggested for the formation of 4, such that the reaction of 5a and 2a should first give the directly coupled product with acetate incorporation at the benzylic carbon (VI, Scheme 4). However, this intermediate can likely undergo a formal [1,3]-acetate migration via nitrogen lone pair-assisted elimination/acetate addition to the indole 3-position. Decarboxylation can then occur via a retro-ene-type mechanism to form a rearomatized acetoxy indole intermediate. Subsequent Mn(III)-mediated oxidations gives the acetyl oxocarbenium ion, which upon water or acetate-mediated cleavage forms product 7a.
Scheme 4.

Proposed mechanism for the formation of 7a
The electronic environment of the intermediate adduct radical (II, Scheme 2) was further perturbed in order to investigate Gribble’s mechanistic hypothesis. Towards this end, the Mn(III) conditions were applied to a series of heteroaromatic-2-carbonyl compounds 8a–d (Table 2). Gratifyingly, in addition to the functionalization of 2-formyl- and 2-methylcarboxyindole, the reaction conditions were amenable to the oxidative coupling of pyrrole-2-carbonyl derivatives similar to the tertiary malonate coupled products reported by Baciocchi and coworkers.9e Both methyl 2-pyrrolecarboxylate and 2-acetylpyrrole gave the 5-alkylated product in good to moderate yields. Neither the 3- or 4-substituted regioisomers were detected by inspection of the 1H NMR. Similar to the previously described indole-3-carbonyl derivatives, the yield of oxidative coupling products correlates to the electron-stabilizing effect of the substituent on C2 of the heteroaromatic moiety.
Table 2.
Oxidative coupling with indole- and pyrrole-2-carbonyl compounds (8)a

| ||
|---|---|---|
| Entry | 8 (heteroaromatic, R1) | Product (Yield %)b |
| 1 | 8a (indole, R1 = H) |
|
| 2 | 8b (indole, R1 = OMe) |
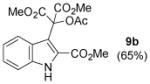
|
| 3 | 8c (pyrrole, R1 = OMe) |
|
| 4 | 8d (pyrrole, R1 = Me) |

|
Reactions were performed by tandem slow addition of indoles 8 (1 equiv.) and malonates 2 (2 equiv.) over 5 h to a stirred slurry of Mn(OAc)3•2H2O (6 equiv.) in AcOH at 85 °C.
Isolated yield.
In addition to the arylation/acetoxylation product of the reaction involving indole-2-carboxaldehyde, a 19% yield of 2-oxindol-3-ylidene (7b) was observed. We speculate that 7b forms from the in situ oxidation of the aldehyde to the carboxylic acid and the product is therefore prone to further oxidation pathways as proposed previously (Scheme 3). Given the highly colored side products observed during purification, it is likely the oxindolylidenes are a common by-product in the reaction of all the indole-carboxaldehyde derivatives studied.
Minimal differences in the yields were obtained from 2- and 3-carbonyl-substituted indoles, though there is a notable disparity in yield between ester-substituted compounds 9b and 4d. Whereas Gribble and coworkers found 3-nitroindole to be unaffected by Mn(III) and dimethyl malonate,13 we found that the less electron withdrawing carbonyl substituents provided the three-component coupling products in moderate to good yields when substituted at C2 or C3 of the indole scaffold. Likely the 3-nitro-substituent significantly impedes the nucleophilicity of the indole core.
3. Conclusions
In summary, we have reported the Mn(OAc)3-mediated oxidative three-component coupling of indole-2-carbonyl, indole-3-carbonyl, and pyrrole-2-carbonyl compounds to dimethyl malonate in acetic acid in moderate to good yields. These oxidative coupling conditions complement the previously reported photoredox-mediated reductive alkylation of indole with bromomalonate derivatives, being tolerant to functional groups sensitive to electron-transfer mediated reduction.14 The congested indole products formed could be applied further in the synthesis of indole alkaloid products bearing C2 quaternary centers through reversible indolidenium ions and nucleophilic capture.
4. Experimental Section
4.1. General experimental methods
The chemicals and reagents were purchased from Sigma-Aldrich, Merck, Combi-Blocks Inc., Chem-Impex Int’l, AK Scientific, Inc., Strem Chemicals Inc. (Manganese (III) acetate) and used without further purification. Infrared spectra were recorded on a Perkin Elmer BX FT-IR fitted with an ATR accessory. Absorptions are given in wavenumbers (cm-1). High resolution mass spectra were obtained at the University of Michigan Mass Spectrometry Centre on a Waters® Micromass® AutoSpec Ultima™ high resolution mass spectrometer. Reactions that were monitored by TLC were visualized by a dual short wave/long wave UV lamp and stained with an ethanolic solution of potassium permanganate. Column flash chromatography was performed using 230–400 mesh silica gel or via automated column chromatography. Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise noted. 1H and 13C NMR spectra were recorded in both 300 MHz NMR spectrophotometer (Bruker, AVANCE) and a 400 MHz spectrophotometer (JEOL, JNM ECS) in CDCl3 using tetramethylsilane (TMS) as the internal standard and coupling constants are expressed in Hertz. Chemical shifts are reported in ppm.
4.2. Synthetic methods
All compounds were synthesized according to a general procedure described herein: Mn(OAc)3 (6 mmol) was added to a 250 mL round bottom flask with a 1″ magnetic stirbar. The flask was then purged with nitrogen gas. The aromatic heterocycle (1 mmol) and malonate (2 mmol) were weighed out separately and subsequently dissolved in 7 mL of acetic acid (glacial), (If the aromatic heterocycle was slow to dissolve the vial was sonicated to facilitate a homogenous solution). Glacial acetic acid (10 mL) was added to the flask containing Mn(OAc)3 and the purge needle was removed. The flask containing the solution of Mn(OAc)3 was heated to 85 °C using a heating block. Once the target temperature was achieved the two solutions of malonate and heterocycle were added to the manganese solution via syringe pump over the course of 5 hours. Once the addition was complete the solution was heated for a further 12–18 hours until the starting material had been consumed as deemed by TLC.
The solution was cooled to room temperature and the acetic acid was removed on a rotary evaporator. To the brown solid was added water (100 mL) and ethyl acetate (50 mL). The two layers were separated and the aqueous phase was further extracted with ethyl acetate. The organic phase was washed with water (x2) and brine (x1), dried over Na2SO4, filtered, concentrated, and subjected to flash column chromatography. Specifics of characterization and purification are described for each compound.
4.3. Physical and spectroscopic data
Dimethyl 2-acetoxy-2-(3-formyl-1H-indol-2-yl)malonate 4a (Table 1, entry 1)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 126.6 mg of a white solid was obtained, 38 % yield; Rf: 0.46 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3302, 3180, 2957, 1753, 1636, 1222;
1H NMR (500 MHz, CDCl3) δ 10.87 (1H, s), 10.47 (1 H, s), 8.38 (1H, d, J = 7.5 Hz), 7.46 (1H, d, J = 7.5 Hz), 7.32 (2H, m), 3.86 (6H, s) and 2.22 (3 H, s).
13C NMR (126 MHz, CDCl3) δ 186.2, 169.3, 165.1, 138.4, 134.5, 125.4, 124.9, 123.5, 122.5, 116.2, 111.9, 77.6, 54.4, 20.7.
m/z: (ESI+) HRMS [M+Na] C16H15NO7Na+: found: 356.0741; calcd: 356.0741.
Dimethyl 2-acetoxy-2-(5-bromo-3-formyl-1H-indol-2-yl)malonate 4b (Table 1, entry 2)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 86.7 mg of a white solid was obtained, 28% yield; Rf: 0.45 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3119, 3068, 3030, 2956, 2850, 2728, 1756, 1631, 1210;
1H NMR (700 MHz, CDCl3) δ 10.97 (1H, s), 10.41 (1H, s), 8.52 (1H, d, J = 1.3 Hz), 7.40 (2H, dd, J = 8.6, 1.8 Hz), 7.32 (1H, d, J = 8.6 Hz), 3.86 (6H, s) and 2.21 (3 H, s).
13C NMR (176 MHz, CDCl3) δ 186.0, 169.3, 164.9, 139.3, 133.2, 127.9, 126.9, 125.1, 117.1, 115.5, 113.4, 77.4, 54.6, 20.7.
m/z: (ESI+) HRMS [M+Na] C16H14BrNO7Na+: found: 433.9844; calcd: 433.9846.
Dimethyl 2-acetoxy-2-(3-formyl-4-(methoxycarbonyl)-1H-indol-2-yl)malonate 4c (Table 1, entry 3)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 227.0 mg of a white solid was obtained, 58 % yield; Rf: 0.43 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3209, 3023, 2955, 2848, 1765, 1747, 1714, 1642, 1237;
1H NMR (500 MHz, CDCl3) δ 11.02 (1H, s), 10.46 (1H, s), 7.64 (1H, dd, J = 7.4, 0.9 Hz), 7.61 (1H, dd, J = 8.2, 0.9 Hz), 7.35 (1H, d, J = 8.2, 7.5 Hz), 3.98 (3H, s) 3.86 (6H, s) and 2.15 (3 H, s).
13C NMR (176 MHz, CDCl3) δ 186.8, 169.7, 169.2, 165.1, 138.5, 135.1, 126.1, 124.4, 123.8, 121.9, 115.7, 115.6, 77.6, 54.4, 52.3, 20.6.
m/z: (ESI+) HRMS [M+Na] C18H17NO9Na+: found: 414.0797; calcd: 414.0796.
Diethyl 2-(3-formyl-1H-indol-2-yl)-2-methylmalonate 4ab (Table 1, entry 4)
Using a gradient eluent from 0% → 30% ethyl acetate in hexanes, 88.5 mg of a light orange colored solid was obtained, 28 % yield; Rf: 0.30 (7:3, hexane:ethyl acetate)
ν max (ATR)/cm−1: 3220, 2985, 2940, 1733, 1627, 1201, 1165;
1H NMR (500 MHz, CDCl3) δ 10.75 (1H, s), 10.20 (1H, s), 8.28–8.19 (1H, m), 7.49–7.40 (1H, m), 7.34–7.27 (2H, m), 4.32 and 4.29 (ABX3, 4H, J = 10.5, 7.5 Hz), 2.02 (3H, s) and 1.28 (6H, dd, J = 7.5 Hz).
13C NMR (126 MHz, CDCl3) δ 184.4, 170.1, 143.6, 133.9, 126.6, 123.8, 123.0, 120.8, 113.8, 111.6, 62.8, 54.2, 25.6, 13.8.
m/z: (ESI+) HRMS [M+Na] C17H19NO5Na+: found: 340.1158; calcd: 340.1155.
Dimethyl 2-acetoxy-2-(3-(methoxycarbonyl)-1H-indol-2-yl)malonate 4d (Table 1, entry 5)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 290.5 mg of a yellow foam was obtained, 80 % yield; Rf: 0.41 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3353, 2954, 1763, 1748, 1690, 1223;
1H NMR (500 MHz, CDCl3) δ 10.54 (1H, s), 8.11 (1H, d, J = 7.8 Hz), 7.44 (1H, d, J = 7.7 Hz), 7.27 (2H, m), 3.91 (3H, s), 3.86 (6H, s) and 2.11 (3 H, s).
13C NMR (126 MHz, CDCl3) δ 170.0, 165.6, 164.9, 136.8, 133.9, 125.8, 124.0, 122.3, 121.8, 112.0, 105.8, 78.0, 54.2, 51.1, 20.7.
m/z: (ESI+) HRMS [M+Na] C17H17NO8Na+: found: 386.0846; calcd: 386.0847.
Dimethyl 2-acetoxy-2-(5-bromo-3-(methoxycarbonyl)-1H-indol-2-yl)malonate 4e (Table 1, entry 6)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 230.0 mg of a white solid was obtained, 52 % yield; Rf: 0.41 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3354, 2953, 1763, 1747, 1734, 1697, 1196;
1H NMR (500 MHz, CDCl3) δ 10.59 (1H, s), 8.25 (1H, d, J = 1.8 Hz), 7.39 (1H, dd, J = 8.7, 1.8 Hz), 7.33 (1H, d, J = 8.7 Hz), 3.91 (3H, s), 3.87 (6H, s) and 2.11 (3 H, s).
13C NMR (176 MHz, CDCl3) δ 170.1, 165.5, 164.4, 137.9, 132.4, 127.3, 127.1, 124.5, 115.9, 113.5, 105.4, 77.7, 54.3, 51.3, 20.7.
m/z: (ESI+) HRMS [M+Na] C17H16BrNO8Na+: found: 453.9952; calcd: 463.9952.
Dimethyl 2-acetoxy-2-(5-methoxy-3-(methoxycarbonyl)-1H-indol-2-yl)malonate 4f (Table 1, entry 7)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 157.4 mg of a white solid was obtained, 40 % yield; Rf: 0.42 (1:1, hexane:ethyl acetate)
ν max (ATR)/cm−1: 3387, 3337, 2952, 1760, 1732, 1701, 1166;
1H NMR (500 MHz, CDCl3) δ 10.46 (1H, s), 7.57 (1H, d, J = 2.3 Hz), 7.33 (1H, d, J = 8.9 Hz), 6.94 (1H, dd, J = 8.9, 2.4 Hz), 3.89 (3H, s), 3.86 (3H, s) 3.85 (6H, s) and 2.10 (3 H, s).
13C NMR (176 MHz, CDCl3) δ 169.9, 165.6, 164.8, 156.0, 136.6, 129.0, 126.8, 114.9, 112.9, 105.4, 102.8, 78.0, 55.7, 54.2, 50.9, 20.7.
m/z: (ESI+) HRMS [M+Na] C18H19NO9Na+: found: 416.0953; calcd: 416.0952.
Diethyl 2-(3-(methoxycarbonyl)-1H-indol-2-yl)-2-methylmalonate 4db (Table 1, entry 8)
Using a gradient eluent from 0% → 30% ethyl acetate in hexanes, 68.3 mg of a light yellow colored oil was obtained, 18 % yield; Rf: 0.53 (7:3, hexane:ethyl acetate)
ν max (ATR)/cm−1: 3361, 2987, 1730, 1692, 1527, 1441, 1188;
1H NMR (500 MHz, CDCl3) δ 10.46 (1H, s), 8.16–8.09 (1H, m), 7.44–7.38 (1H, m), 7.28–7.22 (2H, m), 4.28 (4H, q, J = 7.1 Hz), 3.88 (3H, s), 1.98 (3H, s, J = 7.5 Hz), 1.26 (6H, t, J = 7.1 Hz).
13C NMR (126 MHz, CDCl3) δ 170.6, 165.3, 142.7, 133.7, 126.8, 123.0, 122.0, 121.7, 111.5, 104.2, 62.4, 55.3, 50.8, 23.9, 13.9;
m/z: (EI+) HRMS [M+] C18H21NO6+: found: 347.1375; calcd: 347.1369.
Dimethyl 2-acetoxy-2-(3-acetyl-1H-indol-2-yl)malonate 4g (Table 1, entry 9)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 216.0 mg of a slightly orange colored solid was obtained, 62 % yield; Rf: 0.35 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3328, 2955, 2848, 1752, 1720, 1655, 1224, 1182;
1H NMR (500 MHz, CDCl3) δ 10.70 (1H, s), 7.92–7.84 (1H, m), 7.52–7.45 (1H, m), 7.33–7.24 (2H, m), 3.85 (6H, s), 2.70 (3H, s) 3.85 (6H, s) and 2.09 (3 H, s).
13C NMR (126 MHz, CDCl3) δ 194.0, 170.2, 165.6, 136.4, 134.2, 125.1, 123.7, 122.5, 120.7, 114.9, 112.7, 78.4, 54.1, 31.1, 20.7.
m/z: (ESI+) HRMS [M+Na] C17H17NO7Na+: found: 370.0896; calcd: 370.0897.
Dimethyl 2-(3-oxoindolin-2-ylidene)malonate 7a (Scheme 3)
Using a gradient eluent from 0% → 50% diethyl ether in hexanes, 104.5 mg of bright red colored crystals were obtained, 40 % yield; Rf: 0.26 (1:1, hexane:diethyl ether)
ν max (ATR)/cm−1: 3343, 2990, 2958, 2840, 1744, 1681, 1597, 1188;
1H NMR (500 MHz, CDCl3) δ 9.17 (1H, s), 7.63 (1H, d, J = 7.6 Hz), 7.49 (1H, app t, J = 7.7 Hz), 6.99 (1H, app t, J = 7.5 Hz), 6.92 (1H, d, J = 8.0 Hz), 3.92 (3H, s), and 3.83 (3H, s).
13C NMR (126 MHz, CDCl3) δ 185.6, 166.4, 165.3, 152.1, 142.8, 137.6, 125.7, 122.3, 119.8, 111.9, 101.3, 53.1, 52.6.
m/z: (ESI+) HRMS [M+Na] C13H11NO5Na+: found: 284.0530; calcd: 284.0529.
Dimethyl 2-(2-oxoindolin-3-ylidene)malonate 7b (Scheme 3)
Using a gradient eluent from 0% → 50% diethyl ether in hexanes, 99.0 mg of an orange crystalline solid was obtained, 38 % yield; Rf: 0.27 (1:1, hexane:diethyl ether)
ν max (ATR)/cm−1: 3194, 3163, 3091, 3032, 2948, 2838, 1737, 1713, 1609, 1249;
1H NMR (500 MHz, CDCl3) δ 8.74 (1H, s), 8.35 (1H, d, J = 7.9 Hz), 7.32 (1H, app t, J = 7.9 Hz), 7.01 (1H, app t, J = 7.8 Hz), 6.83 (1H, d, J = 7.8 Hz), 3.94 (3H, s) and 3.91 (3H, s).
13C NMR (126 MHz, CDCl3) δ 167.7, 166.0, 163.2, 143.6, 135.4, 133.4, 129.1, 128.6, 122.9, 119.5, 110.4, 53.2, 53.1.
m/z: (ESI+) HRMS [M+Na] C13H11NO5Na+: found: 284.0530; calcd: 284.0529.
Dimethyl 2-acetoxy-2-(2-formyl-1H-indol-3-yl)malonate 9a (Table 2, entry 1)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 126.5 mg of a white crystalline solid was obtained 38 % yield; Rf: 0.55 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3367, 2952, 1745, 1642, 1223;
1H NMR (500 MHz, CDCl3) δ 10.36 (1H, s), 9.72 (1H, s), 7.88 (1H, d, J = 8.4 Hz), 7.45 (1H, d, J = 8.4 Hz), 7.37 (1H, app t, J = 7.6 Hz), 7.18 (1H, app t, J = 7.6 Hz), 3.80 (6H, s) and 2.35 (3H, s).
13C NMR (126 MHz, CDCl3) δ 183.8, 169.5, 166.2, 136.4, 132.9, 127.1, 125.0, 122.5, 122.0, 116.0, 112.6, 81.3, 53.7, 20.8.
m/z: (ESI+) HRMS [M+Na] C16H15NO7Na+: found: 356.0741; calcd: 356.0741.
Dimethyl 2-acetoxy-2-(2-(methoxycarbonyl)-1H-indol-3-yl)malonate 9b (Table 2, entry 2)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 236.4 mg of a yellow crystalline solid was obtained, 65 % yield; Rf: 0.40 (1:1, hexanes:ethyl acetate)
ν max (ATR)/cm−1: 3384, 3054, 2998, 2953, 1773, 1726, 1208;
1H NMR (500 MHz, CDCl3) δ 9.35 (1H, s), 7.74 (1H, d, J = 8.3 Hz), 7.38 (1H, d, J = 8.2 Hz), 7.29 (1H, app t, J = 7.5 Hz), 7.15 (1H, app t, J = 7.5 Hz), 3.83 (6H, s) 3.82 (3H, s) and 2.22 (3H, s).
13C NMR (126 MHz, CDCl3) δ 169.1, 166.7, 160.7, 135.1, 126.2, 125.5, 124.4, 122.3, 121.4, 114.7, 112.2, 81.4, 53.4, 52.1, 20.9.
m/z: (ESI+) HRMS [M+Na] C17H17NO8Na+: found: 386.0847; calcd: 386.0846.
Dimethyl 2-acetoxy-2-(5-(methoxycarbonyl)-1H-pyrrol-2-yl)malonate 9c (Table 2, entry 3)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 244.9 mg of a yellow oil was obtained, 78 % yield; Rf: 0.5 (1:1, hexane:ethyl acetate)
ν max (ATR)/cm−1: 3429, 3313, 3006, 2956, 1748, 1705, 1206;
1H NMR (500 MHz, CDCl3) δ 9.99 (1H, s), 6.79 (1H, dd, J = 3.8, 2.8 Hz), 6.27 (1H, dd, J = 3.8, 2.7 Hz), 3.82 (3H, s), 3.80 (6H, s) and 2.18 (3H, s).
13C NMR (126 MHz, CDCl3) δ 169.6, 165.2, 160.9, 128.2, 123.8, 115.0, 110.3, 78.2, 53.8, 51.6, 20.7.
m/z: (ESI+) HRMS [M+Na] C13H15NO8Na+: found: 336.0691; calcd: 336.0690.
Dimethyl 2-acetoxy-2-(5-acetyl-1H-pyrrol-2-yl)malonate 9d (Table 2, entry 4)
Using a gradient eluent from 5% → 40% ethyl acetate in hexanes, 124.5 mg of a yellow oil was obtained, 42 % yield; Rf: 0.45 (1:1, hexane:ethyl acetate)
ν max (ATR)/cm−1: 3429, 3252, 3012, 2957, 1746, 1649, 1206;
1H NMR (500 MHz, CDCl3) δ 9.99 (1H, s), 6.80 (1H, dd, J = 3.9, 2.6 Hz), 6.33 (1H, dd, J = 3.9, 2.7 Hz), 3.83 (6H, s), 2.41 (3H, s) and 2.23 (3H, s).
13C NMR (126 MHz, CDCl3) δ 187.8, 169.4, 165.0, 132.8, 129.7, 115.9, 110.3, 78.3, 53.9, 25.4, 20.7.
m/z: (ESI+) HRMS [M+Na] C13H15NO7Na+: found: 320.0739; calcd: 320.0741.
Supplementary Material
Acknowledgments
The authors thank Mr. Joel Beatty, and Dr. Irene Bosque for their helpful discussions and suggestions. This work was partially funded by the Boston University Undergraduate Research Opportunities Program. Financial support for this research from the NIH-NIGMS (R01-GM096129), the Camille Dreyfus Scholar Award Program, Eli Lilly, Novartis, Boston University and the University of Michigan is gratefully acknowledged.
Footnotes
Copies of 1H and 13C NMR spectra for all products. Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/xxxxxxxxxxxxxxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Kaushik NK, Kaushik N, Attri P, Kumar N, Kim CH, Verma AK, Choi EH. Molecules. 2013;18:6620–6662. doi: 10.3390/molecules18066620. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O’Connor SE, Maresh JJ. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/b512615k. [DOI] [PubMed] [Google Scholar]
- 2.Massiot G, Boumedjel A, Nuzillard JM, Richard B, Le Men-Olivier L, David B, Hadi HA. Phytochemistry. 1992;31:1078–1079. [Google Scholar]
- 3.(a) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2008;130:7568–7569. doi: 10.1021/ja803158y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vaswani RG, Day JJ, Wood JL. Org Lett. 2009;11:4532–4535. doi: 10.1021/ol901746c. [DOI] [PubMed] [Google Scholar]; (d) Zaimoku H, Taniguchi T, Ishibashi H. Org Lett. 2012;14:1656–1658. doi: 10.1021/ol300280s. [DOI] [PubMed] [Google Scholar]; (e) Galicia IZ, Maldonado LA. Tetrahedron Lett. 2013;54:2180–2182. [Google Scholar]; (f) Granger BA, Jewett IT, Butler JD, Hua B, Knezevic CE, Parkinson EI, Hergenrother PJ, Martin SF. J Am Chem Soc. 2013;135:12984–12986. doi: 10.1021/ja4070206. [DOI] [PubMed] [Google Scholar]
- 4.(a) Potier P. J Nat Prod. 1980;43:72–86. [Google Scholar]; (b) Langlois N, Gueritte F, Langlois Y, Potier P. J Am Chem Soc. 1976;98:7017–7024. doi: 10.1021/ja00438a046. [DOI] [PubMed] [Google Scholar]; (c) Kutney JP, Hibino T, Jahngen E, Okutani T, Ratcliffe AH, Treasurywala AM, Wunderly S. Helv Chim Acta. 1976;59:2858–2882. doi: 10.1002/hlca.19760590824. [DOI] [PubMed] [Google Scholar]; (d) Kuehne ME, Matson PA, Bornmann WG. J Org Chem. 1991;56:513–528. [Google Scholar]; (e) Bornmann WG, Kuehne ME. J Org Chem. 1992;57:1752–1760. [Google Scholar]; (f) Yokoshima S, Ueda T, Kobayashi S, Sato A, Kuboyama T, Tokuyama H, Fukuyama T. J Am Chem Soc. 2002;124:2137–2139. doi: 10.1021/ja0177049. [DOI] [PubMed] [Google Scholar]; (g) Kuboyama T, Yokoshima S, Tokuyama H, Fukuyama T. Proc Natl Acad Sci USA. 2004;101:11966–11970. doi: 10.1073/pnas.0401323101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Magnus P, Stamford A, Ladlow M. J Am Chem Soc. 1990;112:8210–8212. [Google Scholar]; (i) Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904–4916. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J Am Chem Soc. 2006;128:10596–10612. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org Lett. 2005;7:4539–4542. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904–4916. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For comprehensive reviews on Mn(III)-mediated transformations, see: De Klein WJ. In: In Organic Syntheses by Oxidation with Metal Compounds. Mijs WJ, De Jonge CRHI, editors. Plenum Press; New York: 1986. pp. 261–314.Snider BB. Chem Rev. 1996;96:339–364. doi: 10.1021/cr950026m.Melikyan GG. Synthesis. 1993:833–850.
- 6.Kerr MA, Magolan J. Org Lett. 2006;8:4561–4564. doi: 10.1021/ol061698+. [DOI] [PubMed] [Google Scholar]
- 7.Kraus GA, Dneprovskaia E, Nguyen TH, Jeon I. Tetrahedron. 2003;59:8975–8978. [Google Scholar]
- 8.Kerr MA, Magolan J. Org Lett. 2008;10:1437–1440. doi: 10.1021/ol800259s.for more recent examples see also: Oisaki K, Abe J, Kanai M. Org Biomol Chem. 2013;11:4569–4572. doi: 10.1039/c3ob40855h.Lee HS, Kim SH, Kim YM, Kim JN. Tetrahedron Lett. 2010;51:5071–5075.
- 9.Some notable examples: Nishino H, Cong Z. Synthesis. 2008;17:2686–2694.Tsai AI, Lin CH, Chuang CP. Heterocycles. 2005;65:2381–2394.Wang SF, Chuang CP. Heterocycles. 1997;45:347–359.Chuang CP, Wang SF. Tetrahedron Lett. 1994;35:1283–1284.Baciocchi E, Muraglia E. J Org Chem. 1993;58:7610–7612.Citterio A, Santi R, Fiorani T, Strologo S. J Org Chem. 1989;54:2703–2712.
- 10.Related coupling processes achived using copper: Bao Y-H, Zhu J-Y, Qin W-B, Kong U-B, Chen Z-W, Tang S-B, Liu L-X. Org Biomol Chem. 2013;11:7938–7945. doi: 10.1039/c3ob41589a.
- 11.(a) Levinson AM. Org Lett. 2014;16:4904–4907. doi: 10.1021/ol5024163. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiricek J, Blechert S. J Am Chem Soc. 2004;126:3534–3538. doi: 10.1021/ja0399021. [DOI] [PubMed] [Google Scholar]; (c) Zhong X, Li Y, Han FS. Chem Eur J. 2012;18:9784–9788. doi: 10.1002/chem.201201344. [DOI] [PubMed] [Google Scholar]; (d) Zhong X, Li Y, Zhang J, Han FS. Org Lett. 2015;17:720–723. doi: 10.1021/ol503734x. [DOI] [PubMed] [Google Scholar]
- 12.General procedure: Mn(OAc)3 (6 mmol) was added to a 250 mL round bottom flask with a 1″ magnetic stirbar and purged with nitrogen gas. The aromatic heterocycle (1 mmol) and malonate (2 mmol) were weighed out separately and subsequently dissolved in 7 mL of acetic acid (glacial), (if the aromatic heterocycle was slow to dissolve the vial was sonicated to facilitate formation of a homogeneous solution). Glacial acetic acid (10 mL) was added to the flask containing Mn(OAc)3 and the purge needle was removed. The flask containing the solution of Mn(OAc)3 was heated to 85 °C using a heating block. Once the target temperature was achieved the two solutions of malonate and heterocycle were added to the manganese solution via syringe pump over the course of 5 hours. Once the addition was complete the solution was heated for a further 12–24 hours until the starting material had been consumed as deemed by TLC. The solution was cooled to room temperature and the acetic acid was removed on a rotary evaporator. To the brown solid was added water (100 mL) and ethyl acetate (50 mL). The two layers were separated and the aqueous phase was further extracted with ethyl acetate. The organic phase was washed with water (x2), washed with brine (x1), dried over Na2SO4, filtered, and concentrated. Purification by flash chromatagraphy provided compounds 4a–g, 7a–b, and 9a–d.
- 13.Gribble GW, Kishbaugh TLS, Adrosov DA. Tetrahedron Lett. 2008;49:6621–6623. [Google Scholar]
- 14.(a) Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org Lett. 2010;12:368–371. doi: 10.1021/ol902703k. [DOI] [PubMed] [Google Scholar]; (b) Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org Lett. 2010;12:3104–3107. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]; (d) Devery JJ, III, Douglas JJ, Nguyen JD, Cole KP, Flowers RA, II, Stephenson CRJ. Chem Sci. 2015;6:537–541. doi: 10.1039/c4sc03064h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Swift EC, Williams TM, Stephenson CRJ. Synlett. 2015 accepted. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.