

Abstract
B-lymphocyte migration, directed by chemokine gradients, is essential for homing to sites of antigen presentation. B cells move rapidly, exhibiting amoeboid morphology like other leukocytes, yet quantitative studies addressing B-cell migration are currently lacking relative to neutrophils, macrophages, and T cells. Here, we used total internal reflection fluorescence (TIRF) microscopy to characterize the changes in shape (morphodynamics) of primary, murine B cells as they migrated on surfaces with adsorbed chemokine, CXCL13, and the adhesive ligand, ICAM-1. B cells exhibited frequent, spontaneous dilation and shrinking events at the sides of the leading membrane edge, a phenomenon that was predictive of turning versus directional persistence. To characterize directed B-cell migration, a microfluidic device was implemented to generate gradients of adsorbed CXCL13 gradients. Haptotaxis assays revealed a modest yet consistently positive bias of the cell’s persistent random walk behavior towards CXCL13 gradients. Quantification of tactic fidelity showed that bias is optimized by steeper gradients without excessive midpoint density of adsorbed chemokine. Under these conditions, B-cell migration is more persistent when the direction of migration is better aligned with the gradient.
INTRODUCTION
In the adaptive process by which humoral immunity is achieved, antibody-producing B lymphocytes must first become activated through contact with cognate helper T cells. This process requires trafficking of T and B cells within secondary lymphoid tissues, where lymphocytes dynamically organize to form spatially defined germinal centers. B-lymphocyte homing and trafficking is directed by gradients of attractants known as chemokines (1–3). In particular, the chemokine CXCL13 is important for directing B-cell entry into secondary lymphoid organs and the formation of germinal centers (4). Another chemokine, CXCL12, initially attracts naïve B cells to the so-called dark zone of the germinal center, where they proliferate and interact with follicular dendritic cells (FDCs); thereafter, the B cells lose expression of the CXCL12 receptor, CXCR4, and follow a gradient of CXCL13 to the light zone of the germinal center, where somatic hypermutation takes place (4, 5). Within the germinal center, B-cell adhesion and migration are also mediated by the integrin LFA-1, which binds to ICAM-1 expressed by FDCs (6, 7). LFA-1 is converted to a high-affinity state in response to chemokine stimulation (8). Signaling pathways triggered by ligated chemokine receptors and integrins converge to activate WASP-family proteins, leading to F-actin reorganization and cell polarization (9, 10). F-actin polymerization might, in turn, promote LFA-1 binding and activation (11). The morphological changes exhibited by chemokine-stimulated B cells have also been linked to antigen-dependent B-cell activation (12, 13).
The distribution of CXCL13 in vivo has been examined by antibody staining (14), suggesting a surface-bound distribution. It is known that CXC-family chemokines bind to glycosaminoglycans (GAGs), and therefore it is plausible that CXCL13 is largely immobilized in vivo (15, 16). Therefore, studying B-cell migration on adhesive surfaces coated with CXCL13 is valuable for understanding how B cells move in tissues (12, 17). While functional studies have implicated CXCL13-directed cell migration in B cell maturation (18), detailed characterization of B-cell migration and how it is biased by an immobilized chemokine gradient (haptotaxis) is lacking, in part because methods to characterize the morphologies and behaviors of individual cells have yet to be widely adopted. The use of microfluidic devices to generate gradients of soluble and immobilized factors has yielded insights into the directed migration of various cell types, including leukocytes (19–25) and fibroblasts (26, 27), suggesting a promising application in the characterization of B-cell migration.
Here, we address two quantitative aspects of B-cell migration. First, we used total internal reflection fluorescence (TIRF) microscopy to image the contact areas of randomly migrating B cells, and we analyzed how changes in cell shape (dilation and shrinking of the cell’s leading edge) are related to/predictive of the cell’s directional persistence/turning behavior. Second, we used microfluidic chambers to generate surfaces with gradients of immobilized CXCL13 along with uniformly adsorbed ICAM-1. Analysis of single-cell tracks revealed how haptotactic fidelity and directional persistence are affected by the properties of the CXCL13 gradient.
RESULTS
Migrating B cells exhibit cycles of dilation and shrinking of a broad leading edge
Changes in cell shape (morphodynamics) offer insight into mechanisms that affect the efficiency and directional persistence of cell movement (26, 28, 29). To study the morphodynamics of B-cell migration, a cohort of 30 primary B cells isolated from mouse spleens (13 independent experiments) were labeled and allowed to migrate on surfaces with uniform coatings of CXCL13 and ICAM-1. The cells’ areas of contact with the surface were imaged by total internal reflection fluorescence (TIRF) microscopy and analyzed. We found that mouse B-cell migration is characterized by a widely spread leading edge, which exhibits periods of dilation (red arrows) and shrinking (blue arrow) (Fig. 1A and Movie S1). Comparison of the cell contact area illuminated by TIRF and the projected area imaged by epi-fluorescence indicates that the rear of the cell, typically including the nuclear region, is seldom adhered to the surface (Fig. S1 and Movie S2). This is consistent with the polarized morphology of randomly migrating B cells observed previously (12). Spatiotemporal maps of protrusion/retraction rates and the presence of morphological extensions (Fig. 1B) (28, 30) reveal that the activities occurred most prominently at the two extrema of the leading edge contour (Fig. 1C). To visualize the attendant shape changes, we replotted the protrusion/retraction rates with a moving reference frame, i.e., with a stationary cell centroid (Fig. 1D). The map generated in this fashion reveals frequent waves of leading-edge dilation and shrinking, often as pairs of dilation-shrinking events (Fig. 1E). Their occurrence at the two ends of a persistent leading edge suggests that these events are indicative of turning/steering of the cell and not a transient loss and re-establishment of polarity, which can be considered a separate process (31).
Fig. 1. Morphodynamics of B-lymphocyte migration.

a) Representative TIRF montage of a murine B cell executing a turn (inverted grayscale). Red arrows indicate dilations and blue arrows indicate shrinking events at the leading edge. b) Stacked outlines of a representative B cell. Warmer colors indicate later times, and green dots indicate the cell centroid position. c) Top panel: illustration of protruded (red) and retracted (blue) pixels; bottom panel: representative protrusion/retraction map of a migrating B cell. Pixels associated with structures that are morphologically extended from the cell body are marked as black dots overlaid with protrusion/retraction velocity data as shown in the color map. The direction of cell centroid translocation (green dots) is also shown. d) Stacked cell outlines of the same sequence in b) with a moving reference. e) Top panel: illustration of dilated (red) and shrunk (blue) pixels; bottom panel: dilation/shrinking map of the same sequence depicted in c, constructed as in c except without the morphological extension data. The black boxes indicate two representative dilation-shrinking pairs. All scale bars = 10 μm.
The balance of dilation and shrinking between the two sides of the cell determines the directionality of B-cell migration
To relate turning behavior to the observed dilation and shrinking events, we documented the fates of those events, depicted along with the path of the cell centroid as a graph. We noted 1) whether the site is dilating or shrinking and 2) whether the event occurred on the left or the right side of the apparent migration axis; the length of each segment in the graph is proportional to the recorded lifetime of the event. The graph for a representative cell illustrates that noticeable changes in directionality tend to be associated with an imbalance between the dilation/shrinking activities on the two sides of the cell (Fig. 2A). The two pairs of black arrows in Fig. 2A show examples of prolonged dilation-shrinking pairs on the left side of the migration axis prior to counterclockwise cell turning.
Fig. 2. Linking morphodynamics to directional persistence of B-lymphocyte migration.

a) For the representative sequence depicted in Fig. 1b–e, dilation (red) and shrinking (blue) events are marked as segments on the cell centroid track (green) indicating whether the event occurred on the left or right of the migration axis. The length of each segment is proportional to the recorded lifetime of the event. The pairs of black arrows show examples of prolonged dilation-shrinking pairs prior to cell turning. b) Normalized waiting-time distributions (WTDs) for pairs of all consecutive dilation/shrinking events (n = 846) and pairs following a dilation/shrinking event on the opposite side (n = 428). The red and blue symbols are the corresponding best-fit values assuming an exponential distribution. c) Normalized distributions for the durations of straight (n = 72) and turning (n = 114) periods. d) Normalized distributions of the aggregate lifetimes of dilation-shrinking pairs on the left side minus that on the right side that were initiated during the 0.25 min window before left turning (n = 53), right turning (n = 44), and straight (n = 67) periods.
To define the characteristics of B-cell migration more precisely, we developed a quantitative analysis. The waiting times between consecutive dilation or shrinking waves were determined from the morphodynamic maps, within time intervals of 0.25 min (which we considered the limit of resolution). The normalized waiting-time distributions (WTDs) of the dilation/shrinking events are presented as a histogram and compared to that of a Poisson process, i.e., an exponential distribution (Fig. 2B). The WTD for successive dilation/shrinking events on opposite sides of the leading edge was found to be similar, suggesting that the dilation/shrinking of each side is spontaneous and uncorrelated with that of the other (Fig. 2B). We also note that dilation-shrinking pairs were observed 3 times as frequently as same-type pairs (636 vs. 210).
As all migrating cells tend to execute a persistent random walk in the absence of spatial cues, B cells exhibit distinct periods of straight movement vs. turning. Analysis of the durations of these two states revealed non-exponential distributions, suggesting that a change in migration behavior is not spontaneous but rather the outcome of multiple events (Fig. 2C). Accordingly, the average durations of straight (0.99 min) and turning periods (0.88 min) are much longer than the mean waiting time between dilation/shrinking events (0.20 min). Given that net protrusion/retraction of the two sides of the leading edge determines cell turning behavior, we postulated that dilation-shrinking pairs observed on the same side of the cell might be indicative of straight vs. turning migration states. We examined the 0.25 min interval just before each straight or turning period and recorded the accumulated lifetime of the dilation-shrinking pairs on the left minus that on the right side of the cell within this window. The normalized distributions of this quantity (excluding periods with no dilation-shrinking pair present), for the left-turning (counterclockwise) periods, right-turning (clockwise) periods, and straight periods indicate that an imbalance of dilation-shrinking is predictive of the initiation of cell turning (Fig. 2D). Dilation-shrinking pairs on the left side of the migration axis predict counterclockwise turning and vice-versa, whereas balanced pairs predict a transition from turning to straight movement.
B cells execute biased random walks on surface-immobilized CXCL13 gradients
Having elucidated the nature of random B-cell motility at the level of leading-edge adhesive dynamics, we sought to study the fidelity of primary B-cell migration biased by a gradient of surface-immobilized chemokine. CXCL13 gradients were established by physisorption on glass, using a microfluidic device as described previously (26, 32). To account for the variability of this process, antibody labeling was used to quantify the relative CXCL13 gradient after each experiment (Fig. 3A&B). TIRF imaging confirmed that the morphologies of B cells migrating on a chemokine gradient are qualitatively similar to those of randomly migrating cells (Movie S3). B-cell migration was subtly affected by this directional cue, with some cells moving predominantly up-gradient while others moved without apparent bias (Fig. 3C). To quantify the directional responses for the population of cells tracked in each experiment, we plot as a polar histogram the angles of the cells’ movement vectors relative to the gradient. The result shows a modest yet significant bias, as indicated by the positive 95% confidence interval of the forward migration index (FMI), which is also commonly referred to as the chemotactic index (Fig. 3D, gray). As a random migration control performed for most experiments, we also recorded B cells migrating on the uniform CXCL13-coated surface in the source chamber. The same FMI analysis of randomly migrating cells showed no significant directional bias (Fig. 3D, red).
Fig. 3. Quantifying the haptotactic bias of B-lymphocyte migration on CXCL13 gradients.

a) Surface-bound CXCL13 gradient labeled with fluorescent antibodies and b) quantification of the gradient profile. Linear regression of fluorescence intensity as a function of position is shown in red. Scale bar = 100 μm. c) Centroid tracks of the cells migrating on the surface shown in a. Each circle symbol marks the start, and the corresponding square symbol marks the end of the centroid track. Green arrows indicate representative cells migrating towards higher CXCL13 concentration; red arrows indicate representative cells migrating without apparent bias. The CXCL13 gradient increases from right to left. Scale bar: 10μm. d) Wind rose plot showing the angular histogram (of time intervals with the particular direction of migration relative to the gradient) for the migrating cells in b (gray bars), overlaid with that of the corresponding random migration control (red bars). Zero degrees corresponds to perfectly alignment with the gradient. The mean forward migration index (FMI) and 95% confidence interval are given.
The tactic coefficient of directed B-cell migration is a decreasing function of chemokine density
To assess the reproducibility of B-cell taxis and its dependence on the properties of the CXCL-13 gradient (steepness, midpoint density), the experiment described in the previous section was repeated 65 times with different gradients. The average FMI values are predominantly positive, indicating that B cell migration is biased by the CXCL13 gradient, whereas analysis of the cells migrating on uniform CXCL13 in the source chamber (n = 63, total number of cells = 493) yielded a mean FMI that is indistinguishable from zero; however, the average FMI values do not correlate well with absolute gradient steepness (Fig. 4A). We reasoned that the tactic fidelity also depends on the midpoint density of chemokine (measured in relative terms as intensity units, IU, based on the antibody staining). Accordingly, we found that the gradients with the greatest steepness values also had the highest midpoint intensities, which might explain the lower FMI values in those experiments; considering only those gradients with midpoint intensities below 35 IU (40% of the data), a reasonably positive correlation between FMI and gradient steepness (R = 0.43) was recovered (Fig. 4A, solid line). The distribution of FMI values suggests that CXCL13 gradients with moderate steepness (i.e., without excessive chemokine density) are optimal. We then set gradient steepness cut-offs to group the experiments into nearly equal-sized shallow (n = 20, total cell number = 222), moderate (n = 24, total number of cells = 268), and steep (n = 21, total number of cells = 259) subsets. Consistent with our previous observation, the mean FMI values of the shallow, moderate, and steep gradient groups are all significantly positive and higher than the random migration control group, indicating biased migration elicited by the chemokine gradients (Fig. 4B). To further analyze how properties of the gradient affect tactic fidelity, we tested mathematical models quantifying directed migration responses. To parse the competing effects of gradient steepness and midpoint density of CXCL13, we invoke the definition of the tactic coefficient, χ, which relates the directed migration response to the gradient steepness (33). For each experiment, χ is calculated as the adjusted mean FMI value (with an offset constant of 0.0335 added based on the line fit shown in Fig. 4A, which has a y-intercept of −0.0335) divided by the gradient steepness (Fig. 4C). We explored three models of how χ varies with chemokine density, C: 1) absolute sensing based on receptor occupancy, with saturation of the ligand-receptor interaction (22, 24, 34), 2) relative sensing, in which the response depends on the fractional difference in chemokine density across the cell (35), and 3) mixed sensing, in which both absolute and relative sensing contribute to overall fidelity (Fig. 4C). For the experiments with mean FMI values above the 95% confidence interval for random migration the (n = 56), all three of the phenomenological models fit the dependence of χ on C reasonably well (Fig. 4D), with similar sums of squared deviations (Model 1: 9.22; Model 2: 9.21; Model 3: 8.88); although Model 3 yielded the best fit, it requires 2 fitting parameters compared with 1 for Model 2. In the absolute sensing with saturation model (Model 1), the estimated difference in receptor occupancy across a typical cell body length (10 μm) is on the order of 1% of the cell surface receptors (Supplemental Fig. S2A). Considering instead the relative sensing model (Model 2), the relative difference in chemokine density (corresponding to receptor occupancy if there were negligible saturation) is in the range of 1.5–5.5% (Fig. S2B). Based on this analysis, we conclude that there are multiple interpretations that plausibly explain the dependence of B-cell tactic fidelity on CXCL13 density.
Fig. 4. Dependence of B-cell tactic bias on CXCL13 gradient steepness and midpoint surface density.
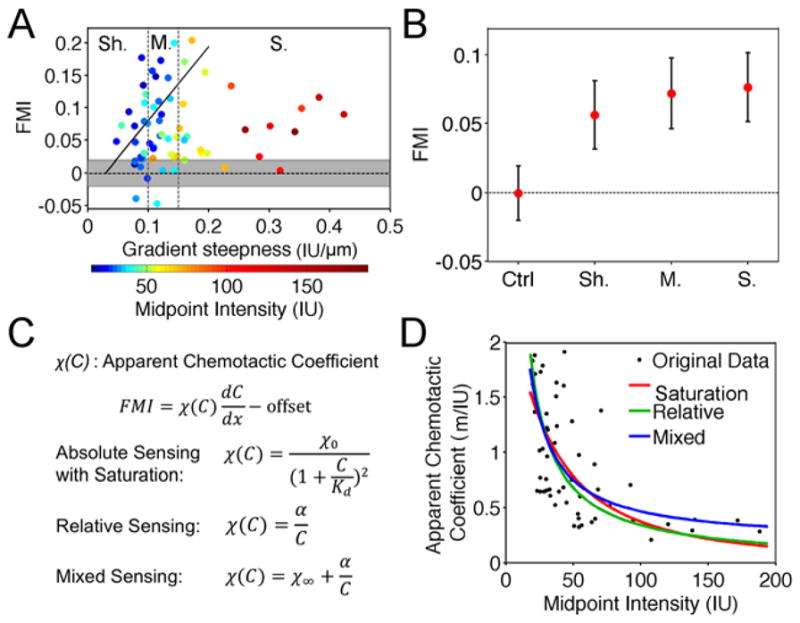
a) Mean FMI values versus gradient slope for all haptotaxis experiments (n = 65, total number of cells = 749). The corresponding midpoint intensities are indicated by the color map. The trend line is the best linear fit to the data for gradients with midpoint intensities below 35 IU. The shaded area is the 95% confidence interval for the random migration control experiments (n = 63, total number of cells = 493). Sh.: shallow (n = 20, total number of cells = 222); M.: moderate (n = 24, total number of cells = 268); S.: steep (n = 21, total number of cells = 259). b) Mean of the mean FMI values for each data subset: random control (Ctrl), shallow gradient (Sh.), moderate gradient (M), and steep gradient (S.). Error bars: 95% confidence intervals. c) Specification of 3 different gradient sensing models. d) Apparent chemotactic coefficient plotted versus the gradient midpoint intensity for each experiment (see text for details).
Optimal CXCL13 gradients bias the directional persistence of B-cell migration
The analysis described in the previous section suggests that moderately steep CXCL13 gradients tend to elicit stronger haptotactic responses. We tested this further by analyzing the directional persistence of cells in relation to their orientation with the gradient. To ensure unbiased analysis of directional persistence, we excluded cell tracks shorter than 3 minutes. To assess the extent to which the initial movement direction affects persistence, we further grouped the individual cell tracks based on the average direction of movement during the first 30 seconds. The corresponding normalized distributions of angular movement direction at each time point are shown as heat maps. The results suggest that B cells tend to maintain their direction of migration regardless of their initial orientation, though the persistence of up-gradient movement is stronger for cells in the shallow and moderate gradient subsets (Fig. 5A, D, G). To quantify this tendency, we calculated the autocorrelation coefficient of the cell movement vector for each time interval, as a function of increasing duration, and we binned the values by the direction of cell movement at the start of the interval. We reasoned that a bias in persistence would manifest as a slower decay of the autocorrelation coefficient with time. As expected, the moderate gradient subset yielded a slower decay of the autocorrelation coefficient for migration vectors that are better aligned with the gradient, indicating a bias in directionality (Fig. 5E&F). The trend is less prominent for the cells migrating on shallow gradients (Fig. 5B&C) or on steep gradients with high midpoint density of CXCL13 (Fig. 5H&I).
Fig. 5. Optimal CXCL13 gradients foster a bias in directional persistence.

a) Normalized distribution of the centroid movement angles at each time point relative to the beginning of the track for cells that initiated migration away from (top, n = 129) and towards (bottom, n = 79) the gradient, including cells from the shallow gradient subset with tracks longer than 3 min. b) Mean autocorrelation coefficient of track segments binned by the angle of movement at the start of the segment and by the time interval. The bin size is 45°. c) Autocorrelation coefficient as a function of time interval for the indicated angle bins from b (mean ± 95% confidence interval). d–f) Same as a–c except for the moderate gradient group: n = 147 cells initially moving away from the gradient, n = 102 towards. g–i) Same as a–c except for the steep gradient group: n = 155 cells initially moving away from the gradient, n = 83 towards.
DISCUSSION
Despite the broadly appreciated significance of B-lymphocyte migration in the actuation of adaptive immunity and the role of the CXC-family chemokines in directing B-cell trafficking, quantitative studies focusing on details of how B cells move and respond to chemokine gradients have lagged. B cells migrating on CXCL13 and ICAM-1 showed the characteristic amoeboid morphology and fast movement (average translocation speed ≈ 14 μm/min), consistent with previous studies (10, 12); however, the dilation/shrinking dynamics reported here is a new insight revealed using TIRF microscopy. This is distinct from the leading-edge dynamics reported for other fast-moving amoeboid cells, such as Dictyostelium discoideum and T lymphocytes (29, 36–38). Our analysis showed independent dilation/shrinking waves on the two sides of the cell front. Our conclusion is that the balance between dilation/shrinking at the edges determines cell migration directionality, which stands in contrast to the bifurcation of protrusions that characterizes migration of primary T lymphocytes on CXCL12/ICAM-1 (29); the leading edge dynamics in B cells is more reminiscent of the instabilities associated with turning of fish keratocytes (39).
Since the murine T and B lymphocyte populations we have studied were isolated from the same source, it would be interesting to elucidate the reasons for this dichotomy of phenotype. One possibility is that there are unique signaling pathways at play, such as those mediated by interleukin-2-inducible T-cell kinase in T cells (40, 41) and by Bruton’s tyrosine kinase in B cells (42). The need to activate the B cells ex vivo certainly introduces major differences in cell physiology, as BCR signaling probably interacts with both the chemokine and LFA-1 signaling networks through activation of protein kinases such as Syk and ZAP70 (43).
Quantification of B-cell haptotaxis revealed diverse responses, with a substantial number of B cells that were apparently insensitive to the chemokine gradient. Although some of this variability was attributed to differences in gradient steepness, heterogeneity in the cell population is likely another cause. Indeed, a mixed population of different B-cell subtypes is to be expected. Flow cytometry results (Fig. S3) confirmed the variable expression of CXCR5, consistent with the different types of B cells found in spleen (1, 44, 45). Another intriguing consideration is that higher chemokine doses can elicit chemorepulsion rather than chemotaxis of individual B cells (46). Such chemorepulsive responses have also been reported for neutrophils and T cells (47–49). Evidence suggests that cell-cell interactions might promote collective chemotaxis of B cells in a chemokine gradient (46).
Although we did not find any evidence of chemorepulsion on our gradients, we observed that tactic fidelity was reduced for gradients with higher midpoint density of chemokine. Two opposing explanations for this dependence were found to be equally plausible. On the one hand, gradient sensing might depend on the absolute difference in receptor occupancy across the cell, which is saturable. With saturation of receptor binding at high chemokine density, steeper gradients can yield shallower gradients in receptor ligation, as assumed in previous analyses of leukocyte gradient sensing (22, 24, 34). In the context of this model, we estimate that the absolute difference in occupancy is ~ 1% of the total cell-surface density of receptor. Alternatively, gradient sensing might depend on the relative difference or ratio of receptor occupancy across the cell, consistent with adaptation of the signaling circuit as proposed for other chemotactic cells (35, 50, 51). In the context of this alternative model, in which receptor saturation does not need to be invoked to explain the data, B-cell taxis is sensitive to relative gradients in the range of 2–5% across the cell.
The modest bias of persistent but otherwise random walk behavior might reflect a beneficial search strategy employed by B cells in secondary lymphoid tissues (52, 53). Conceptual and theoretical models of the search strategies employed by lymphocytes in vivo have been widely considered (54–57). A central concept is the balance between persistent migration and local ‘diffusive’ search (58). It is generally accepted that purely Brownian movement is suboptimal. Periods of high persistence (i.e. Lévy walks or a two-state model) improves the efficacy of the search, and chemokines provide a spatial cue that biases the cell movement towards the target (56, 58, 59). However, if the bias were too strong, erroneous decisions due to noisy sensing (60) might prove costly, while more random migration would allow B cells to prioritize among multiple sources of attraction, as expected in vivo.
MATERIALS AND METHODS
Isolation, activation, and culture of primary B cells
Mostly naïve B lymphocytes were isolated from C57BL/6 mouse spleens, kindly provided by the laboratory of Garnett Kelsoe (Duke University Medical Center). Mouse spleens were cut in halves and ground with frost slides in culture medium. The cell mixture was filtered through a 130 μm mesh, and B cells were isolated following the standard protocol of the Dynabeads® Mouse CD43 (Untouched™ B Cells) kit (Invitrogen). In brief, the cell mixture was resuspended to 5×107 cells/ml and mixed with 125 μl pre-washed Dynabeads for every 1 ml of the mixture. The cells and beads were incubated at room temperature with gentle tilting and rotation for 20 minutes prior to magnetic selection. The unbound B cells in the supernatant were collected and resuspended in B-cell culture medium [RPMI1640 supplemented with 25 mM HEPES, 10% fetal bovine serum, 1% sodium pyruvate, 1% non-essential amino acids, 0.1% 2-Mercaptoethanol, and 1% penicillin/streptomycin, all from Invitrogen] at 2×106 cells/ml. B cells were then transferred to a 12-well cell culture plate, activated by stimulation with 20 μg/ml anti-mouse IgM (Jackson ImmunoResearch) and 2 μg/ml anti-CD40 (BD Biosciences) for B-cell activation, and maintained at 37°C, 5% CO2 without changing the medium. The activation step is necessary for optimizing B-cell mobility as shown previously (61). The cells were collected for tests 24 h, 48 h, and 72 h after isolation.
TIRF microscopy
Glass-bottom dishes (MatTek) were coated first with 10 μg/ml Protein A (Invitrogen) and 5 μg/ml recombinant mouse CXCL13/BLC/BCA-1 (R&D Systems) in PBS at room temperature for 2 h. The surfaces were washed once with 10 mg/ml bovine serum albumin (BSA, fatty-acid free, Sigma) in PBS. Then, 10 μg/ml mouse ICAM-1/CD54 Fc chimera (R&D Systems) was added and incubated at room temperature for 2 h. The surfaces were washed once and blocked with 10 mg/ml BSA in PBS at 4°C overnight. The surfaces were washed with warm migration medium (phenol red-free RPMI1640, 10 mg/ml BSA) before adding cells. B cells were labeled with Vybrant® DiO (Invitrogen), following the manufacturer’s protocol. In brief, B cells were resuspended in warm migration medium at 106 cells/ml and mixed with DiO solution in the ratio of 5 μl DiO to 1 ml cell solution. The cells were incubated at 37°C for 2 minutes, pelleted, and resuspended in migration medium. The cells rested for 10 minutes at 37°C prior to seeding. Approximately 2.5×105 cells were seeded onto the migration surface and allowed to adhere for 5 minutes at 37°C. The cells were imaged using a prism-based total internal reflection fluorescence (TIRF) microscope (62, 63) at 37°C in a humidified chamber. Images were acquired at a rate of 20 frames/min with a 40X, 0.8 NA Achroplan water-dipping objective (Carl Zeiss), ORCA-ER cooled charge-coupled device (Hamamatsu Photonics), and MetaMorph software (Universal Imaging).
Haptotaxis assay
The microfluidic device master plate, using a design described previously (26, 32), was fabricated on silicon by UV crosslinking of SU-8 substrate (MicroChem). A mixture of Polydimethylsiloxane (PDMS) substrate and crosslinking agent (Sylgard® 184 Silicone Elastomer Kit, Dow Corning; mixed at a ratio of 10:1) is poured onto the master plate and cured at 95°C for 1 h. The PDMS devices are detached, cut out, and cleaned, and the outlet channels are punched using flat-tip needles. The device and a glass-bottom dish is plasma-treated and attached together to make the chamber. To establish chemokine gradients, a solution of 1, 5, or 10 μg/ml CXCL13 in PBS, supplemented with 1:1000 Texas Red®-dextran, 10,000 MW (Invitrogen) to visualize the gradient during the process, was injected into the source chamber and incubated for 15 min at 37°C. The chambers were flushed with PBS, then 10 μg/ml Protein A in PBS was added into all chambers and incubated for 15 min at 37°C. The chambers were flushed with PBS again, followed by the incubation with 10 μg/ml mouse ICAM-1 in PBS for 45 min at 37°C. Approximately 5×105 B cells were resuspended in 100 μl pre-warmed migration medium (RPMI1640 supplemented with 10 mg/ml BSA). The chambers were flushed with warm migration medium, and finally the cell suspension was injected into the chambers. 4 ml migration medium was added into the glass-bottom dish, immersing the device. The cells were allowed to adhere at 37°C for 5min. The dish was imaged under bright field illumination at 37°C in a humidified chamber. Images were acquired at a rate of 6 frames/min as described in the previous section.
Quantification of chemokine gradients
After each experiment, the device was washed once with PBS. The surface was then incubated with biotinylated anti-mCXCL13/BLC/BCA-1 antibody (R&D Systems), diluted 1:200 in PBS at 37°C for 30 min. The chamber was washed with PBS and then incubated in streptavidin-Alexa Fluor® 488 conjugate (Life Technologies), diluted 1:200 in PBS at 37°C for 30 min. The chambers were washed twice with PBS before imaging. The images were acquired using an Axio Observer Z1 microscope with 10X, 0.3 NA Achroplan objective (Carl Zeiss). The same acquisition settings were used for all experiments. Image analysis was performed using ImageJ. The background-subtracted fluorescence was measured, and the linear portion of the fluorescence profile was fit to a line. The slope of the line and its midpoint intensity, taken as the predicted intensity located 300 μm from the cell chamber border on the source end, were determined.
Computational image analysis
Morphodynamic analyses were performed using MATLAB software (MathWorks). Codes for identification and spatiotemporal mapping of protruded/retracted areas and extended morphological structures were described previously (28). The dilation/shrinking maps were constructed by computationally shifting and overlapping the cell centroids for each frame. For all morphometric maps, the angular position of each protruded/retracted pixel was binned (rounded to the nearest whole angle in degrees, relative to the vector pointed from the cell centroid in the negative x-direction). Protrusion/retraction velocity was calculated as the net change in the number of protruded/retracted pixels along the indicated angle (multiplied by the pixel size, 0.25 μm), divided by the time interval. For analysis of haptotaxis, cell centroid tracks were obtained using the Manual Tracking Plugin in ImageJ. The forward migration index (FMI), often referred to as the chemotactic index, is calculated for each centroid track; it is defined as the overall translocation of the centroid in the direction of the gradient divided by the total path length. Defined mathematically, if the segments of the centroid path were expressed as an array of vectors V (which could be cast as velocities, since the centroid position was measured at equal time intervals), with the x-direction oriented up-gradient, FMI would be calculated as follows.
The summations are over all segments in the track.
Waiting-time distributions
The waiting time and lifetime of each phenotypic event were manually documented with a temporal resolution of 0.25 minute. To compare the waiting time distribution to that of a Poisson process, comparison to an exponential distribution was performed as previously described (29). All fits were obtained using the curve fitting toolbox of MATLAB.
Supplementary Material
INSIGHT STATEMENT.
In the adaptive immune response to an infection, cognate B cells are activated and produce antibodies to eradicate the pathogen and provide immunological memory. Central to this process is the directed migration, or homing, of B cells within secondary lymphoid organs, where intercellular signaling occurs. The chemokine CXCL13 is secreted and deposited in those tissues to attract B cells. In this paper, we combine microscopy, computational image analysis, and microfluidics to study migration of primary B cells and how that process is biased by CXCL13 gradients. The integration of these methods offers insights regarding the mechanics and relevant timescales of B-cell crawling and the conditions that yield optimal bias of B-cell migration. Finally, we speculate on the balance of biased versus random migration as a search strategy for B cells in vivo.
Acknowledgments
This work was supported under contract # HHSN272201000053C from the National Institute of Allergy and Infectious Diseases. Partial support under National Institutes of Health grant R01-GM110155 to JEB and JMH is also acknowledged. We are grateful to the laboratory of Dr. Garnett Kelsoe (Duke University Medical Center) for providing primary B cells and expertise in B-cell culture.
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58–71. doi: 10.1034/j.1600-065x.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 3.Stachowiak AN, Wang Y, Huang YC, Irvine DJ. Homeostatic lymphoid chemokines synergize with adhesion ligands to trigger T and B lymphocyte chemokinesis. J Immunol. 2006;177:2340–2348. doi: 10.4049/jimmunol.177.4.2340. [DOI] [PubMed] [Google Scholar]
- 4.Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 5.Gatto D, Brink R. The germinal center reaction. J Allergy Clin Immunol. 2010;126:898–907. doi: 10.1016/j.jaci.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Lo CG, Lu TT, Cyster JG. Integrin-dependence of lymphocyte entry into the splenic white pulp. J Exp Med. 2003;197:353–361. doi: 10.1084/jem.20021569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goval JJ, Greimers R, Boniver J, de Leval L. Germinal center dendritic cells express more ICAM-1 than extrafollicular dendritic cells and ICAM-1/LFA-1 interactions are involved in the capacity of dendritic cells to induce PBMCs proliferation. J Histochem Cytochem. 2006;54:75–84. doi: 10.1369/jhc.5A6740.2005. [DOI] [PubMed] [Google Scholar]
- 8.Hogg N, Patzak I, Willenbrock F. The insider’s guide to leukocyte integrin signalling and function. Nat Rev Immunol. 2011;11:416–426. doi: 10.1038/nri2986. [DOI] [PubMed] [Google Scholar]
- 9.Ramesh N, Geha R. Recent advances in the biology of WASP and WIP. Immunol Res. 2009;44:99–111. doi: 10.1007/s12026-008-8086-1. [DOI] [PubMed] [Google Scholar]
- 10.Banon-Rodriguez I, Saez de Guinoa J, Bernardini A, Ragazzini C, Fernandez E, Carrasco YR, Jones GE, Wandosell F, Anton IM. WIP regulates persistence of cell migration and ruffle formation in both mesenchymal and amoeboid modes of motility. PloS One. 2013;8:e70364. doi: 10.1371/journal.pone.0070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comrie WA, Babich A, Burkhardt JK. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J Cell Biol. 2015;208:475–491. doi: 10.1083/jcb.201406121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sáez de Guinoa J, Barrio L, Mellado M, Carrasco YR. CXCL13/CXCR5 signaling enhances BCR-triggered B-cell activation by shaping cell dynamics. Blood. 2011;118:1560–1569. doi: 10.1182/blood-2011-01-332106. [DOI] [PubMed] [Google Scholar]
- 13.Yuseff MI, Pierobon P, Reversat A, Lennon-Duménil AM. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat Rev Immunol. 2013;13:475–486. doi: 10.1038/nri3469. [DOI] [PubMed] [Google Scholar]
- 14.Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, Ngo VN. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. 2000;176:181–193. doi: 10.1034/j.1600-065x.2000.00618.x. [DOI] [PubMed] [Google Scholar]
- 15.Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J Biol Chem. 2007;282:10018–10027. doi: 10.1074/jbc.M608796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–744. doi: 10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schumann K, Lämmermann T, Bruckner M, Legler DF, Polleux J, Spatz JP, Schuler G, Förster R, Lutz MB, Sorokin L, Sixt M. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity. 2010;32:703–713. doi: 10.1016/j.immuni.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 18.Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Jeon N, Baskaran H, Dertinger SKW, Whitesides GM, Van de Water L, Toner M. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat Biotechnol. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 20.Abhyankar VV, Lokuta MA, Huttenlocher A, Beebe DJ. Characterization of a membrane-based gradient generator for use in cell-signaling studies. Lab Chip. 2006;6:389–393. doi: 10.1039/b514133h. [DOI] [PubMed] [Google Scholar]
- 21.Lin F, Butcher EC. T cell chemotaxis in a simple microfluidic device. Lab Chip. 2006;6:1462–1469. doi: 10.1039/b607071j. [DOI] [PubMed] [Google Scholar]
- 22.Herzmark P, Campbell K, Wang F, Wong K, El-Samad H, Groisman A, Bourne HR. Bound attractant at the leading vs the trailing edge determines chemotactic prowess. Proc Natl Acad Sci U S A. 2007;104:13349–13354. doi: 10.1073/pnas.0705889104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim D, Lokuta MA, Huttenlocher A, Beebe DJ. Selective and tunable gradient device for cell culture and chemotaxis study. Lab Chip. 2009;9:1797–1800. doi: 10.1039/b901613a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haessler U, Pisano M, Wu M, Swartz MA. Dendritic cell chemotaxis in 3D under defined chemokine gradients reveals differential response to ligands CCL21 and CCL19. Proc Natl Acad Sci U S A. 2011;108:5614–5619. doi: 10.1073/pnas.1014920108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung HH, Chan CK, Khire TS, Marsh GA, Clark A, Waugh RE, McGrath JL. Highly permeable silicon membranes for shear free chemotaxis and rapid cell labeling. Lab Chip. 2014;14:2456–2468. doi: 10.1039/c4lc00326h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asokan SB, Johnson HE, Rahman A, King SJ, Rotty JD, Lebedeva IP, Haugh JM, Bear JE. Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCγ/PKCα pathway. Dev Cell. 2014;31:747–760. doi: 10.1016/j.devcel.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welf ES, Ahmed S, Johnson HE, Melvin AT, Haugh JM. Migrating fibroblasts reorient directionality by a metastable, PI3K-dependent mechanism. J Cell Biol. 2012;197:105–114. doi: 10.1083/jcb.201108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Welf ES, Haugh JM. Linking morphodynamics and directional persistence of T lymphocyte migration. J R Soc Interface. 2015:12. doi: 10.1098/rsif.2014.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Machacek M, Danuser G. Morphodynamic profiling of protrusion phenotypes. Biophys J. 2006;90:1439–1452. doi: 10.1529/biophysj.105.070383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devreotes P, Janetopoulos C. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J Biol Chem. 2003;278:20445–20448. doi: 10.1074/jbc.R300010200. [DOI] [PubMed] [Google Scholar]
- 32.Johnson HE, King SJ, Asokan SB, Rotty JD, Bear JE, Haugh JM. F-actin bundles direct the initiation and orientation of lamellipodia through adhesion-based signaling. J Cell Biol. 2015;208:443–455. doi: 10.1083/jcb.201406102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lauffenburger DA, Linderman JJ. Receptors: models for binding, trafficking, and signaling. New York: Oxford University Press; 1993. [Google Scholar]
- 34.Tranquillo RT, Zigmond SH, Lauffenburger DA. Measurement of the chemotaxis coefficient for human neutrophils in the under-agarose migration assay. Cell Motil Cytoskeleton. 1988;11:1–15. doi: 10.1002/cm.970110102. [DOI] [PubMed] [Google Scholar]
- 35.Schneider IC, Haugh JM. Mechanisms of gradient sensing and chemotaxis: conserved pathways, diverse regulation. Cell Cycle. 2006;5:1130–1134. doi: 10.4161/cc.5.11.2770. [DOI] [PubMed] [Google Scholar]
- 36.Andrew N, Insall RH. Chemotaxis in shallow gradients is mediated independently of PtdIns 3-kinase by biased choices between random protrusions. Nat Cell Biol. 2007;9:193–200. doi: 10.1038/ncb1536. [DOI] [PubMed] [Google Scholar]
- 37.Bosgraaf L, Van Haastert PJM. The ordered extension of pseudopodia by amoeboid cells in the absence of external cues. PloS One. 2009;4:e5253. doi: 10.1371/journal.pone.0005253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Driscoll MK, McCann C, Kopace R, Homan T, Fourkas JT, Parent C, Losert W. Cell shape dynamics: from waves to migration. PLoS Comput Biol. 2012;8:e1002392. doi: 10.1371/journal.pcbi.1002392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lacayo CI, Pincus Z, VanDuijn MM, Wilson CA, Fletcher DA, Gertler FB, Mogilner A, Theriot JA. Emergence of large-scale cell morphology and movement from local actin filament growth dynamics. PLoS Biol. 2007;5:e233. doi: 10.1371/journal.pbio.0050233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain N, Miu B, Jiang J, McKinstry KK, Prince A, Swain SL, Greiner DL, Thomas CJ, Sanderson MJ, Berg LJ, Kang J. CD28 and ITK signals regulate autoreactive T cell trafficking. Nat Med. 2013;19:1632–1637. doi: 10.1038/nm.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannan AK, Kim DG, August A, Bynoe MS. Itk signals promote neuroinflammation by regulating CD4+ T-cell activation and trafficking. J Neurosci. 2015;35:221–233. doi: 10.1523/JNEUROSCI.1957-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Gorter DJJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, Pals ST, Spaargaren M. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26:93–104. doi: 10.1016/j.immuni.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 43.Klasen C, Ohl K, Sternkopf M, Shachar I, Schmitz C, Heussen N, Hobeika E, Levit-Zerdoun E, Tenbrock K, Reth M, Bernhagen J, El Bounkari O. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. J Immunol. 2014;192:5273–5284. doi: 10.4049/jimmunol.1302209. [DOI] [PubMed] [Google Scholar]
- 44.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 45.Brandes M, Legler DF, Spoerri B, Schaerli P, Moser B. Activation-dependent modulation of B lymphocyte migration to chemokines. Int Immunol. 2000;12:1285–1292. doi: 10.1093/intimm/12.9.1285. [DOI] [PubMed] [Google Scholar]
- 46.Malet-Engra G, Yu W, Oldani A, Rey-Barroso J, Gov NS, Scita G, Dupré L. Collective cell motility promotes chemotactic prowess and resistance to chemorepulsion. Curr Biol CB. 2015;25:242–250. doi: 10.1016/j.cub.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 47.Poznansky MC, I, Olszak T, Foxall R, Evans RH, Luster AD, Scadden DT. Active movement of T cells away from a chemokine. Nat Med. 2000;6:543–548. doi: 10.1038/75022. [DOI] [PubMed] [Google Scholar]
- 48.Tharp WG, Yadav R, Irimia D, Upadhyaya A, Samadani A, Hurtado O, Liu SY, Munisamy S, Brainard DM, Mahon MJ, Nourshargh S, van Oudenaarden A, Toner MG, Poznansky MC. Neutrophil chemorepulsion in defined interleukin-8 gradients in vitro and in vivo. J Leukoc Biol. 2006;79:539–554. doi: 10.1189/jlb.0905516. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, Xu J. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol. 2012;13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Artemenko Y, Lampert TJ, Devreotes PN. Moving towards a paradigm: common mechanisms of chemotactic signaling in Dictyostelium and mammalian leukocytes. Cell Mol Life Sci. 2014;71:3711–3747. doi: 10.1007/s00018-014-1638-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Byrne MB, Kimura Y, Kapoor A, He Y, Mattam KS, Hasan KM, Olson LN, Wang F, Kenis PJA, Rao CV. Oscillatory behavior of neutrophils under opposing chemoattractant gradients supports a winner-take-all mechanism. PloS One. 2014;9:e85726. doi: 10.1371/journal.pone.0085726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- 53.Cahalan MD, Parker I. Choreography of cell motility and interaction dynamics imaged by two-photon microscopy in lymphoid organs. Annu Rev Immunol. 2008;26:585–626. doi: 10.1146/annurev.immunol.24.021605.090620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chavali AK, Gianchandani EP, Tung KS, Lawrence MB, Peirce SM, Papin JA. Characterizing emergent properties of immunological systems with multi-cellular rule-based computational modeling. Trends Immunol. 2008;29:589–599. doi: 10.1016/j.it.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 55.Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD, Liu AJ, Hunter CA. Generalized Lévy walks and the role of chemokines in migration of effector CD8+ T cells. Nature. 2012;486:545–548. doi: 10.1038/nature11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tokarski C, Hummert S, Mech F, Figge MT, Germerodt S, Schroeter A, Schuster S. Agent-based modeling approach of immune defense against spores of opportunistic human pathogenic fungi. Front Microbiol. 2012;3:129. doi: 10.3389/fmicb.2012.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan C, Billard M, Ramirez SA, Schmidl H, Monson E, Kepler TB. A model for migratory B cell oscillations from receptor down-regulation induced by external chemokine fields. Bull Math Biol. 2013;75:185–205. doi: 10.1007/s11538-012-9799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li L, Nørrelykke SF, Cox EC. Persistent cell motion in the absence of external signals: a search strategy for eukaryotic cells. PloS One. 2008;3:e2093. doi: 10.1371/journal.pone.0002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Haastert PJM, Bosgraaf L. Food searching strategy of amoeboid cells by starvation induced run length extension. PloS One. 2009;4:e6814. doi: 10.1371/journal.pone.0006814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tranquillo RT, Lauffenburger DA, Zigmond SH. A stochastic model for leukocyte random motility and chemotaxis based on receptor binding fluctuations. J Cell Biol. 1988;106:303–309. doi: 10.1083/jcb.106.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vernekar VN, Wallace CS, Wu M, Chao JT, O’Connor SK, Raleigh A, Liu X, Haugh JM, Reichert WM. Bi-ligand surfaces with oriented and patterned protein for real-time tracking of cell migration. Colloids Surf B Biointerfaces. 2014;123:225–235. doi: 10.1016/j.colsurfb.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schneider IC, Haugh JM. Quantitative elucidation of a distinct spatial gradient-sensing mechanism in fibroblasts. J Cell Biol. 2005;171:883–892. doi: 10.1083/jcb.200509028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson HE, Haugh JM. Quantitative analysis of phosphoinositide 3-kinase (PI3K) signaling using live-cell total internal reflection fluorescence (TIRF) microscopy. Curr Protoc Cell Biol. 2013;61:14.14.1–14.14.24. doi: 10.1002/0471143030.cb1414s61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.