

Abstract
We used deep sequencing technology to identify transcriptional adaptation of the euryhaline unicellular cyanobacterium Synechococcus sp. PCC 7002 and the marine facultative aerobe Shewanella putrefaciens W3-18-1 to growth in a co-culture and infer the effect of carbon flux distributions on photoautotroph–heterotroph interactions. The overall transcriptome response of both organisms to co-cultivation was shaped by their respective physiologies and growth constraints. Carbon limitation resulted in the expansion of metabolic capacities, which was manifested through the transcriptional upregulation of transport and catabolic pathways. Although growth coupling occurred via lactate oxidation or secretion of photosynthetically fixed carbon, there was evidence of specific metabolic interactions between the two organisms. These hypothesized interactions were inferred from the excretion of specific amino acids (for example, alanine and methionine) by the cyanobacterium, which correlated with the downregulation of the corresponding biosynthetic machinery in Shewanella W3-18-1. In addition, the broad and consistent decrease of mRNA levels for many Fe-regulated Synechococcus 7002 genes during co-cultivation may indicate increased Fe availability as well as more facile and energy-efficient mechanisms for Fe acquisition by the cyanobacterium. Furthermore, evidence pointed at potentially novel interactions between oxygenic photoautotrophs and heterotrophs related to the oxidative stress response as transcriptional patterns suggested that Synechococcus 7002 rather than Shewanella W3-18-1 provided scavenging functions for reactive oxygen species under co-culture conditions. This study provides an initial insight into the complexity of photoautotrophic–heterotrophic interactions and brings new perspectives of their role in the robustness and stability of the association.
Keywords: cyanobacterium, heterotroph, co-culture, microbial interactions, transcriptome, SOLiD sequencing
Introduction
Experimental evidence suggests the existence of strong positive and negative interactions between photoautotrophic and heterotrophic microorganisms (Caldwell and Caldwell, 1978; Cole, 1982; Carpenter and Foster, 2002; Amin et al., 2012). In aquatic environments, an important class of interactions is based on cross-feeding and metabolite exchange, whereby photosynthetically fixed dissolved organic carbon can elicit chemotactic responses (Paerl and Gallucci, 1985; Seymour et al., 2010) that lead to the development of spatial associations (Paerl and Pinckney, 1996; Bertilsson et al., 2007). The spectrum of behavioral responses varies greatly, as the origin of excreted material ranges from targeted secretion of photosynthetic intermediates (for example, glycolate, osmolytes and fatty acids) and extracellular polymeric substance (Seymour et al., 2010; Bruckner et al., 2011), to the products of cell lysis that can include sugars, proteins, lipids and nucleic acids (Grossart, 1999; Stevenson and Waterbury, 2006; Shen et al., 2011). In exchange, heterotrophic bacteria are thought to provide essential micronutrients, such as vitamins, amino acids and bioavailable trace metals (Amin et al., 2009; Hayashi et al., 2011; Kazamia et al., 2012; Xie et al., 2013), necessary to maintain high photosynthetic productivity.
Furthermore, tight associations, which involve complementation of physiological or biochemical functions, may undergo evolutionary selection emphasizing optimal metabolic performance and enhanced productivity (Overmann, 2006; Stevenson and Waterbury, 2006). For example, stimulation of cyanobacterial N2 fixation in association of Anabaena sp. with heterotrophic bacteria is well known and attributed to the respiratory O2 removal during periods of high ambient oxygen concentrations (Paerl and Kellar, 1978). Similarly, co-cultivation of marine heterotrophs with Prochlorococcus ecotypes has been shown to enhance photosynthetic growth (Morris et al., 2008; Sher et al., 2011). This positive effect was attributed to the decrease of oxidative stress by heterotrophs through catalase-mediated scavenging of peroxide and other reactive oxygen species (ROS) (Morris et al., 2008). Such interactions have ecosystem-scale consequences because ROS may directly affect nutrient bioavailability, photosynthetic productivity and carbon fluxes (Amin et al., 2009).
As we move toward a system-level understanding of community organization and function (Zengler and Palsson, 2012), the behavioral complexity increases with the number of interacting organisms (Wintermute and Silver, 2010). To that end, application of genome-scale approaches to dissect subcellular pathways and regulatory networks involved in governing interspecies interactions is dependent, at least initially, on the availability of model biological systems and genomic information (Tai et al., 2009). Here, we investigated the growth of the euryhaline unicellular cyanobacterium Synechococcus sp. PCC 7002 (hereafter Synechococcus 7002) in a co-culture with a marine facultative aerobe Shewanella putrefaciens W3-18-1 (hereafter Shewanella W3-18-1) under different trophic conditions. Although recognizing the opportunistic nature of interactions between these two strains, there is ample evidence that certain Shewanella species live in association with photoautotrophic prokaryotes (Simidu et al., 1990; Bowman et al., 1997; Salomon et al., 2003). Through the application of next-generation sequencing technology in conjunction with controlled cultivation and metabolomic profiling, we were able to delineate specific transcriptional responses of each organism to co-cultivation. Building upon previous genome-scale investigations of phototrophic consortia (Tai et al., 2009; Wenter et al., 2010), the functional annotation of the differentially regulated transcripts leads to the development of a conceptual model of interactions between cyanobacteria and heterotrophs as a function of carbon sources and flux directions.
Materials and methods
Strains and growth conditions
Axenic batch cultures of Synechococcus 7002 were routinely grown in modified basal medium A containing: 18 g l–1 NaCl, 0.6 g l–1 KCl, 0.9 g l–1 NH4Cl, 5.0 g l–1 MgSO4 7 H2O, 50 mg l–1 KH2PO4, 266 mg l–1 CaCl2, 30 mg l–1 Na2EDTA 2 H2O, 3.89 mg l–1 FeCl3 6 H2O, 1 g l–1 Tris HCl (pH 8.2), 34.26 mg l–1 H3BO3, 4.32 mg l–1 MnCl2 4 H2O, 0.315 mg l–1 ZnCl2, 0.03 mg l–1 MoO3, 12.15 μg l–1 CoCl2 6 H2O, 3 μg l–1 CuSO4 5 H2O and 4 μg l–1 vitamin B12 (Stevens and Porter, 1980; Ludwig and Bryant, 2011). The cultivation was carried out at room temperature in 1-L Roux bottles sparged with 2% CO2-enriched air under continuous 50 μmol m−2 s−1 illumination. Axenic Shewanella W3-18-1 cultures were maintained at 30 °C in tryptic soy broth or M1 defined medium (Pinchuk et al., 2008) amended with 20 mM D,L-Na lactate and 10 ml each of 10X Wolfe's vitamin and 10X mineral solutions (Kieft et al., 1999).
Continuous cultivation experiments were carried out in a custom-made photobioreactor built using the Bioflo 3000/310 fermenter platform (New Brunswick Scientific, New Brunswick, NJ, USA). The system consisted of a 7.5-l borosilicate glass vessel (49.5 cm height, 13.4 cm inner diameter) and a cage-like light enclosure carrying eight 14-W Pentron 3500K tubular fluorescent lights (model FP14/835/ECO, Osram Sylvania, Danvers, MA, USA) mounted in pairs at each quadrant of the vessel. Irradiance was controlled using a custom dimmer integrated within the BioFlo 310 bioreactor controller module. A solar blanket was used to isolate the bioreactor from ambient light. Control over other cultivation parameters including temperature, agitation, pH and dissolved O2 tensions was achieved using built-in BioFlo 310 controls. Temperature was set at 30 °C, agitation at 150 r.p.m. and pH of 7.5 was maintained throughout the run by automatic bases or acid addition. Dissolved O2 tension, expressed as percent of O2 saturation, was continuously monitored with an InPro 6110/320 DO probe (Mettler-Toledo Inc., Toledo, OH, USA) and maintained at a desired value by automatically altering the proportion of O2 and air.
Chemostat cultivation
All growth studies, expression profiling and metabolomics measurements were conducted in axenic and mixed cultures grown under steady-state conditions. Chemostat cultivation was specifically chosen to provide a well-defined physiological state, which eliminates confounding variables that often complicate transcriptome analyses (Wu et al., 2004; Bull, 2010). In chemostats, Synechococcus 7002 and Shewanella W3-18-1 were grown in the basal medium A in the absence of Tris HCl using 8 mM NaHCO3 and 5 mM D,L-Na lactate, respectively, as the sole source of carbon. Co-culture chemostats were initially established by adding 0.5 l of lactate-grown Shewanella W3-18-1 cells to a 5.5-l chemostat culture of Synechococcus 7002 and a gradual transition from NaHCO3 to D,L-Na lactate as the sole source of carbon. Transition of the co-culture back to growth with NaHCO3 was carried out in a step-wise manner by gradually reducing lactate concentration in the feed to minimize cell lysis and wash-out. The steady state was inferred from stability of the following growth readouts: OD730 (⩽3% variation between measurements), pH and dissolved O2 concentration. Samples for all the analyses were only taken after at least five volume changes under steady-state conditions. The details of media delivery rate, biomass concentration and dissolved O2 tensions for each steady-state condition are specified in the text and in Table 1. The irradiance values for each steady state are reported as quanta incident to the center of the reactor. The measurements were carried out using LI193S underwater spherical quantum sensor (LI-COR, Lincoln, NE, USA) and determined to be radially isotropic. The steady-state chemostat conditions used for transcriptome analyses were determined to be light saturated and only carbon limited. Light saturation was assessed by the fact that biomass (OD730) did not respond to small increases or decreases in incident irradiance.
Table 1. Baseline cultivation conditions and physiological outputs of Synechococcus 7002 and Shewanella W3-18-1 grown axenically and in co-cultures.
| Organism(s) and chemostat conditiona | Carbon source | Dilution rate, h−1 | Irradiance, μmol photons m–2 s–1b | DOT, % of O2 saturation | OD730 | Biomass, AFDW, mg l–1 |
|---|---|---|---|---|---|---|
| Shewanella W3-18-1 carbon-limited chemostat | D,L-lactate, 5 mM | 0.075 | 1720 | 50.1 | 0.105 | 131 |
| Synechococcus 7002 carbon-limited chemostat | HCO3−, 8 mM | 0.075 | 1720 | 50.2 | 0.327 | 150 |
| Synechococcus 7002, Shewanella W3-18-1 carbon-limited chemostat | D,L-lactate, 5 mM | 0.075 | 1720 | 49.5 | 0.431 | 172 |
| Synechococcus 7002, Shewanella W3-18-1 carbon-limited chemostat | HCO3−, 8 mM | 0.075 | 1720 | 47.8 | 0.373 | 149 |
| Synechococcus 7002, Shewanella W3-18-1 light-limited chemostat | HCO3−, 8 mM | 0.075 | 640 | 48.2 | 0.289 | 116 |
Abbreviation: AFDW, ash-free dry weight.
The cells were determined to be light saturated and carbon limited when steady-state biomass concentration did not respond to any changes in incident irradiance. Conversely, under light-limited conditions, the biomass concentration changed as a function of irradiance.
The irradiance values for each steady state are reported as quanta incident to the center of the reactor.
Analytical procedures and metabolite detection
Nuclear magnetic resonance spectroscopy (NMR) analysis was used to quantitatively measure excreted organic metabolites in axenic and mixed chemostat cultures of Synechococcus 7002. Twenty-milliliter aliquots from each culture were passed through 0.2 μm membranes (EMD Millipore Corporation, Billerica, MA, USA) and spiked with 1/10 volume of a solution containing 5 mM 2,2-dimethyl-2-silapentane-5-sulfonate and 0.2% sodium azide (Sigma-Aldrich, St Louis, MO, USA) dissolved in 100% deuterium oxide (D2O, Cambridge Isotope Laboratories, Tewksbury, MA, USA). All samples were stored at 6 °C until data collection. NMR data were acquired on a Varian Direct Drive (VNMRS) 600 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with a Dell Precision T3500 Linux workstation (Dell, Plano, TX, USA) running VNMRJ 3.2 (Agilent Technologies). The spectrometer system was outfitted with a Varian triple resonance salt-tolerant cold probe with a cold carbon preamplifier. A Varian standard one-dimensional proton nuclear overhauser effect spectroscopy (NOESY) with presaturation (TNNOESY) was collected on each sample, using the following data collection protocol: nonselective 90° excitation pulse, a 100 ms mixing time, acquisition time of 4 s, a presaturation delay of 1.5 s, spectral width of 12 p.p.m. and temperature control set to 25 °C. Collected spectra were analyzed using Chenomx 7.6 software (Edmonton, Alberta, Canada), with quantifications based on spectral intensities relative to 0.5 mM 2,2-dimethyl-2-silapentane-5-sulfonate.
Growth stoichiometry and theoretical carbon balance
Maximum theoretical yields were calculated from stoichiometric equations formulated on a Cmol basis (see Supplementary Material Equations). Equations were derived for the oxidation of lactate and subsequent reduction of inorganic carbon (represented as CO2) and accounted for both biomass and extracellular metabolite production. Net theoretical yields were calculated from stoichiometric carbon balances.
Flow cytometry and imaging
The flow cytometry data were obtained using a BD Influx Fluorescence-Activated Cell Sorter (BD Biosciences, San Jose, CA, USA). Upon harvesting, the cells were immediately treated with 50 mM Na2EDTA (Sigma-Aldrich) and gently pipetted to disrupt large aggregates and then fixed with 2% paraformaldehyde. Using the 488-nm excitation from a Sapphire LP laser (Coherent Inc., Santa Clara, CA, USA) at 100 mW, samples were analyzed using a 70-μm nozzle. Optimization and calibration of the fluorescence-activated cell sorting was performed before each analysis using 3-μm Ultra Rainbow Fluorescent Particles (Spherotech, Lake Forest, IL, USA). The ratio of the two distinct populations of cells within a mixed microbial community were identified from 50 000 recorded cells using size and complexity gates using FCS Express (Los Angeles, CA, USA) flow cytometry software.
Microscopic images were acquired on a Zeiss LSM 710 Scanning Confocal Laser Microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany) equipped with a W Plan-Apochromatic 63x/1.0 M27 objective. Shewanella W3-18-1 cells were visualized by acridine orange (Invitrogen, Grand Island, NY, USA) at 490–577 nm. Synechococcus 7002 was visualized by phycocyaninauto fluorescence measured at 640 nm. Images were processed with Volocity (Perkin Elmer, Waltham, MA, USA) and used to obtain the cell size measurements made along the major and minor axis. Cell volume calculations for each organism were carried out using the equation for a prolate ellipsoid, 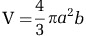 , where a is the diameter of the minor axis, and b is the diameter of the major axis. The relative biomass content of each organism in the co-culture was calculated using the equation,
, where a is the diameter of the minor axis, and b is the diameter of the major axis. The relative biomass content of each organism in the co-culture was calculated using the equation, 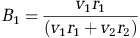 , where v1 and v2 are the cell volumes of each organism, r1 and r2 are the measured population ratios.
, where v1 and v2 are the cell volumes of each organism, r1 and r2 are the measured population ratios.
RNA isolation and sequencing
Cells for transcriptomic analysis were harvested from duplicate chemostat cultures by centrifugation at 7000 r.p.m. for 5 min at 4 °C, flash frozen in liquid nitrogen and stored at −80 °C. Total RNA extraction was carried out according to previously published protocols (Beliaev et al., 2005). The quality and integrity of the RNA was assessed on an Agilent 2100 Bioanalyzer and only samples with integrity numbers between 8 and 10 were selected for further analysis (Schroeder et al., 2006). Template complementary DNA was prepared using the Applied Biosystems SOLiD Total RNA-Seq Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocol. Sequencing was carried out using the SOLiD 5500XL protocol (Life Technologies). The 50-base sequence reads were mapped to the genomes of Synechococcus 7002 (GenBank #: NC_010475) and Shewanella W3-18-1 (GenBank #: NC_008750_1) using SOLiDTM LifeScope v. 2.5 software. The normalization of the RNA-seq data was carried out using the RPKM (reads per kilobase per million) calculation (Mortazavi et al., 2008). To allow comparisons between conditions, additional normalization relative to the total number of reads across different samples was performed.
Functional enrichment analysis
The transcription data were analyzed using pathway genome databases (PGDBs; http://pathways.mbl.edu/) developed for Synechococcus 7002 (SynechococcusPCC7002Cyc) and Shewanella W3-18-1 (Shew3181Cyc). These databases contain manually curated gene products, regulon predictions available in RegPrecise (Novichkov et al., 2012), and classification of gene products into BioCyc pathways and GO: Biological Process terms (Caspi et al., 2012), which include functions not covered by the BioCyc pathway schema. Previously developed software (Karp et al., 2010) was used to extract gene products, pathways and regulators for sets of differentially expressed genes. Calculations were done to identify enriched pathways, GO: Biological Processes and transcription factors in the data set using the Fisher's exact parent–child method, which was applied to avoid overrepresentation of parent terms (Grossmann et al., 2007).
Results and Discussion
Metabolic coupling leads to co-culture growth on either inorganic or organic carbon
Synechococcus 7002 is an oxygenic photoautotroph that cannot use any exogenous organic carbon compounds other than glycerol for growth under photoheterotrophic or dark respiratory conditions (Lambert and Stevens, 1986). It uses nitrate, ammonium or urea as a nitrogen source, and is auxotrophic for vitamin B12, which is required for methionine biosynthesis and photosynthetic activity at low temperatures (Batterton and Vanbaalen, 1971; Rippka et al., 1979). Conversely, Shewanella W3-18-1 (Nealson and Scott, 2003) is a facultative anaerobe that possesses a versatile heterotrophic metabolism and can utilize a wide array of carbon sources including various carboxylic acids, carbohydrates, amino acids and nucleic acids, a range of di-peptides as nitrogen sources, and DNA as a phosphorus source (Pinchuk et al., 2008; Rodionov et al., 2010; Rodrigues et al., 2011). Shewanella W3-18-1 also requires B12 for glycerol utilization but is able to circumvent the requirement for methionine biosynthesis by employing B12-independent methionine synthase.
To serve as a basis for comparison with interacting populations, both organisms were grown axenically in carbon-limited chemostats using 8 mM NaHCO3− (Synechococcus 7002) or 5 mM D,L-Na lactate (Shewanella W3-18-1) (Table 1). Under these conditions, Synechococcus 7002 accumulated 143 mg l–1 of ash-free dry weight (AFDW) biomass, whereas aerobic chemostat cultures of Shewanella W3-18-1 accumulated 131 mg l–1 AFDW. Co-culture growth was established upon addition of 0.5 l of lactate-grown Shewanella W3-18-1 cells to a 5.5-l continuous culture of Synechococcus 7002 and a gradual transition from HCO3− to lactate as the sole source of carbon. The co-culture grown with lactate was dependent on continuous illumination of the reactor vessel as no growth occurred in the dark when using 100% N2 as the sparging gas. In the light, there was positive selection for both organisms via the production of CO2 by Shewanella W3-18-1 from lactate. At steady state, the lactate-supplemented co-culture accumulated 172.4 mg l–1 AFDW biomass, 60% of which was Synechococcus 7002 and 40% was Shewanella W3-18-1 (Table 1; Supplementary Figure S1). Theoretical yield calculations, performed in consideration of both biomass and extracellular metabolite production, confirmed that 5 mM lactate feed was sufficient to support the measured biomass via CO2 exchange (Supplementary Material Equations). Notably, biomass yield of Shewanella W3-18-1 on lactate was twofold lower during co-cultivation than in the axenic state, thus suggesting changes in C flux distributions.
When the C source was shifted from lactate to bicarbonate, the direction of carbon flux was reversed. That is, growth of Shewanella W3-18 was driven by the release of photosynthetically fixed organic carbon by Synechococcus 7002. At photon flux of 1720 μmol photons m−2 s−1, the bicarbonate-limited co-culture accumulated 149 mg l–1 AFDW biomass, of which Synechococcus 7002 comprised 99% (Table 1; Supplementary Figure S2). An irradiance decrease from 1720 to 640 μmol photons m−2 s−1 led to onset of light limitation and drop of biomass yield to 116 mg l–1 AFDW (Table 1), although the ratio of Synechococcus 7002 to Shewanella W3-18 population remained unchanged.
Identification of putative interactions through metabolite profiling
The total organic carbon concentrations were similar in filtrates of HCO3− grown axenic Synechococcus 7002 cultures (3.9±0.2 mg l–1) and Synechococcus 7002–Shewanella W3-18-1 co-cultures (4.7±0.5 mg l–1). NMR analysis of dissolved metabolites excreted by axenic Synechococcus 7002 cells identified formate, acetate, lactate and alanine as major extracellular metabolites (Table 2). O2 concentrations positively affected the extracellular accumulation of amino acids with concentrations of alanine, isoleucine, leucine and valine increasing 1.5- to 3-fold. In co-cultures, formate concentrations remained high, while decreased concentrations of acetate, pyruvate, lactate and glycolate suggested their potential utilization by Shewanella W3-18-1 (Table 2). These findings are consistent with the genome annotation and phenotype profiling of Shewanella W3-18-1, which utilizes formate only under anoxic conditions using the nitrate-inducible formate dehydrogenase (Rodrigues et al., 2011). Nonetheless, measured decreases in steady-state concentrations of excreted organic acids between the axenic Synechococcus 7002 and co-cultures grown on HCO3− can account for only 185 μg l–1 of total organic carbon, which constitutes <20% of the Shewanella W3-18-1 biomass. Thus, cross-feeding between Synechococcus 7002 and Shewanella W3-18-1 may occur via metabolic intermediates that are not easily detected by NMR, which include partial breakdown products from complex polymers such as exopolysaccharides and other extracellular polymeric substance components (Belenguer et al., 2006).
Table 2. NMR analysis of excreted metabolites by photoautotrophically grown axenic and mixed cultures of Synechococcus 7002.
| Excreted metabolite |
Metabolite concentrations under different growth conditions, μMa |
|||
|---|---|---|---|---|
| Synechococcus 7002, dilution rate: 0.075 h−11720 μmol photons m–2 s–15% DOTb | Synechococcus 7002, dilution rate: 0.075 h−11720 μmol photons m–2 s–150% DOTb | Synechococcus 7002, dilution rate: 0.075 h−11720 μmol photons m–2 s–180% DOTb | Synechococcus 7002, Shewanella W3-18-1 dilution rate: 0.075 h−11720 μmol photons m–2 s–150% DOTb | |
| Lactate | ND | 0.9 | 0.7 | 0.4 |
| Pyruvate | 0.5 | 1.5 | 0.7 | ND |
| Acetate | 2.9 | 6.7 | 8.4 | 2.9 |
| Formate | 14.0 | 191.1 | 134.1 | 241.7 |
| Glycolate | ND | 3.1 | 1.2 | 2.3 |
| Alanine | 3.5 | 8.9 | 4.0 | 9.2 |
| Isoleucine | 0.6 | 1.0 | 1.6 | 1.2 |
| Leucine | 0.8 | 0.9 | 0.9 | 1.3 |
| Methionine | 0.1 | 0.6 | 0.3 | 0.4 |
| Threonin | ND | 0.2 | 0.1 | ND |
| Valine | 0.5 | 0.9 | 0.8 | 1.3 |
Abbreviations: DOT, dissolved O2 tension; ND, not determined; NMR, nuclear magnetic resonance spectroscopy.
Each measurement represents an average of four replicates. The s.d. for each data point does not exceed 5%.
DOT value represents % of O2 saturation.
Transcriptome patterning reveals metabolite exchange and downregulation of oxidative stress response during co-cultivation
A total of 473 and 234 transcripts in Synechococcus 7002 and Shewanella W3-18-1, respectively, displayed ⩾2-fold change in relative mRNA abundances as a result of co-cultivation irrespective of the carbon source. In Synechococcus 7002, a large fraction of these transcripts was affiliated with the transport (14%), energy metabolism (8%) and cellular processes (7%) role categories (Figure 1a, Supplementary Table S1). Significant decreases in transcript levels of Fe uptake and acquisition genes (for example, synechobactin biosynthesis) indicated higher availability of this important micronutrient for Synechococcus 7002 during co-cultivation. At the same time, increased mRNA abundances for genes encoding functions associated with carbon fixation (that is, RuBisCo, carboxysome components, bicarbonate transporter, the H+-translocating NADH-quinone dehydrogenase) were likely due to the inorganic carbon limitation in the co-cultures. Other potentially significant responses to co-cultivation in Synechococcus 7002 were associated with programmed cell death and detoxification, as manifested by the decreased transcript levels of a substantial number of one- and two-component toxin–antitoxin systems as well as three oxidative stress-related genes (SYNPCC7002_A0320, SYNPCC7002_A2422 and SYNPCC7002_G0082).
Figure 1.

Functional assignments of (a) Synechococcus 7002 and (b) Shewanella W3-18-1 genes displaying ⩾2-fold change in mRNA abundances under all co-culture conditions relative to the axenic state. The transcriptome comparisons were conducted using co-cultures and axenic cells grown in carbon-limited chemostats at a photon flux of 1720 μmol m−2 s−1.
In Shewanella W3-18-1, the largest group of transcripts displaying ⩾2-fold change in relative abundance under all co-culture conditions mapped to the transport and amino-acid biosynthesis categories (Figure 1b, Supplementary Table S2). The specific downregulation of methionine and tryptophan biosynthesis pathways in Shewanella W3-18-1 supported that these amino acids are likely to be produced and excreted into the medium by Synechococcus 7002 (Table 2). Furthermore, the decrease in transcripts encoding key steps of thiamine pyrophosphate formation (thiDEG) and thiamine pyrophosphate-dependent branched chain amino-acid biosynthesis (ilvI) in Shewanella W3-18-1, across all co-culture conditions suggested the presence of other metabolite exchange pathways.
Interestingly, the co-cultivation led to a decrease in transcript abundance within the oxidative stress response subcategory of Shewanella W3-18-1 (Supplementary Table S2), which included two putative redoxin domain genes (ohrA, bcp) and previously uncharacterized gene cluster (SputW3181_1296- SputW3181_1306) that displays a high degree of similarity to the singlet oxygen (1O2) protection ChrR regulon of Rhodobacter sphaeroides (Dufour et al., 2008). Other significantly downregulated transcripts, whose presence was previously linked to stress response in Shewanella (Qiu et al., 2005; McLean et al., 2008), encoded lambda-like bacteriophage proteins and the LrgAB system that controls autolysis via modulating murein hydrolase activity (Groicher et al., 2000). Notably, this coincided with transcriptional upregulation of several surface attachment factors (pilA, pilM, pilXWV, bpfA and csgA) of Shewanella W3-18-1, which are typically associated with biofilm lifestyle (Schembri et al., 2003; Beloin and Ghigo, 2005).
Transcriptional response to carbon limitation in a co-culture leads to the maximization of metabolic capacity
As the growth-limiting carbon source determined flux directions and densities of interacting populations, it exerted a major effect on the global transcriptome patterns in both organisms (Figure 2). During lactate-supported growth, 1184 Synechococcus 7002 genes and 456 Shewanella W3-18-1 genes showed ⩾2-fold change in relative mRNA abundances (Supplementary Tables S3 and S4). Conversely, in HCO3− grown co-cultures, 690 Synechococcus 7002 genes and 1342 Shewanella W3-18-1 genes were differentially expressed (Supplementary Tables S5 and S6). The inversed patterns likely reflect substrate availability and flux partitioning as carbon limitation, and to a larger degree carbon starvation, lead to competitive inter-specific interactions in mixed cultures (Kjelleberg et al., 1993; Garbeva and de Boer, 2009).
Figure 2.

Venn diagram of the global transcriptome changes in Synechococcus 7002 and Shewanella W3-18-1 in response to co-cultivation. Arrows indicate the direction of the carbon flux in lactate (blue) and HCO3− (red)-supplemented co-cultures. The area of each circle represents the total number of genes displaying ⩾2-fold change in mRNA abundances in co-cultures relative to the axenic state. The transcriptome comparisons were conducted using co-cultures and axenic cells grown in carbon-limited chemostats at a photon flux of 1720 μmol m−2 s−1.
Pathway enrichment analysis comparing global transcript levels in lactate-grown co-cultures relative to axenic controls indicated that both Synechococcus 7002 and Shewanella W3-18-1 had maximized the uptake and utilization of specific nutrients during co-cultivation. In Synechococcus 7002 (Table 3, Supplementary Table S3), the increase in mRNA levels of putative glyoxylate metabolism enzymes, for example, D-3-phosphoglycerate dehydrogenase (SYNPCC7002_A1246) and phosphoglycolate phosphatase (SYNPCC7002_A0506), suggested occurrence of photorespiration, which was likely due to the elevated O2/CO2 ratio compared with bicarbonate-grown cultures (Table 1). Interestingly, despite the obvious excess of ammonium in the growth medium, a broad transcriptional upregulation of putative nitrogen assimilation machinery in Synechococcus 7002 was observed, which included glutamine synthetase (glnA) and assimilatory ferredoxin-nitrite reductase (nirA) genes. In addition, increased transcript abundance of the L-arginine deiminase (arcA), and L-asparagine amidopeptidase (asnA1) genes, which are involved in cyanophycin utilization, also suggests utilization of nitrogen-containing organic compounds as an additional source of carbon by the cyanobacterium.
Table 3. Pathway and regulator enrichment for Synechococcus 7002 grown in carbon-limited chemostat co-culture with lactate as the sole source of carbon.
| Pathway/regulon enrichment | P-valuea |
|---|---|
| Genes displaying ⩾2-fold increase in mRNA abundance relative to the axenic state | |
| Cell structures biosynthesis: ddl, glmM, lpxA, mraY, murA, murC, murE, murF, murG, murI, SYNPCC7002_A0138 | 0.0075 |
| Fatty acid biosynthesis, initiation and elongation: fabB, fabF, fabZ | 0.0220 |
| UDP-N-acetylmuramoyl-pentapeptide biosynthesis: ddl, murA, murC, murE, murF, murI | 0.0221 |
| Photorespiration: gcvT, glcE, rbcS, serA, SYNPCC7002_A0506 | 0.0228 |
| Vitamin biosynthesis: bioD, bioF, coaE, cobP, cobT, fabB, fabF, fabZ, folE, folQ, gcvT, hggT, ilvC, panB, pdxH, plr1, purU, pyrR, ribAB, serA, SYNPCC7002_A0363, vte1, ygfA | 0.0240 |
| Nucleic acid processing: queA, queF, tgt | 0.0277 |
| Genes displaying ⩾2-fold decrease in mRNA abundance relative to the axenic state | |
| Siderophore biosynthesis: sybA, sybB, sybC, sybD, sybE, sybF, sybL | 0.0000 |
| Oxygenic photosynthesis: petG, petH, petM, prk, psaE, psaK, psbD, psbE, psbF, psbJ, psbL, psbO, psbT, psbY, rpiA | 0.0062 |
| Photosynthesis, light reactions: petG, petH, petM, psaE, psaK, psbD, psbE, psbF, psbJ, psbL, psbO, psbT, psbY | 0.0083 |
| Generation of precursor metabolites and energy: ald, ctaCI, fumC, gap, hoxH, hoxY, ndhD1, ndhM, nifJ, petG, petH, petM, ppsA, prk, psaE, psaK, psbD, psbE, psbF, psbJ, psbL, psbO, psbT, psbY, rpiA, sdhB, talA, talC, zwf | 0.0240 |
| Ferric uptake, Fur regulon: chlL, chlN, exbB, exbD, fecB, fecC, fecD, fecE, fhuA, futC, hik20, nifJ, pchR, schT, SYNPCC7002_A2346, SYNPCC7002_G0005, SYNPCC7002_G0006, SYNPCC7002_G0089, SYNPCC7002_G0099, tonB | 0.0006 |
P-value represents the probability that the number of genes associated with a specific pathway/regulon occurs by chance.
In Shewanella W3-18-1 (Table 4; Supplementary Table S4), pathway enrichment analysis revealed that growth in lactate-supplemented co-cultures was also associated with elevated transcription of nitrogen transport and assimilation genes. Specifically, the relative mRNA abundances of nitrogen regulator (glnK), glutamine synthetase (glnA) and ammonia transporter (amt) genes increased 25-, 5- and 8-fold, respectively, relative to the axenic state. Broad decrease in transcript abundance was also displayed by pathways involved in biosynthesis of amino acid, such as methionine, tryptophan, isoleucine and valine. At the same time in lactate-supplemented co-cultures, Shewanella W3-18-1 downregulated transcripts for genes encoding D,L-lactate (lldP, lldEF and SputW3181_2834) utilization machinery (Pinchuk et al., 2009), suggesting preferential utilization of organic compounds excreted or released by the cyanobacterium. Supporting this notion were elevated mRNA levels of alanine dehydrogenase (Ald) and putative nucleotide deaminase (W3181_3499), which can provide Shewanella W3-18-1 with additional sources of carbon and nitrogen (Supplementary Table S4). Upregulation of extracellular phospholipase A (pldA), lipase (W3181_1613) and malate synthase (aceB) genes can potentially increase carbon availability via the degradation of extracellular lipopolysaccharides into acetyl-CoA and subsequent incorporation into the tricarboxylic acid cycle.
Table 4. Pathway and regulator enrichment for Shewanella W3-18-1 in carbon-limited chemostat co-culture with lactate as the sole source of carbon.
| Pathway/regulon enrichment | P-valuea |
|---|---|
| Genes displaying ⩾2-fold increase in mRNA abundance relative to the axenic state | |
| Amino acids biosynthesis: argC, aroF, asnB, cysD, cysJ, glnA, hisD, hisG, lysC, tyrA | 0.0001 |
| Metabolism of inorganic nutrients: cysD, cysJ, glnA, nrfA | 0.0063 |
| Phospholipase: pldA | 0.0130 |
| Alanine degradation: ald | 0.0133 |
| Nitrogen assimilation, NtrC regulon: amtB, glnA, glnK, SputW3181_0223 | 0.0001 |
| Tryptophan biosynthesis, TrpR regulon: aroF, tyrA | 0.0043 |
| Genes displaying ⩾2-fold decrease in mRNA abundance relative to the axenic state | |
| Aspartate super-pathway: asd, lysA, metA, metB, metC, metE, metH, metK, metL | 0.0001 |
| Threonine degradation: ilvA, ilvC, ilvD, ilvG, ilvH, ilvI | 0.0002 |
| Tryptophan biosynthesis: trpA, trpB, trpCF, trpD, trpE, trpG | 0.0002 |
| Fatty acid and lipids degradation: fadA, fadB, fadE, fadI, fadJ, glpD, SputW3181_1978, SputW3181_2433 | 0.0004 |
| Homoserine and methionine biosynthesis: asd, metA, metB, metC, metE, metE, metH, metL | 0.0069 |
| Isoleucine and valine biosynthesis: asd, ilvA, ilvC, ilvD, ilvG, ilvH, ilvI, metL | 0.0106 |
| B12 biosynthesis and uptake: cobC, cobS, cobT, cobU, btuC, btuD | 0.0237 |
| Thiamin biosynthesis thiC, thiDE, thiF, thiH | 0.0237 |
| Superpathway of chorismate metabolism: aroE, pheA, trpA, trpB, trpCF, trpD, trpE, trpG, ubiC | 0.0276 |
| Methylglyoxal detoxification: dldD, gloB | 0.0286 |
| Methionine biosynthesis, MetJ regulon: btuB, metA, metB, metE, metF, metH, metK, metL, metR, metT, metY, msrA, SputW3181_0088, SputW3181_0089, SputW3181_1636, SputW3181_2707 | 0.0000 |
| Fatty acid degradation, PsrA regulon: etfQ, fadA, fadB, fadE, fadH, fadI, fadJ, psrA, SputW3181_1978, SputW3181_2146 | 0.0004 |
| Methionine biosynthesis, MetR regulon: metC, metE, metR | 0.0058 |
| Fatty acid degradation, FadR regulon: fadI, fadJ, fadL | 0.01319 |
| Lactate utilization, LlldR regulon: lldE, lldF | 0.03802 |
P-value represents the probability that the number of genes associated with a specific pathway/regulon occurs by chance.
The opportunistic nature of interactions was even more evident during co-culturing using HCO3− as the sole source of C. Under these conditions, the only influx of organic carbon for heterotrophic growth of Shewanella W3-18-1 was provided by the cyanobacterium, either by excretion, cell lysis or exoenzyme-mediated release from the cyanobacterial cell surfaces. When compared with lactate-grown co-cultures (Supplementary Table S7), Synechococcus 7002 transcription profiles displayed downregulation of photorespiration and carbohydrate metabolism genes during co-culturing with HCO3−. These patterns as well as elevated transcript levels of Fe acquisition genes (Table 5) are more likely to be linked to the increased production of light-capturing and photosynthetic machinery (Ludwig and Bryant, 2012; Morrissey and Bowler, 2012) and higher CO2 availability (Table 1). Similarly, lower mRNA levels of putative nitrile/cyanate, dipeptide and amino acids transporter genes suggested decreased need for additional (organic) carbon sources during photoautotrophic growth of Synechococcus 7002.
Table 5. Pathway and regulator enrichment for Synechococcus 7002 grown in carbon-limited chemostat co-culture with HCO3 − as the sole source of carbon.
| Pathway/regulon enrichment | P-valuea |
|---|---|
| Genes displaying ⩾2-fold increase in mRNA abundance relative to the lactate co-culture | |
| Histidine biosynthesis: hisA, hisB, hisF | 0.0431 |
| Ferric uptake, Fur regulon: exbB, fecB, fhuA, hik20, pchR, SYNPCC7002_G0005, SYNPCC7002_G0006 | 0.0718 |
| Heat shock response, HrcA regulon: groES, SYNPCC7002_A0107 | 0.0220 |
| Genes displaying ⩾2-fold decrease in mRNA abundance relative to the lactate co-culture | |
| Photorespiration: gcvT, rbcS, serA, SYNPCC7002_A0506 | 0.0033 |
| Carbohydrates metabolism: glgA2, gpmB, rbcS, sppA, SYNPCC7002_A0448, SYNPCC7002_A1492, SYNPCC7002_A1554 | 0.0407 |
| Serine biosynthesis: serA | 0.0045 |
P-value represents the probability that the number of genes associated with a specific pathway/regulon occurs by chance.
Increased transcription levels of multiple catabolic pathways and the putative carbon starvation protein (cstA) involved in peptide uptake was indicative that Shewanella W3-18-1 experienced severe C-limitation upon shift of the co-culture from lactate to HCO3− (Table 6; Supplementary Table S8). Elevated transcript levels were detected for genes involved in lipid, amino acid (Phe, Tyr and Met), polyamine, putrescine and organic acids (acetate, acetoacetate and propionate) utilization. Furthermore, enrichment analysis of predicted regulons verified that metabolism of fatty acids (fadR, fabR and psrA), and degradation of histidine, tyrosine and propionate (hutC, tyrR and prpR) were significantly upregulated in Shewanella W3-18-1 during growth in HCO3−-supplemented co-cultures (Table 6).
Table 6. Pathway and regulator enrichment for Shewanella W3-18-1 in carbon-limited chemostat co-culture with HCO3 − as the sole source of carbon.
| Pathway/regulon enrichment | P-valuea |
|---|---|
| Genes displaying ⩾2-fold increase in mRNA abundance relative to the lactate co-culture | |
| Amino acids biosynthesis: aroA, aroE, carA, hisA, hisB, hisC, hisD, hisF, hisG, hisH, hisIE, ilvA, ilvD, ilvE, ilvG, ilvM, kbl, metE, phhA, serB, thrA, thrB, thrC, trpB, trpG | 0.0000 |
| Degradation/utilization/assimilation: acnD, acs, aguA, can, deoD, fadA, fadB, fadD, fadE, fadI, fadJ, gabD, gloB, hmgA, hppD, hutH, hutU, ilvA, ilvD, ilvE, ilvG, ilvM, kbl, liuF, liuG, mdeA, phhA, phhB, prpB, prpC, prpF, puuA, puuB, puuC, speC, SputW3181_1978, SputW3181_2433, SputW3181_4006, SputW3181_4017 | 0.0001 |
| Fatty acid and lipids degradation: fadA, fadB, fadD, fadE, fadI, fadJ, liuF, liuG, SputW3181_1978, SputW3181_2433, SputW3181_4017 | 0.0001 |
| Glyoxylate cycle: aceA, aceB, gltA, sdhC, sdhD | 0.0003 |
| Guanosine nucleotides de novo biosynthesis: gmk, guaB, ndk, nrdA | 0.0207 |
| Fatty acid degradation, PsrA regulon: aceA, aceB, etfQ, fadA, fadB, fadD, fadE, fadH, fadI, fadJ, gltA, psrA, sdhC, sdhD, SputW3181_1978, SputW3181_2146 | 0.0001 |
| Fatty acid biosynthesis repressor, FabR regulon: desC, fabR, fadL, plsC, SputW3181_4017, yqfA | 0.0001 |
| Methyl citrate utilization repressor, PrpR regulon: acnD, prpB, prpC, prpF | 0.0004 |
| Histidine utilization repressor, HutC regulon: hutC, hutH, hutU | 0.0104 |
| Amino acid (Tyr/Phe) utilization activator, TyrR regulon: aceA, aceB, aroA, can, liuF, liuG, mdeA, phhA, phhB, SputW3181_1883 | 0.0153 |
| Fatty acid degradation, FadR regulon: fadI, fadJ, fadL | 0.0233 |
| Genes displaying ⩾2-fold decrease in mRNA abundance relative to the lactate co-culture | |
| Thiamin biosynthesis: sufS, thiC, thiDE, thiF, thiH, thiL | 0.0001 |
| Inorganic nutrients metabolism: cysC, cysD, cysH, cysI, cysJ, cysN, nrfA, phsB | 0.0029 |
| Secondary metabolites degradation: aceE, aceF, lldE, lldF, lldG, nagA | 0.0077 |
| Pentose phosphate pathway, oxidative branch: gnd, pgl, zwf | 0.0286 |
| Methionine biosynthesis: cysC, cysD, cysH, cysI, cysJ, cysN, metE, metK, metL | 0.0287 |
| Arginine biosynthesis: argB, argC, argF, argG, argH | 0.0332 |
| Rhamnose biosynthesis: rbfC, rmlC, rmlD | 0.0350 |
| Lactate utilization, LlldR regulon: lldE, lldF, lldG | 0.0057 |
| Pyruvate metabolism repressor, PdhR regulon: aceE, aceF, oadA, oadB, pflA, flB, pflX | 0.0018 |
| Tryptophan biosynthesis, TrpR regulon: aroF, trpR, tyrA | 0.0057 |
P-value represents the probability that the number of genes associated with a specific pathway/regulon occurs by chance.
Cell envelope modifications and evidence of cell–cell interactions
During co-culture growth on lactate, where Shewanella W3-18-1 accounted for >80% of the co-culture cell population (Supplementary Figure S1), microscopic observations revealed the presence of multicellular aggregates comprised of cyanobacterial and proteobacterial cells (Figure 3). It is important to note that, while heterogeneity across the aggregates may exert some effects on the co-culture transcriptome patterns, these effects are likely to be minimal. For Synechococcus 7002, only a substantially smaller portion of the cells were attached to the aggregates of Shewanella W3-18-1, and then were located at the surface (Figure 3). For Shewanella W3-18-1, which also aggregated when grown axenically under identical conditions (data not shown), cells in the interior of the flocs may be growing more slowly than those at the periphery or those suspended singly; however, to the extent that this is true, chemostat theory predicts a compensation in which other cells are growing faster than the dilution rate.
Figure 3.

Representative micrograph of Synechococcus 7002 and Shewanella W3-18-1 cell aggregates formed in a co-culture grown under carbon-limited aerobic chemostat conditions using 5 mM lactate as the sole source of carbon. Synechococcus 7002 cells (red) were visualized by phycocyanin autofluorescence measured at 640 nm. Shewanella W3-18-1 cells (green) were visualized by acridine orange staining and detection at 490–577 nm.
In lactate-grown co-cultures, enrichment of pathways involved in biosynthesis of fatty acids, peptidoglycan and lipopolysaccharides indicated remodeling of the cell surface in Synechococcus 7002. Transcripts encoding the N-acetyl muramoyl-pentapeptide biosynthesis reactions (ddl, murA, murC, murE, murF and murI; Table 3; Supplementary Table S3) were upregulated, while those encoding putative transcarboxypeptidases (dacB, dacC, dacD and SynPCC7002_A0082) and lipopolysaccharide biosynthesis enzymes were broadly downregulated in Synechococcus 7002. In Shewanella W3-18-1, a number of biofilm formation factors including type IV pili (pilAB, pilWWV and pilM), curli (csgAB, csgD and csgEFG) and extracellular proteins predicted to promote adhesion (pilY, bpfA, Sputw3181_3687 and SputW3181_1756) also displayed elevated mRNA levels under co-culture conditions (Supplementary Table S4).
Furthermore, the observed cell–cell interactions between the two organisms could facilitate the uptake and re-utilization of complex exopolymers or organic substrates released from the surface. In HCO3− grown co-cultures (Supplementary Table S4), Shewanella W3-18-1 displayed increased transcript abundances of genes encoding secreted or outer membrane proteins including a lipase (SputW3181_1613), several proteases (sapSH, Sputw3181_3341, Sputw3181_3384 and SputW3181_0531) and a polymorphic toxin (Sputw3181_0994), which together may be involved in cell envelope lysis and degradation of surface structures (Zhang et al., 2012). Similarly, the elevated mRNA levels of 4-alpha-glucanotransferase gene (malQ) can potentially allow for carbohydrate utilization, while DNA can be broken down by the putative extracellular endonuclease (exeS). Proteins/peptides appeared also to be used by Shewanella W3-18-1 as well, perhaps directly as amino acids or after further catabolism as a nitrogen, carbon and energy source (Supplementary Table S4).
Conclusions
Development of predictive, system-level understanding of microbial consortia requires integration of authentic field investigation with well-controlled laboratory-scale studies. Here, we applied deep transcriptome sequencing to a laboratory-based model system consisting of a euryhaline cyanobacterium, Synechococcus 7002 and a marine heterotroph, Shewanella W3-18-1, to investigate the effect of co-cultivation and carbon flux directions on the interactions between these organisms. Although the opportunistic nature of this association constrains, to some degree, the ecological implications of observed transcriptional responses, the results provide a number of novel and relevant insights into the physiological basis of microbial interactions.
The overall transcriptional response of both organisms to co-cultivation was shaped by their respective physiologies and the growth constraints. Carbon limitation resulted in the expansion of metabolic capacity, which was manifested through transcriptional upregulation of transport and catabolic pathways (Tables 3, 4, 5, 6). As metabolic coupling occurred either via lactate oxidation or secretion of photosynthetically fixed carbon, NMR (Table 2) and transcriptome analyses (Tables 3, 4, 5, 6) suggested specific metabolite exchange during co-cultivation. On one hand, the detection of methionine and alanine excretion by the cyanobacterium leads us to hypothesize that downregulation of the corresponding biosynthetic machinery of Shewanella W3-18-1 reflects utilization of these amino acids by the heterotroph. On the other hand, the broad and consistent decrease of mRNA levels for many Fe-regulated Synechococcus 7002 genes during co-cultivation, including synechobactin biosynthesis pathway and the Fur regulon (Table 2 and Supplementary Table S4), suggested increased Fe availability in the cyanobacterium during co-cultivation.
The latter observation is in agreement with a previous study, in which downregulation of Fe-acquisition machinery was reported in Synechococcus sp. WH8102 during co-cultivation with Vibrio parahaemolyticus (Tai et al., 2009). However, the causative effect of co-cultivation on Fe homeostasis in cyanobacteria is yet to be determined. Although increased Fe availability can be a result of lower demand—for instance, owing to declining photosynthetic activity (Ludwig and Bryant, 2012), the mRNA abundance of Synechococcus 7002 genes involved in photosynthetic metabolism was high across all experimental conditions. A more compelling explanation may be linked to the documented capacity of aerobic heterotrophic microorganisms to produce large quantities of extracellular Fe chelators (Wandersman and Delepelaire, 2004), which can be recognized and transported intracellularly by the cyanobacterium. Bacteria of the genus Shewanella were also shown to carry out Fe reduction aerobically inside cell aggregates (McLean et al., 2008) thus providing more facile and energy-efficient mechanisms for Fe acquisition by the cyanobacterium.
Furthermore, this study provides evidence of potentially novel interactions between oxygenic photoautotrophs and heterotrophs related to the oxidative stress response. Our results are distinct from those reported in a previous Prochlorococcus spp. study, where heterotrophs were hypothesized to decrease oxidative stress in the cyanobacterium via catalase-dependent ROS scavenging (Morris et al., 2008). In Shewanella W3-18-1, transcript levels of genes involved in oxidative stress response and scavenging of ROS radicals displayed broad decrease under co-culture conditions, which included putative 1O2 protection (chrR regulon) (Dufour et al., 2008) and methylglyoxal degradation (Hoque et al., 2010) pathways (Table 4 and Supplementary Table S4). Because of the propensity of marine heterotrophs to produce large quantities of extracellular superoxide and related ROS (Diaz et al., 2013), our observations indicate that some photoautotrophs may provide community-level protection against oxidative stress as opposed to being protected by other members. In that regard, our study not only measures the responses at the system-level to explore the interactions between the partners more holistically but also formulates new hypotheses about cyanobacterial–heterotrophic interactions.
Acknowledgments
The research was supported by the Genomic Science Program (GSP), Office of Biological and Environmental Research (BER), US Department of Energy (DOE) and is a contribution of the PNNL Foundational Scientific Focus Area (FSFA). A significant portion of the research was performed using the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by DOE BER and located at PNNL. We acknowledge PNNL staff who helped to support this work, specifically Oleg Geydebrekht and Thomas Wietsma for assistance with the analytical measurements. We are also grateful to Dr William Nelson for help with the functional genome annotation and Dr Sergey Stolyar for valuable advice and critical discussions. PNNL is operated for the DOE by Battelle Memorial Institute under Contract DE-AC05-76RLO 1830.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Database access to the sequencing data
The RNA sequencing data are available online at the Gene Expression Ominbus (GEO) database (http://www.ncbi.nlm.nih.gov/gds/) under accession number GSE53360.
Supplementary Material
References
- Amin SA, Green DH, Hart MC, Kupper FC, Sunda WG, Carrano CJ. (2009). Photolysis of iron-siderophore chelates promotes bacterial-algal mutualism. Proc Natl Acad Sci USA 106: 17071–17076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SA, Parker MS, Armbrust EV. (2012). Interactions between diatoms and bacteria. Microbiol Mol Biol R 76: 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterton JC, Vanbaalen C. (1971). Growth responses of blue-green algae to sodium chloride concentration. Arch Mikrobiol 76: 151–165. [DOI] [PubMed] [Google Scholar]
- Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE et al. (2006). Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbiol 72: 3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliaev AS, Klingeman DM, Klappenbach JA, Wu L, Romine MF, Tiedje JM et al. (2005). Global transcriptome analysis of Shewanella oneidensis MR-1 exposed to different terminal electron acceptors. J Bacteriol 187: 7138–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C, Ghigo JM. (2005). Finding gene-expression patterns in bacterial biofilms. Trends Microbiol 13: 16–19. [DOI] [PubMed] [Google Scholar]
- Bertilsson S, Eiler A, Nordqvist A, Jorgensen NO. (2007). Links between bacterial production, amino-acid utilization and community composition in productive lakes. ISME J 1: 532–544. [DOI] [PubMed] [Google Scholar]
- Bowman JP, McCammon SA, Brown MV, Nichols DS, McMeekin TA. (1997). Diversity and association of psychrophilic bacteria in Antarctic sea ice. Appl Environ Microbiol 63: 3068–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner CG, Rehm C, Grossart HP, Kroth PG. (2011). Growth and release of extracellular organic compounds by benthic diatoms depend on interactions with bacteria. Environ Microbiol 13: 1052–1063. [DOI] [PubMed] [Google Scholar]
- Bull AT. (2010). The renaissance of continuous culture in the post-genomics age. J Ind Microbiol Biotechnol 37: 993–1021. [DOI] [PubMed] [Google Scholar]
- Caldwell DE, Caldwell SJ. (1978). A Zoogloea sp. associated with blooms of Anabaena flos-aquae. Can J Microbiol 24: 922–931. [DOI] [PubMed] [Google Scholar]
- Carpenter EJ, Foster RA. (2002). Marine cyanobacterial symbioses. In: Rai AN, Bergman B, Rasmussen U (eds) Cyanobacteria in Symbiosis. Kluwer Academic Publishers: The Netherlands. [Google Scholar]
- Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM et al. (2012). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 40: D742–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JJ. (1982). Interactions between bacteria and algae in aquatic ecosystems. Annu Rev Ecol Syst 13: 291–314. [Google Scholar]
- Diaz JM, Hansel CM, Voelker BM, Mendes CM, Andeer PF, Zhang T. (2013). Widespread production of extracellular superoxide by heterotrophic bacteria. Science 340: 1223–1226. [DOI] [PubMed] [Google Scholar]
- Dufour YS, Landick R, Donohue TJ. (2008). Organization and evolution of the biological response to singlet oxygen stress. J Mol Biol 383: 713–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbeva P, de Boer W. (2009). Inter-specific interactions between carbon-limited soil bacteria affect behavior and gene expression. Microb Ecol 58: 36–46. [DOI] [PubMed] [Google Scholar]
- Groicher KH, Firek BA, Fujimoto DF, Bayles KW. (2000). The Staphylococcus aureus lrgAB operon modulates murein hydrolase activity and penicillin tolerance. J Bacteriol 182: 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossart HP. (1999). Interactions between marine bacteria and axenic diatoms (Cylindrotheca fusiformis, Nitzschia laevis, and Thalassiosira weissflogii) incubated under various conditions in the lab. Aquat Microb Ecol 19: 1–11. [Google Scholar]
- Grossmann S, Bauer S, Robinson PN, Vingron M. (2007). Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics 23: 3024–3031. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Itoh K, Suyama K. (2011). Growth of the cyanobacterium Synechococcus leopoliensis CCAP1405/1 on agar media in the presence of heterotrophic bacteria. Microbes Environ 26: 120–127. [DOI] [PubMed] [Google Scholar]
- Hoque MA, Uraji M, Banu MN, Mori IC, Nakamura Y, Murata Y. (2010). The effects of methylglyoxal on glutathione S-transferase from Nicotiana tabacum. Biosci Biotechnol Biochem 74: 2124–2126. [DOI] [PubMed] [Google Scholar]
- Karp PD, Paley SM, Krummenacker M, Latendresse M, Dale JM, Lee TJ et al. (2010). Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform 11: 40–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazamia E, Czesnick H, Nguyen TT, Croft MT, Sherwood E, Sasso S et al. (2012). Mutualistic interactions between vitamin B12-dependent algae and heterotrophic bacteria exhibit regulation. Environ Microbiol 14: 1466–1476. [DOI] [PubMed] [Google Scholar]
- Kieft TL, Fredrickson JK, Onstott TC, Gorby YA, Kostandarithes HM, Bailey TJ et al. (1999). Dissimilatory reduction of Fe(III) and other electron acceptors by a Thermus isolate. Appl Environ Microbiol 65: 1214–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjelleberg S, Albertson N, Flardh K, Holmquist L, Jouper-Jaan A, Marouga R et al. (1993). How do non-differentiating bacteria adapt to starvation? Antonie Van Leeuwenhoek 63: 333–341. [DOI] [PubMed] [Google Scholar]
- Lambert DH, Stevens SE Jr.. (1986). Photoheterotrophic growth of Agmenellum quadruplicatum PR-6. J Bacteriol 165: 654–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Bryant DA. (2011). Transcription profiling of the model cyanobacterium Synechococcus sp. Strain PCC 7002 by next-gen (SOLiD) sequencing of cDNA. Front Microbiol 2: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Bryant DA. (2012). Acclimation of the global transcriptome of the cyanobacterium Synechococcus sp. strain PCC 7002 to nutrient limitations and different nitrogen sources. Front Microbiol 3: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JS, Pinchuk GE, Geydebrekht OV, Bilskis CL, Zakrajsek BA, Hill EA et al. (2008). Oxygen-dependent autoaggregation in Shewanella oneidensis MR-1. Environ Microbiol 10: 1861–1876. [DOI] [PubMed] [Google Scholar]
- Morris JJ, Kirkegaard R, Szul MJ, Johnson ZI, Zinser ER. (2008). Facilitation of robust growth of Prochlorococcus colonies and dilute liquid cultures by "helper" heterotrophic bacteria. Appl Environ Microbiol 74: 4530–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey J, Bowler C. (2012). Iron utilization in marine cyanobacteria and eukaryotic algae. Front Microbiol 3: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Nealson KH, Scott J. (2003). Ecophysiology of the genus Shewanella. In: Dworkin M (ed) The Prokaryotes: An Evolving Electronic Resource for the Microbiological Community. Springer-NY, LLC: New York, NY. [Google Scholar]
- Novichkov PS, Brettin TS, Novichkova ES, Dehal PS, Arkin AP, Dubchak I et al. (2012). RegPrecise web services interface: programmatic access to the transcriptional regulatory interactions in bacteria reconstructed by comparative genomics. Nucleic Acids Res 40: W604–W608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmann J. (2006). Symbiosis between non-related bacteria in phototrophic consortia. Prog Mol Subcell Biol 41: 21–37. [DOI] [PubMed] [Google Scholar]
- Paerl HW, Kellar PE. (1978). Significance of bacterial Anabaena (Cyanophyceae) associations with respect to N2 fixation in freshwater. J Phycol 14: 254–260. [Google Scholar]
- Paerl HW, Gallucci KK. (1985). Role of chemotaxis in establishing a specific nitrogen-fixing cyanobacterial-bacterial association. Science 227: 647–649. [DOI] [PubMed] [Google Scholar]
- Paerl HW, Pinckney JL. (1996). A mini-review of microbial consortia: their roles in aquatic production and biogeochemical cycling. Microb Ecol 31: 225–247. [DOI] [PubMed] [Google Scholar]
- Pinchuk GE, Ammons C, Culley DE, Li SM, McLean JS, Romine MF et al. (2008). Utilization of DNA as a sole source of phosphorus, carbon, and energy by Shewanella spp.: ecological and physiological implications for dissimilatory metal reduction. Appl Environ Microbiol 74: 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinchuk GE, Rodionov DA, Yang C, Li X, Osterman AL, Dervyn E et al. (2009). Genomic reconstruction of Shewanella oneidensis MR-1 metabolism reveals a previously uncharacterized machinery for lactate utilization. Proc Natl Acad Sci USA 106: 2874–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Sundin GW, Wu L, Zhou J, Tiedje JM. (2005). Comparative analysis of differentially expressed genes in Shewanella oneidensis MR-1 following exposure to UVC, UVB, and UVA radiation. J Bacteriol 187: 3556–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. (1979). Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol 111: 1–61. [Google Scholar]
- Rodionov DA, Yang C, Li X, Rodionova IA, Wang Y, Obraztsova AY et al. (2010). Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genomics 11: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues JL, Serres MH, Tiedje JM. (2011). Large-scale comparative phenotypic and genomic analyses reveal ecological preferences of Shewanella species and identify metabolic pathways conserved at the genus level. Appl Environ Microbiol 77: 5352–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon PS, Janson S, Graneli E. (2003). Molecular identification of bacteria associated with filaments of Nodularia spumigena and their effect on the cyanobacterial growth. Harmful Algae 2: 261–272. [Google Scholar]
- Schembri MA, Kjaergaard K, Klemm P. (2003). Global gene expression in Escherichia coli biofilms. Mol Microbiol 48: 253–267. [DOI] [PubMed] [Google Scholar]
- Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M et al. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour JR, Ahmed T, Durham WM, Stocker R. (2010). Chemotactic response of marine bacteria to the extracellular products of Synechococcus and Prochlorococcus. Aquat Microb Ecol 59: 161–168. [Google Scholar]
- Shen H, Niu Y, Xie P, Tao M, Yang X. (2011). Morphological and physiological changes in Microcystis aeruginosa as a result of interactions with heterotrophic bacteria. Freshwater Biol 56: 1065–1080. [Google Scholar]
- Sher D, Thompson JW, Kashtan N, Croal L, Chisholm SW. (2011). Response of Prochlorococcus ecotypes to co-culture with diverse marine bacteria. ISME J 5: 1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simidu U, Kita-Tsukamoto K, Yasumoto T, Yotsu M. (1990). Taxonomy of four marine bacterial strains that produce tetrodotoxin. Int J Syst Bacteriol 40: 331–336. [DOI] [PubMed] [Google Scholar]
- Stevens SE, Porter RD. (1980). Transformation in Agmenellum quadruplicatum. Proc Natl Acad Sci USA 77: 6052–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson BS, Waterbury JB. (2006). Isolation and identification of an epibiotic bacterium associated with heterocystous Anabaena cells. Biol Bull-US 210: 73–77. [DOI] [PubMed] [Google Scholar]
- Tai V, Paulsen IT, Phillippy K, Johnson DA, Palenik B. (2009). Whole-genome microarray analyses of Synechococcus-Vibrio interactions. Environ Microbiol 11: 2698–2709. [DOI] [PubMed] [Google Scholar]
- Wandersman C, Delepelaire P. (2004). Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol 58: 611–647. [DOI] [PubMed] [Google Scholar]
- Wenter R, Hutz K, Dibbern D, Li T, Reisinger V, Ploscher M et al. (2010). Expression-based identification of genetic determinants of the bacterial symbiosis 'Chlorochromatium aggregatum'. Environ Microbiol 12: 2259–2276. [DOI] [PubMed] [Google Scholar]
- Wintermute EH, Silver PA. (2010). Dynamics in the mixed microbial concourse. Genes Dev 24: 2603–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhang N, Hayes A, Panoutsopoulou K, Oliver SG. (2004). Global analysis of nutrient control of gene expression in Saccharomyces cerevisiae during growth and starvation. Proc Natl Acad Sci USA 101: 3148–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B, Bishop S, Stessman D, Wright D, Spalding MH, Halverson LJ. (2013). Chlamydomonas reinhardtii thermal tolerance enhancement mediated by a mutualistic interaction with vitamin B12-producing bacteria. ISME J 7: 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengler K, Palsson BO. (2012). A road map for the development of community systems (CoSy) biology. Nat Rev Microbiol 10: 366–372. [DOI] [PubMed] [Google Scholar]
- Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. (2012). Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.