

Abstract
Salt-inducible kinases (SIKs) are promising therapeutic targets for modulating cytokine responses during innate immune activation. The study of SIK inhibition in animal models of disease has been limited by the lack of selective small-molecule probes suitable for modulating SIK function in vivo. We used the pan-SIK inhibitor HG-9-91-01 as a starting point to develop improved analogs, yielding a novel probe 5 (YKL-05-099) that displays increased selectivity for SIKs versus other kinases and enhanced pharmacokinetic properties. Well-tolerated doses of YKL-05-099 achieve free serum concentrations above its IC50 for SIK2 inhibition for > 16 hours and reduce phosphorylation of a known SIK substrate in vivo. While in vivo active doses of YKL-05-099 recapitulate the effects of SIK inhibition on inflammatory cytokine responses, they did not induce metabolic abnormalities observed in Sik2 knockout mice. These results identify YKL-05-099 as a useful probe to investigate SIK function in vivo, and further support the development of SIK inhibitors for treatment of inflammatory disorders.

Salt-inducible kinases (SIK) 1–3 are serine/threonine kinases in the adenosine monophosphate-activated protein kinase (AMPK) family first recognized for their role in energy metabolism where they link G protein-coupled receptor (GPCR)/cAMP signaling to gene expression programs that increase gluconeogenesis in hepatocytes and regulate lipid metabolism in adipose tissue(1-4). Under basal conditions, SIKs phosphorylate the CREB-regulated transcriptional coactivators (CRTCs) and class IIa histone deacetylases (HDAC4, 5, 7 and 9), resulting in their cytosolic sequestration by phosphorylation-dependent interactions with 14-3-3 proteins(4, 5). Inhibitory phosphorylation of SIKs by protein kinase A (PKA) in response to elevated intracellular cAMP enables CRTCs and class IIa HDACs to enter the nucleus and coordinately regulate gene expression(6-8). As such, SIKs are critical mediators of signaling induced by hormones like glucagon or catecholamine that activate GPCRs in metabolic tissues.
More recently, SIKs have been identified as key regulators of GPCR-modulated cytokine responses in innate immune cells like macrophages and dendritic cells. For instance, PKA-dependent suppression of SIK activity is observed in innate immune cells treated with the prostanoid receptor agonist prostaglandin E2 (PGE2)(9, 10). Inhibiting SIK activity converts innate immune cells to a more tolerogenic state characterized by increased CREB-dependent expression of the anti-inflammatory cytokine interleukin-10 (IL-10), as well as reduced inflammatory cytokine expression due to deacetylation of NF-κB subunits by class IIa HDACs(9-11). Of note, directly targeting SIKs with small-molecule inhibitors recapitulates many of the immunomodulatory effects induced by elevated intracellular cAMP(12-14).
The physiological role of SIKs has been studied in knockout mice. Sik3−/− mice are born below Mendelian ratios and survivors display a malnourished phenotype, insulin handling defects, and increased sensitivity to lipopolysaccharide (LPS)-induced production of IL-6 and death(15, 16). In contrast, Sik2 knockout mice are grossly normal, but have enlarged fat cells, increased macrophage infiltration of adipose tissue, hypertriglyceridemia and decreased plasma adiponectin levels(3). Consistent with the role of adiponectin in promoting glucose utilization, Sik2−/− mice display modestly elevated blood glucose levels following glucose or insulin challenge(3). Although these studies provide insight into the physiological roles of SIKs, acutely inhibiting SIK activity with small molecules may not recapitulate these developmental and metabolic defects. To address this question, we developed SIK-targeting small-molecule inhibitors with pharmacokinetic (PK) properties suitable for use in vivo, and applied these tools to study the impact of acute SIK inhibition on specific immunomodulatory and metabolic responses regulated by these kinases.
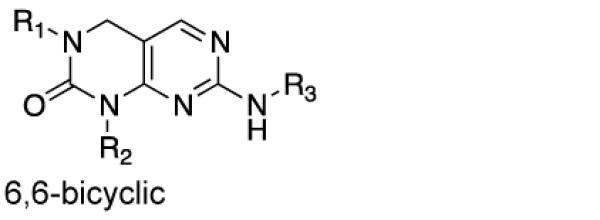
We first determined if the SIK-targeting inhibitor 1 (HG-9-91-01; Figure 1A) has PK properties amenable for use in vivo. HG-9-91-01 was > 99% serum bound and rapidly degraded by mouse liver microsomes (t1/2 = 11 min) making this compound unsuitable for direct injection into animals (Table 1). To guide efforts to develop HG-9-91-01 derivatives with improved PK properties, we docked the inhibitor to a homology model built using the available crystal structure of MARK3/Par-1 kinase domain (PDB ID: 3FE3) (Figure 1B), which shares > 60% sequence identity to the protein kinase domains of SIK1–3 (Supplementary Figure 1). This model predicts that the 4,6-diaminopyrimidine core of HG-9-91-01 forms a pseudo-bicyclic ring that orients the urea carbonyl to interact with Lys-49; this finding suggests that replacing the pseudo-bicyclic ring with a 6,6-bicyclic pyrimidopyrimidinone core (e.g., 2; Table 1), a modification expected to improve microsomal stability, might be tolerated. While 2 exhibits improved microsomal stability (t1/2 = 20 min) and retains the ability to inhibit SIK2 and enhance IL-10 production by activated bone marrow-derived dendritic cells (BMDCs), it is highly toxic in this cell type. Substitution of the 2,4-dimethoxy-phenyl side chain with a 3-methoxypyridine group (3; YKL-05-095) further increased microsomal stability (t1/2 = 27 min), but again resulted in significantly elevated toxicity. These results suggest the need to incorporate additional modifications to mitigate the toxicity observed with the more stable fused pyrimidopyrimidinone core.
Figure 1.

Molecular modeling informs development of SIK inhibitors with improved pharmacokinetic properties. A) Chemical structure of HG-9-91-01. B) HG-9-91-01 docked into a model of the protein kinase domain of SIK2. B) Chemical structure and summary of structure-activity relationship data for development of 5 (YKL-05-099). C) The activity of 141 kinases were measured in the presence of 3 (YKL-05-095; 1 μM) or 5 (YKL-05-099; 1 μM) and is reported as the percent untreated activity that remains in the presence of the inhibitor (mean, n = 2 from 1 independent experiment). Table displays kinases inhibited by YKL-05-099 (1 μM) with < 10% activity remaining (mean, n = 2 from 1 independent experiment).
Table 1.
Activity profile and microsomal stability of HG-9-91-01 derivatives. Inhibition of SIK2 (IC50) as well as effects on IL-10 production (EC50) and cellular cytotoxicity (CC50) in Zymosan A-stimulated BMDCs are presented as the mean ± SD from ≥ 2 independent experiments. Mouse liver microsome (MLM) t1/2's are from 1 independent experiment. N.D., not determined.
| Core |
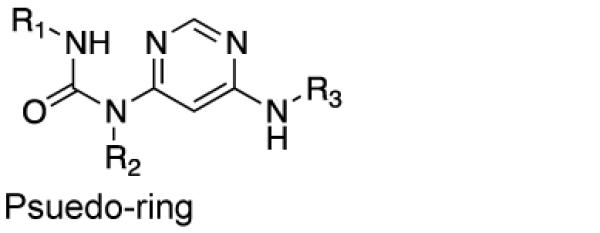
|
|
||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Core | R1 | R2 | R3 | SIK2 IC50 (nM) |
IL-10 EC50 (nM) |
Toxicit y CC50 (μM) |
MLM t1/2 (Min) |
|
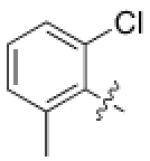
1 (HG-9-91- 01) |
Pseudo -ring |
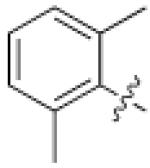
|
|
|
3.5 ± 0.5 |
220 ± 90 |
5.0 ± 2.5 |
11 |
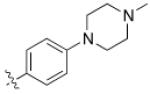
| 2 | 6,6- bicyclic |
|
|
|
19.5 ± 0.6 |
3 ± 2 | 0.007 ± 0.005 |
20 |
|
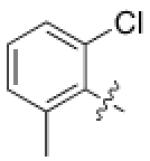
3 (YKL-05- 095) |
6,6- bicyclic |
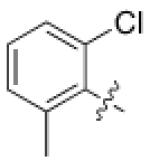
|
|
|
6 ± 3 | 11 ± 1 | 0.19 ± 0.06 |
27 |
|
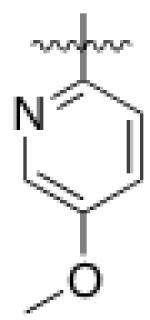
4 (YKL-05- 096) |
6,6- bicyclic |
|
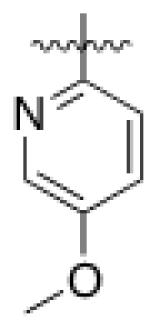
|
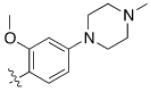
|
34 ± 14 |
70 ± 40 | 6 ± 1 | 37 |
|
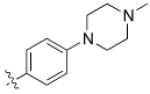
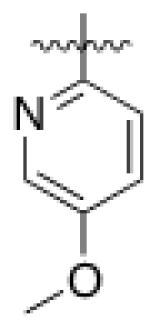
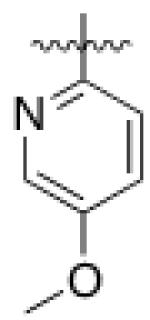
5 (YKL-05- 099) |
6,6- bicyclic |

|
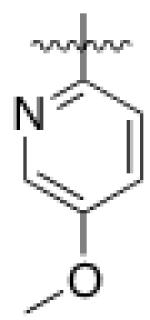
|
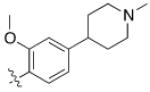
|
40 ± 25 |
460 ± 110 |
>10 | >120 |
| 6 | 6,6- bicyclic |
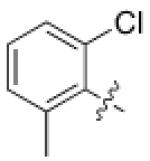
|
|
|
150 ± 20 |
>10,00 0 |
>10 | N.D. |
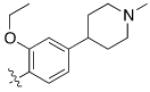
| 7 | 6,6- bicyclic |

|
|
|
>700 0 |
>10,00 0 |
>10 | N.D. |
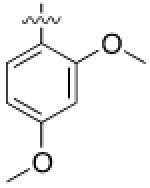
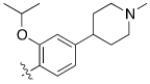
As HG-9-91-01 and its derivatives potently inhibit number of kinases including many members of the Src-family kinases(12), we hypothesized that improving the kinase selectivity towards SIKs might reduce the cellular toxicity. To accomplish this we explored substitution of 2-anilino substituent, which improved the kinase selectivity of the structurally similar Alk inhibitor ceritinib(17). Methyl ether substitution of the ortho position yielded 4 (YKL-05-096), a promising analog that retains potent SIK2-inhibitory (IC50 = 34 ± 14 nM) and IL-10-enhancing (EC50 = 70 ± 40 nM) activities while only displaying cell-based toxicity at concentrations > 5 μM (Table 1). Ethyl or isopropyl ether substitution of the ortho position of the 2-anilino substituent (analogs 6 and 7) progressively impaired SIK2-inhibitory activity (Table 1), which suggests that this site may be sterically limited in SIK2. Hence, methyl ether substitution of the aniline tail appears to effectively maintain potent SIK2-inhibitory and IL-10-potentiating activities, while mitigating toxicity associated with the fused core.
Converting the terminus of the 2-aniline substituent to a 1-methylpiperidine group yielded 5 (YKL-05-099) (Figure 1C), which has slightly less potent SIK2-inhibitory (IC50 = 40 ± 25 nM) and IL-10-enhancing activities (EC50 = 460 ± 110 nM), but is non-toxic at concentrations ≤ 10 μM and stable in mouse liver microsomes for > 2 hours (Table 1). In addition, YKL-05-099 is highly soluble (PBS solubility = 428 ± 11 μM) and present in an unbound state at appreciable levels in mouse plasma (Free fraction = 6 ± 1%). Consistent with the observations for ceritinib, methyl ether substitution of YKL-05-099’s aniline tail improved kinase selectivity for SIK2 and 3 relative to the unsubstituted analog YKL-05-095 (Figure 1D and Table S1), and overall selectivity against a panel of 468 kinases (Table S2). In addition, we confirmed that YKL-05-099 binds to SIK1 and SIK3 with IC50’s ~10 and ~30 nM, respectively, in a competitive binding assay. The activity profile, in vitro PK properties, and improved SIK kinase selectivity highlights YKL-05-099 as a promising probe for exploring the functional consequences of SIK inhibition in cells and in vivo.
We next determined whether, in addition to up-regulating IL-10 (Figure 2A), YKL-05-099 induces cell-based phenotypes consistent with SIK inhibition. First, we found that pre-incubating bone marrow-derived macrophages (BMDMs) with YKL-05-099 reduced LPS stimulated phosphorylation of HDAC5 at the SIK-specific phosphorylation site Ser259(7, 8) (Figure 2B). Given that potentiation of CREB activity is a known consequence of inhibiting SIKs in many cell types(2, 9, 12, 13), we asked whether YKL-05-099 up-regulates expression of the CREB target genes Il10 and Nurr77(9). Indeed, levels of both transcripts were increased by pre-incubation of BMDCs with YKL-05-099 prior to LPS stimulation (Figure 2C). Finally, as observed for other SIK inhibitors(12-14), YKL-05-099 suppressed production of the inflammatory cytokines TNFα, IL-6 and IL-12p40, and only modestly enhanced IL-1β release in BMDCs stimulated with the yeast cell wall extract Zymosan A (Figure 2D). Collectively, these data indicate that YKL-05-099 affects responses previously linked to SIK inhibition in activated innate immune cells.
Figure 2.

YKL-05-099 displays cell-based activities consistent with SIK inhibition. A) Pre-treatment with the indicated concentrations of YKL-05-099 for 24 hr potentiates IL-10 production by Zymosan A-stimulated BMDCs (mean ± SD; n = 3 from 1 independent experiment; Quantified as % of the response elicited by PGE2). B) Pre-incubation of BMDMs with YKL-05-099 (1 μM) or HG-9-91-01 (1 μM) for 6 hr prior to stimulation with LPS for 30 min reduces p-HDAC5 (Ser259) levels. Ratio of p-HDAC5 / HDAC5 quantified as mean ± SD, n = 2 from 1 independent experiment. C) Effect of pre-treatment with YKL-05-099 (1 μM) for 24 hr on Il10 and Nurr77 transcripts in BMDCs stimulated with LPS for the indicated time points (mean ± SD, n = 6 from 1 independent experiment). D) Pre-incubation of BMDCs with YKL-05-099 (1 μM) for 24 hr modulates inflammatory cytokine production induced by stimulation with Zymosan A for 18 hr (mean ± SD, n = 3 from 1 independent experiment). ***, P < 0.001; **, P < 0.01 using unpaired Student’s t tests. All data are representative of ≥ 2 independent experiments.
Given that YKL-05-099 has biochemical and cell-based activities consistent with SIK inhibition and favorable in vitro PK properties, we next asked whether well-tolerated doses of YKL-05-099 achieve free serum concentrations sufficient to inhibit SIKs in vivo. Following IP administration at 20 mg/Kg, YKL-05-099 is rapidly absorbed followed by slow clearance (t1/2 > 7 hour) such that serum concentrations exceeding the IC50 for SIK2 inhibition are maintained for > 16 hour (Figure S2A). Of note, no obvious changes in viability, activity or grooming were induced by YKL-05-099 during the 24 hour treatment period. To assess SIK engagement in vivo, we pre-treated mice with 5 – 50 mg/Kg YKL-05-099 for 15 min prior to stimulation with LPS for 1 hour, and then generated lysates from total splenic leukocytes. In this experiment, YKL-05-099 dose dependently decreased phosphorylation of HDAC5 at the SIK-regulated site Ser259; reduced phosphorylation was observed at the lowest dose (5 mg/Kg) and was below the limit of detection by immunoblotting beginning at the 20 mg/Kg dose (Figure 3A). While total HDAC5 levels are reduced by the higher doses of YKL-05-099, normalizing the change in HDAC5 phosphorylation at Ser259 to total HDAC5 confirms the YKL-05-099-induced decrease in this phosphorylation event (Figure S2B). These data suggest that well tolerated doses of YKL-05-099 are able to inhibit SIKs in vivo.
Figure 3.

YKL-05-099 modulates inflammatory cytokine responses in vivo. A) and B) IP administration of the indicated doses of YKL-05-099 for 15 min prior to stimulation with LPS (0.5 mg/Kg) for 1 hr reduces p-HDAC5 (Ser259) levels in total splenic leukocytes and modulates serum IL-10 and TNFα levels (bars = mean; n = 3 mice; data is representative of 2 independent experiments). ***, P < 0.001; *, P < 0.05 using two-way ANOVA with Dunnett post-test. C) and D) IP administration of YKL-05-099 (20 mg/Kg) for 15 min prior to stimulation with LPS (0.5 mg/Kg) modulates serum IL-10 and TNFα abundance and colonic mRNA levels without affecting IL-6 responses (mean ± SD; n = 3 mice per time point; data is from 1 independent experiment). ***, P < 0.001; **, P < 0.05; *, P < 0.05 using unpaired Student’s t tests.
To determine whether SIK inhibition affects systemic inflammatory responses in vivo, we quantified serum IL-10 and TNFα levels from mice briefly pre-treated with YKL-05-099 prior to stimulation with LPS for 1 hour. Consistent with the cellular consequences of SIK inhibition, YKL-05-099 dose-dependently reduced abundance of TNFα in serum beginning at 5 mg/Kg, and increased IL-10 levels at the 20 mg/Kg dose by > 2-fold (Figure 3B). The differential effects of 5 mg/Kg YKL-05-099 on TNFα suppression versus IL-10 potentiation may reflect different sensitivities of class IIa HDACs versus CRTC proteins to SIK inhibition. In a separate experiment, we found that YKL-05-099 up-regulated IL-10 and reduced TNFα in serum from LPS-stimulated mice for up to 6 hours, suggesting its immunomodulatory effects are sustained over time (Figure 3C). Of note, treatment with YKL-05-099 moderately decreased serum IL-6 levels (Figure 3C), which contrasts with the reported effects of LPS-stimulation in Sik3 KO mice(16). This contrasting phenotype may reflect the difference between pan-SIK inhibition by YKL-05-099 in our experiment versus Sik3-specific deletion in the genetic knockout model in which SIK1 and 2 may compensate for loss of SIK3. In addition to serum measurements, we sought to determine whether SIK inhibition alters cytokine responses in disease-relevant tissues by quantifying abundance of Il10, Tnf and Il6 mRNA in the colon of mice treated as above. As observed for serum cytokine levels, YKL-05-099 pre-treatment significantly enhanced the LPS-induced increase in Il10 mRNA at 1 hour, while Tnf transcript was reduced throughout the experiment (Figure 3D). Again, the LPS-induced increase in Il6 mRNA was not affected by YKL-05-099 (Figure 3D).
As a first step to determining whether acutely inhibiting SIKs with small molecules yields similar metabolic phenotypes as chronic SIK inhibition by genetic deletion, we treated mice with 20 mg/Kg YKL-05-099 daily for 1 week and we observed no changes in weight (Figure S3A). At the end of the treatment course, we measured fasting levels of serum triglycerides as well as high- and low-molecular-weight adiponectin. In contrast to Sik2−/− animals(3), daily dosing with YKL-05-099 (20 mg/Kg, IP) for 1 week did not alter serum levels of these adipocyte-derived hormones and metabolites (Figure S3B and S3C).
In this report, we describe optimization of the SIK-targeting inhibitor HG-9-91-01 to generate analogs suitable for use in animals. Docking HG-9-91-01 into a model of the SIK protein kinase domain based on the closely related AMPK-family member MARK3/Par-1 enabled these efforts by: 1) suggesting that HG-9-91-01’s core could be substituted with a fused pyrimidopyrimidinone in its bound conformation, and 2) identifying the ortho position on the aniline tail as a solvent-exposed site that could be substituted to improve SIK kinase selectivity. Applying these insights yielded YKL-05-099, a pan-SIK inhibitor that displays activity consistent with SIK inhibition (i.e., reduced SIK-specific phosphorylation of HDAC5) in cells and following direct injection into animals. As such, YKL-05-099 and related compounds may represent useful probes for study of SIK function in vivo.
In culture, SIK inhibition converts activated innate immune cells to an anti-inflammatory phenotype marked by up-regulation of IL-10 and reduced production of several inflammatory cytokines including TNFα(12, 13). Our data in mice pre-treated with YKL-05-099 prior to LPS stimulation indicates that SIK inhibition similarly affects inflammatory responses in vivo. In particular, the ability of YKL-05-099 to potentiate IL-10 and suppress TNFα in the colon argues for exploration of SIK inhibitors for treatment of inflammatory disorders affecting the gastrointestinal tract such as Crohn’s disease(18). However, because aberrant production of IL-10 and TNFα are hallmarks of other autoimmune and auto-inflammatory disorders such as rheumatoid arthritis, psoriasis and psoriatic arthritis, SIK inhibition may be a broadly applicable therapeutic strategy(19, 20).
A potential liability for therapeutic development of SIK inhibitors is the role of these kinases in glucose and fat metabolism, adrenal steroidogenesis as well as regulation of microtubule function(2, 3, 21, 22). In a preliminary experiment, daily injections of YKL-05-099 in wild-type mice for 1 week was well tolerated and did not acutely recapitulate the effects of Sik2 deletion on serum triglyceride and adiponectin levels. At the 20 mg/Kg dose, free concentrations of YKL-05-099 are predicted to exceed the IC50 for SIK inhibition for > 16 hours, such that SIK activity may adequately recover between doses to normalize lipid handling in adipocytes. Nevertheless, this data suggests the possibility of a therapeutic window that would allow for repeated dosing of SIK inhibitors to achieve the desirable immunomodulatory effects while preserving normal glucose metabolism. Further studies with in vivo active SIK inhibitors like YKL-05-099 will be critical to fully elucidate the physiological consequences of targeting this family of kinases.
METHODS
In vivo methods
All animal studies were conducted under protocols approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital. YKL-05-099 was diluted in 5% N-methyl-2-pyrrolidinone, 5% Solutol HS15 and 90% normal saline and administered IP to male 8–10 week-old C57BL/6 mice. Serum and tissue samples were collected after euthanizing mice by CO2 inhalation overdose followed by cervical dislocation. Details on the animal handling and tissue collection procedures are available in the Supplementary Information.
Cell culture and biochemical assays
Bone marrow-derived DCs or macrophages were differentiated from C57BL/6 bone marrow in the presence of GM-CSF-conditioned media or recombinant M-CSF, respectively. Inhibition of purified, recombinant hSIK2 was measured using a Caliper-based kinase activity assay (Perkin Elmer). Concentrations of IL-10, TNF-α, IL-6, IL-12p40 and IL-1β in culture media or serum were detected using a FlexSet Cytometric Bead Array assay (BD Biosciences). Alternatively, IL-10 secretion was quantified using an AlphaLISA assay (PerkinElmer). qPCR was conducted using iTaq Universal SYBR Green Supermix (Bio-Rad) on total RNA prepared from cultured cells or distal colon sections. Details on reagents, chemical synthesis and full experimental protocols are available in the Supplementary Information.
Supplementary Material
AWKNOWLEDGEMENTS
We thank the Broad Compound Management and Analytical Chemistry Groups for providing compound plates and conducting in vitro PK studies, respectively. We acknowledge the Nancy Lurie Marks Medicinal Chemistry Core for assistance with compound preparation. We thank P. Cohen and K. Clark (MRC Protein Phosphorylation and Ubiquitylation Unit, University of Dundee) for advice and kinase activity profiling. Lastly, we thank K. Conway and N. Desch (Massachusetts General Hospital) for expert advice on design of the animal studies.
FUNDING SOURCES
Research reported in this publication was supported by funding from the: National Institutes of Health Grants K08DK104021 (B.K.), U01DK062432 and P30DK043351 (R.J.X.), and R01AI095499 (N.S.G.); Michael J. Fox Foundation for Parkinson’s Research (N.S.G); Claudia Adams Barr Program for Innovative Cancer Research (N.S.G); Crohn’s & Colitis Foundation of America Grant 500229 (to R.J.X.); and The Leona M. and Harry B. Helmsley Charitable Trust Grant 500203 (to S.L.S. and R.J.X.). S.L.S. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
- Supplementary Tables 1 and 2
- Supplementary Figures 1 and 2
- Detailed description of synthetic chemistry, compound characterization and biological methodology
REFERENCES
- 1.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel K, Foretz M, Marion A, Campbell DG, Gourlay R, Boudaba N, Tournier E, Titchenell P, Peggie M, Deak M, Wan M, Kaestner KH, Goransson O, Viollet B, Gray NS, Birnbaum MJ, Sutherland C, Sakamoto K. The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat. Commun. 2014;5 doi: 10.1038/ncomms5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J, Yoon YS, Han HS, Kim YH, Ogawa Y, Park KG, Lee CH, Kim ST, Koo SH. SIK2 Is Critical in the Regulation of Lipid Homeostasis and Adipogenesis In Vivo. Diabetes. 2014;63:3659–3673. doi: 10.2337/db13-1423. [DOI] [PubMed] [Google Scholar]
- 4.Henriksson E, Sall J, Gormand A, Wasserstrom S, Morrice NA, Fritzen AM, Foretz M, Campbell DG, Sakamoto K, Ekelund M, Degerman E, Stenkula KG, Goransson O. SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. J. Cell Sci. 2015;128:472–486. doi: 10.1242/jcs.153932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walkinshaw DR, Weist R, Kim GW, You LY, Xiao L, Nie JY, Li CS, Zhao SP, Xu MH, Yang XJ. The Tumor Suppressor Kinase LKB1 Activates the Downstream Kinases SIK2 and SIK3 to Stimulate Nuclear Export of Class IIa Histone Deacetylases. J. Biol. Chem. 2013;288:9345–9362. doi: 10.1074/jbc.M113.456996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henriksson E, Jones HA, Patel K, Peggie M, Morrice N, Sakamoto K, Goransson O. The AMPK-related kinase SIK2 is regulated by cAMP via phosphorylation at Ser358 in adipocytes. Biochem. J. 2012;444:503–514. doi: 10.1042/BJ20111932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walkinshaw DR, Weist R, Kim GW, You L, Xiao L, Nie J, Li CS, Zhao S, Xu M, Yang XJ. The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J. Biol. Chem. 2013;288:9345–9362. doi: 10.1074/jbc.M113.456996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacKenzie KF, Clark K, Naqvi S, McGuire VA, Noehren G, Kristariyanto Y, van den Bosch M, Mudaliar M, McCarthy PC, Pattison MJ, Pedrioli PG, Barton GJ, Toth R, Prescott A, Arthur JS. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J. Immunol. 2013;190:565–577. doi: 10.4049/jimmunol.1202462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc. Natl. Acad. Sci. U. S. A. 2015;112:15642–15647. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luan B, Goodarzi MO, Phillips NG, Guo XQ, Chen YDI, Yao J, Allison M, Rotter JI, Shaw R, Montminy M. Leptin-Mediated Increases in Catecholamine Signaling Reduce Adipose Tissue Inflammation via Activation of Macrophage HDAC4. Cell Metab. 2014;19:1058–1065. doi: 10.1016/j.cmet.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark K, MacKenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, Peggie M, Plater L, Pedrioli PG, McIver E, Gray NS, Arthur JS, Cohen P. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sundberg TB, Choi HG, Song JH, Russell CN, Hussain MM, Graham DB, Khor B, Gagnon J, O'Connell DJ, Narayan K, Dancik V, Perez JR, Reinecker HC, Gray NS, Schreiber SL, Xavier RJ, Shamji AF. Small-molecule screening identifies inhibition of salt-inducible kinases as a therapeutic strategy to enhance immunoregulatory functions of dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 2014;111:12468–12473. doi: 10.1073/pnas.1412308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozanne J, Prescott AR, Clark K. The clinically approved drugs dasatinib and bosutinib induce anti-inflammatory macrophages by inhibiting the salt-inducible kinases. Biochem. J. 2015;465:271–279. doi: 10.1042/BJ20141165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uebi T, Itoh Y, Hatano O, Kumagai A, Sanosaka M, Sasaki T, Sasagawa S, Doi J, Tatsumi K, Mitamura K, Morii E, Aozasa K, Kawamura T, Okumura M, Nakae J, Takikawa H, Fukusato T, Koura M, Nish M, Hamsten A, Silveira A, Bertorello AM, Kitagawa K, Nagaoka Y, Kawahara H, Tomonaga T, Naka T, Ikegawa S, Tsumaki N, Matsuda J, Takemori H. Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS One. 2012;7:e37803. doi: 10.1371/journal.pone.0037803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanosaka M, Fujimoto M, Ohkawara T, Nagatake T, Itoh Y, Kagawa M, Kumagai A, Fuchino H, Kunisawa J, Naka T, Takemori H. Salt-inducible kinase 3 deficiency exacerbates lipopolysaccharide-induced endotoxin shock accompanied by increased levels of pro-inflammatory molecules in mice. Immunology. 2015;145:268–278. doi: 10.1111/imm.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw AT, Kim DW, Mehra R, Tan DSW, Felip E, Chow LQM, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, Solomon BJ, Wolf J, Thomas M, Schuler M, Liu G, Santoro A, Lau YY, Goldwasser M, Boral AL, Engelman JA. Ceritinib in ALK-Rearranged Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, Muise AM, Snapper SB. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv. Immunol. 2014;122:177–210. doi: 10.1016/B978-0-12-800267-4.00005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Souto A, Gomez-Reino JJ. Apremilast for the treatment of psoriatic arthritis. Expert Rev. Clin. Immunol. 2015;11:1281–1290. doi: 10.1586/1744666X.2015.1102057. [DOI] [PubMed] [Google Scholar]
- 20.Kopf M, Bachmann MF, Marsland BJ. Averting inflammation by targeting the cytokine environment. Nat. Rev. Drug Discovery. 2010;9:703–718. doi: 10.1038/nrd2805. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed AA, Lu Z, Jennings NB, Etemadmoghadam D, Capalbo L, Jacamo RO, Barbosa-Morais N, Le XF, Vivas-Mejia P, Lopez-Berestein G, Grandjean G, Bartholomeusz G, Liao W, Andreeff M, Bowtell D, Glover DM, Sood AK, Bast RC, Grp AOCS. SIK2 Is a Centrosome Kinase Required for Bipolar Mitotic Spindle Formation that Provides a Potential Target for Therapy in Ovarian Cancer. Cancer Cell. 2010;18:109–121. doi: 10.1016/j.ccr.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takemori H, Katoh Y, Horike N, Doi J, Okamoto M. ACTH-induced nucleocytoplasmic translocation of salt-inducible kinase. Implication in the protein kinase A-activated gene transcription in mouse adrenocortical tumor cells. J. Biol. Chem. 2002;277:42334–42343. doi: 10.1074/jbc.M204602200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.