

Abstract
Biliary atresia, the most common indication for pediatric liver transplantation, is a fibrotic disease of unknown etiology affecting the extrahepatic bile ducts of newborns. The recently described toxin biliatresone causes lumen obstruction in mouse cholangiocyte spheroids and represents a new model of biliary atresia. The goal of this study was to determine the cellular changes caused by biliatresone in mammalian cells that ultimately lead to biliary atresia and extrahepatic fibrosis. We treated mouse cholangiocytes in three‐dimensional (3D) spheroid culture and neonatal extrahepatic duct explants with biliatresone and compounds that regulate glutathione (GSH). We examined the effects of biliatresone on SOX17 levels and determined the effects of Sox17 knockdown on cholangiocytes in 3D culture. We found that biliatresone caused disruption of cholangiocyte apical polarity and loss of monolayer integrity. Spheroids treated with biliatresone had increased permeability as shown by rhodamine efflux within 5 hours compared with untreated spheroids, which retained rhodamine for longer than 12 hours. Neonatal bile duct explants treated with the toxin showed lumen obstruction with increased subepithelial staining for α‐smooth muscle actin and collagen, consistent with fibrosis. Biliatresone caused a rapid and transient decrease in GSH, which was both necessary and sufficient to mediate its effects in cholangiocyte spheroid and bile duct explant systems. It also caused a significant decrease in cholangiocyte levels of SOX17, and Sox17 knockdown in cholangiocyte spheroids mimicked the effects of biliatresone. Conclusion: Biliatresone decreases GSH and SOX17 in mouse cholangiocytes. In 3D cell systems, this leads to cholangiocyte monolayer damage and increased permeability; in extrahepatic bile duct explants, it leads to disruption of the extrahepatic biliary tree and subepithelial fibrosis. This mechanism may be important in understanding human biliary atresia. (Hepatology 2016;64:880‐893)
Abbreviations
- 2D
two‐dimensional
- 3D
three‐dimensional
- BA
biliary atresia
- BSO
DL‐buthionine sulfoximine
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DMSO
dimethyl sulfoxide
- D‐NAC
N‐acetyl‐d‐cysteine
- EHBD
extrahepatic bile duct
- GSH
glutathione
- L‐NAC
N‐acetyl‐l‐cysteine
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- siRNA
small interfering RNA
- TUNEL
terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling
- α‐SMA
α‐smooth muscle actin
Biliary atresia (BA) causes rapidly progressive liver fibrosis and cirrhosis in neonates and is the primary indication for liver transplantation in children.1 Unlike in most other forms of biliary fibrosis, the initial injury in biliary atresia is most severe in the extrahepatic bile ducts (EHBDs), which appear to undergo damage when fully formed, becoming completely obstructed and impeding bile flow. Surgery directly connecting the liver hilum to the bowel (i.e., Kasai portoenterostomy) slows the progression of fibrosis in some children, demonstrating that the critical defect in BA is EHBD obstruction. BA occurs in 1:8000‐1:15,000 neonates, but its etiology is poorly understood; familial cases are rare, and there is only limited evidence that genetic factors can cause BA. Nonrandom clustering of BA cases in space and time suggests that infections or toxins2, 3, 4 may contribute to disease pathogenesis. Whereas recent data strongly suggest that the insult to the EHBDs is prenatal,5, 6 the nature of the insult and the associated cholangiocyte damage are unknown. If the nongenetic factors that cause BA could be identified, prevention might be possible. Similarly, a better understanding of the cellular mechanisms of BA might lead for the first time to targeted therapies.
Several animal models of BA partially recapitulate the human phenotype, but all have significant limitations. The most commonly used model, infection of newborn BALB/c mice with rhesus rotavirus,3 has many histological and biochemical features characteristic of extrahepatic BA, but there is no definitive evidence that links rotavirus infection to human BA.7 Neonatal rat bile duct ligation also results in liver parenchymal disease similar to human BA,8, 9 but the model is technically difficult and surgical injury to EHBDs makes it difficult to extrapolate the resulting extrahepatic pathology to human BA. Most interestingly, Sox17 haploinsufficient mice in certain backgrounds develop a perinatal BA‐like disease and hepatitis, suggesting that reduced SOX17 levels predispose to BA10 and raising the possibility that nongenetic factors causing such reductions in the EHBD of neonates could lead to BA.
In 1990, Australian veterinary scientists reported that outbreaks of a BA‐like disease had occurred in newborn lambs in New South Wales in 1964 and 1988, both years being notable for severe droughts and the grazing of pregnant livestock on atypical flora.11, 12 Based on this work, we hypothesized that neonatal livestock were poisoned by a plant toxin ingested by their mothers. We recently reported the purification of a previously undescribed isoflavonoid, termed biliatresone, from the suspect Australian plants (Dysphania species), and demonstrated that this toxin causes a BA‐like syndrome in larval zebrafish, with selective destruction of the EHBDs.13 In mouse cholangiocytes but not hepatocytes, biliatresone also caused destabilization of microtubules, and in cholangiocyte spheroids in three‐dimensional (3D) culture, biliatresone disrupted apical polarity and caused luminal obstruction, mimicking the pathology seen in the EHBDs of BA patients.13
How biliatresone caused selective injury to the EHBDs was not obvious from our initial studies. This is a critical point: because it is unlikely that pregnant women ingest biliatresone, defining the molecular mechanisms underlying the BA‐like disease in zebrafish and livestock might lead to the identification of potential human teratogens that work by way of similar mechanisms to cause BA. We observed that biliatresone reacted strongly with glutathione (GSH). Because reduced levels of GSH cause microtubule destabilization in some cell lines,14, 15, 16, 17 and since microtubules are essential to the maintenance of apical polarity,18 we hypothesized that biliatresone acts by lowering GSH levels, resulting in microtubule and polarity abnormalities, loss of cholangiocyte monolayer integrity, and extravasation of toxin and bile into the periepithelial space. Remarkably, we found that biliatresone causes transient decreases in GSH and in SOX17 in cholangiocytes in culture, and that reduced GSH is both necessary and sufficient for cholangiocyte injury and subepithelial fibrosis in neonatal EHBD explants. These observations suggest that any maternal toxin or stressor that reduces GSH and is excreted in neonatal bile could lead to EHBD injury.
Materials and Methods
CELL CULTURE
Unless noted otherwise, a small cholangiocyte cell line was used for spheroid culture as described previously.19 Primary neonatal extrahepatic cholangiocytes were isolated by outgrowth from 0‐ to 3‐day‐old BALB/c mouse pup bile ducts as described previously and were used for spheroids when specified.20 Primary cells were used at passages 1‐3 for all experiments. Cells from a single litter of pups (with at least five animals) were used for each individual experiment with neonatal cholangiocytes. All animal experiments were performed in accordance with National Institutes of Health policy and were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Primary mouse pancreatic ductal cells were obtained from Basil Bakir and Anil Rustgi (University of Pennsylvania) and cultured in 3D as described.21 Mammary epithelial MCF‐10A cells were obtained from Mauricio Reginato (Drexel University) and kidney epithelial IMCD3 cells were obtained from Katalin Susztak (University of Pennsylvania). Both cell types were cultured in 3D in a manner similar to that done for cholangiocytes. Primary mouse enterocytes were obtained from Mary Ann Crissey and John Lynch (University of Pennsylvania) and cultured in 3D as described previously.22, 23
BILIATRESONE AND COMPOUND TREATMENTS
Biliatresone was isolated as described previously13, 24 from Dysphania species plants harvested in Australia in 2008 and shipped frozen to the United States. Cells were treated with vehicle (dimethyl sulfoxide [DMSO]) or biliatresone at 2 μg/mL for up to 24 hours and with DL‐buthionine sulfoximine (BSO; 100 μM, Sigma, catalog #19176), N‐acetyl‐l‐cysteine (L‐NAC; 5 μM, Sigma, catalog #A9165), N‐acetyl‐d‐cysteine (D‐NAC; 5 μM, Princeton BioMolecular Research, catalog #117600), and sulforaphane (20 μM, Sigma, catalog #S4441) as described in the text.
Spheroid Culture and Analysis
Primary neonatal mouse extrahepatic cholangiocytes and the small cholangiocyte cell line were cultured in 3D in a collagen‐Matrigel mixture as described previously.13 Cholangiocytes in 3D culture replicate, polarize, and form hollow spheroids with apical markers on the luminal side and basolateral markers on the external side after 7 or 8 days (Supporting Fig. 1). Spheroids were used for experiments at day 8 after plating. After treatments, cells were fixed with 4% formalin and stained for F‐actin (1:1000; phalloidin‐tetramethylrhodamine B isothiocyanate; Santa Cruz Biotechnology, catalog #301530) or immunostained with antibodies against the integrin β‐1 subunit (1:100; Abcam, catalog #95623), E‐cadherin (1:100; Cell Signaling, catalog #24E10 3195S), ZO‐1 (1:50; Invitrogen, catalog #40‐2200), cellular tubulin (α‐tubulin 1:500; Sigma‐Aldrich, catalog #T9026), and Ki67 (1:200; Abcam, catalog #ab16667); the spheroids were imaged at the times noted. In some cases, spheroids were treated with Sox17 small interfering RNA (siRNA; Qiagen, catalog #SI01429533) or scrambled siRNA (Qiagen, catalog #1027281) as directed by the manufacturer on day 7 after plating and were evaluated on day 9. Images were obtained using a confocal microscope with ×40 magnification (Leica). Photographs were taken at the level of the midsection of each spheroid, where the luminal diameter was greatest. Inserts had between 20 and 80 spheroids for all experiments, and a minimum of five photographs were taken (representative images are shown in the figures).
For biliatresone washout experiments, spheroids were treated with biliatresone for 24 hours followed by washes and a change to media without biliatresone for an additional 24 hours. For quantification of the effect of the different compounds on spheroid lumens, spheroids were stained for F‐actin, and multiple confocal images were acquired and analyzed by two independent observers (O.W.‐Z and H.K). Lumens were assessed as being open, closed (no lumen visible), or partially open or narrowed (small visible lumen). Only spheroids imaged at a level with a clearly visible midsection were counted.
Neonatal Bile Duct Explant Culture
Intact EHBDs were isolated from 0‐ to 3‐day‐old BALB/c mice according to a protocol modified from that described previously.20 Instead of embedding the ducts in a collagen layer, ducts were placed in ice‐cold V‐7 cold preservation buffer (Vitron) followed by incubation with cholangiocyte media, for 30 minutes at 37°C. Biliatresone or other compounds as noted were added, and ducts were then placed on roller inserts and cultured at 37°C in 95% O2/5% CO2 in a Vitron Dynamic Organ Culture Incubator for 1‐3 days. Ducts were stained as described previously25 using antibodies against the cholangiocyte marker keratin 19 (K19, 1:10, Developmental Studies Hybridoma Bank, TROMAIII), α‐smooth muscle actin (α‐SMA; 1:1000, Sigma, catalog #a2547), collagen I (1:200; Southern Biotech, catalog #1310‐01), and EDA fibronectin (1:1000; Santa Cruz Biotechnology, catalog #sc‐59826). Images were taken on a confocal microscope (Leica) at ×40 magnification. For the terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) assay, bile ducts were embedded in paraffin tissue and sectioned. The TUNEL stain, with a DNase‐treated positive control, was performed per the manufacturer's instructions (Millipore, catalog #S7110).
GSH MEASUREMENT
GSH in cells was measured with the GSH‐Glo Glutathione Assay (Promega). Cells were harvested by centrifugation, washed, resuspended, and diluted in phosphate‐buffered saline (PBS; 4000‐8000 cells per well). Tissue extracts (from mouse liver and bile ducts) were washed with PBS and homogenized in PBS with ethylene diamine tetraacetic acid (2 mM). The assay was performed per the manufacturer's protocol. Luminescence measurements (RLU) were performed with the GloMax multi‐detection system (Promega). Net GSH‐dependent luminescence (net RLU) was calculated by subtracting the average luminescence of the negative control reactions (PBS). Detection of GSH by the kit is not disrupted by the interaction between biliatresone and GSH (Supporting Fig. 2).
Rhodamine Efflux Assay
Assay of rhodamine efflux from spheroids, a measure of monolayer permeability, was modified from a published protocol.26 On day 8, spheroids were incubated with rhodamine 123 (100 μM; Sigma) for 15 minutes followed by five washes with phenol‐free DMEM (Invitrogen) and were then treated with either biliatresone or DMSO; live cells were imaged every 20 minutes 1‐12 hours after treatment using a spinning disk confocal microscope with a wide ×40 lens. Permeability was quantified by examining 17 biliatresone‐treated spheroids and 18 DMSO‐treated spheroids imaged over the 12‐hour span with visual determination of when greater than 50% of the rhodamine was lost from the lumen. An unpaired t test was used to compare the two groups.
Real‐time polymerase chain reaction (PCR) and western blotting were performed for the small cholangiocyte cell line treated with vehicle (DMSO) or biliatresone at 2 μg/mL for 24 hours and Sox17 siRNA and scrambled siRNA (Qiagen) for 48 hours. Quantitative real‐time PCR was performed with a Qiagen kit with the primers RPS12 (forward, ACGTCAACACTGCTCTACAAG; reverse, CATCACAGTTGGATGCAAGC); and Sox17 (forward, CTCGGGGATGTAAAGGTGAA; reverse, GCTTCTCTGCCAAGGTCAAC), by the relative standard curve method. RPS12 was used as a control. For all experiments, there were two technical replicates, and experiments were repeated three times. For western blots, cells were lysed in radioimmunoprecipitation assay lysis buffer (Sigma, catalog #R0278) and 50 μg of protein from each sample was separated via sodium dodecyl sulfate‐polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane (0.2 μm; Bio‐Rad, catalog #162‐0176), blocked with an Odyssey blocking buffer (Odyssey, catalog #927‐40000), and incubated with primary antibodies against SOX17 (1:1000, R&D Systems, catalog #AF1924) and glyceraldehyde 3‐phosphate dehydrogenase (1:1000, Cell Signaling, catalog #2118) as a housekeeping gene. Signals were detected using florescence on a Gel Doc system.
Statistics
Statistical significance was calculated by either the one‐tailed Student t test or one‐way analysis of variance. All experiments were done with at least two technical replicates and repeated at least three times.
Results
In our zebrafish model of biliatresone‐induced BA, the intrahepatic bile ducts were spared, whereas the EHBD were destroyed.13 Similarly, whereas previous work showed that symptomatic neonatal livestock with BA‐like disease had cirrhosis,11, 12 in the present study, minimally symptomatic animals necropsied shortly after birth had marked obstruction and fibrosis of the extrahepatic bile ducts, but few changes within the liver parenchyma (Supporting Fig. 3). This result suggests that the BA‐like disease in lambs, which ultimately results in cirrhosis, begins with injury to the EHBDs.
We were interested, therefore, in how biliatresone affects cholangiocytes, and whether its effects are specific for these cells. Our published data show that secretion of biliatresone into bile is required for toxicity.13 Consistent with localization in bile (as opposed to cholangiocyte‐specific mechanisms of injury) as an explanation for selective biliary toxicity, we found that both intrahepatic and extrahepatic cholangiocytes—as well as multiple other ductal epithelial cells (but not enterocytes) cultured as spheroids—were equally sensitive to biliatresone, demonstrating monolayer disruption, lumen obstruction, and loss of the apical F‐actin ring13 (Fig. 1), similar to the pathology observed in the EHBDs of BA patients.27 Spheroid abnormalities were not associated with changes in cholangiocyte proliferation (Supporting Fig. 4A) and were reversible after biliatresone washout (Supporting Fig. 5), suggesting that biliatresone did not primarily target cholangiocyte viability.
Figure 1.
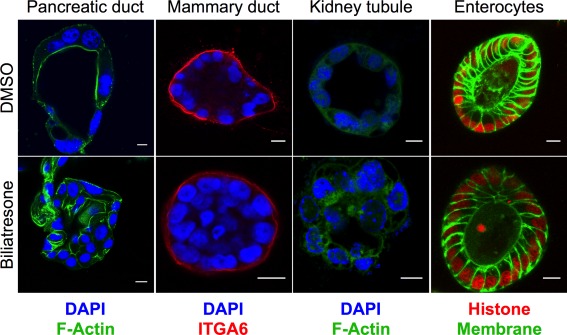
Multiple ductular cells are sensitive to biliatresone. Primary mouse pancreatic ductal cells, MCF‐10A mammary ductal epithelial cells, IMCD3 kidney tubule epithelial cells, and primary mouse enterocytes were cultured in 3D until they formed spheroids and were then treated with vehicle (DMSO) or biliatresone for 24 hours and stained as noted for F‐actin (green) or integrin α6 (ITGA6; red). Enterocytes from transgenic mice were labeled with green fluorescent protein (membrane targeted; green) and mCherry (labeled histone; red). Nuclei in pancreatic ductal cells, mammary ductal cells, and kidney tubule cells were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). Treatment of all cell types except enterocytes with biliatresone resulted in loss of the spheroid monolayer and lumens, similar to what was observed for cholangiocytes. Images are representative of three independent experiments, each with two technical replicates.
Lumen obstruction and changes in F‐actin in spheroids are associated with mislocalization of the apical markers ZO‐1 and E‐cadherin (Fig. 2). Time course studies demonstrated that this follows decreased staining for cellular tubulin, which likely reflects destabilization or depolymerization of microtubules (with the decreased staining a function of fixating and permeabilization); reduced tubulin staining is observed within 3 hours of biliatresone treatment, whereas changes in apical polarity markers and obstruction of spheroid lumens are not seen until 6‐12 hours of treatment (Fig. 2C).
Figure 2.
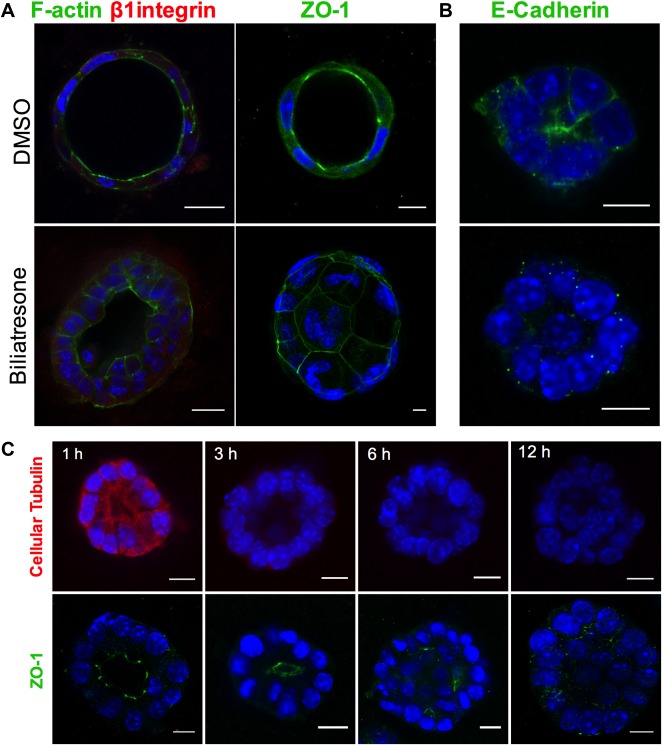
Biliatresone causes destabilization of cellular tubulin and loss of cholangiocyte polarity. (A) Primary neonatal mouse cholangiocytes in spheroid culture were treated with vehicle (DMSO) or biliatresone for 24 hours and immunostained for F‐actin (green), the β1 integrin subunit (red), or ZO‐1 (green) as indicated. (B) A cholangiocyte cell line was treated similarly and immunostained for E‐cadherin (green). (C) Cholangiocyte cell line spheroids were treated with biliatresone for 1‐12 hours and immunostained for cellular tubulin (red) or ZO‐1 (green). In all cases, nuclei were stained with DAPI (blue). Scale bars: 10 μm. Images are representative of three independent experiments, each of which was performed in duplicate.
To determine whether changes in cell‐cell adhesion markers and loss of polarity are associated with increased epithelial permeability, we assayed rhodamine efflux from cholangiocyte spheroid lumens. Rhodamine is taken up from the media at the cholangiocyte basolateral surface, transported across the cells, and then excreted apically, resulting in accumulation in the lumen. We preloaded spheroids equally with rhodamine, and then treated with either biliatresone or vehicle. Spheroids treated with vehicle retained the majority of preloaded rhodamine in the lumen even after 12 hours. Spheroids treated with biliatresone, however, demonstrated rapid loss of luminal rhodamine, particularly within the first hour, with 50% loss in 4.4 ± 0.6 hours compared with 11.4 ± 0.4 hours for the DMSO‐treated spheroids (P < 0.0001). This suggests that biliatresone increased cholangiocyte monolayer permeability (Fig. 3).
Figure 3.
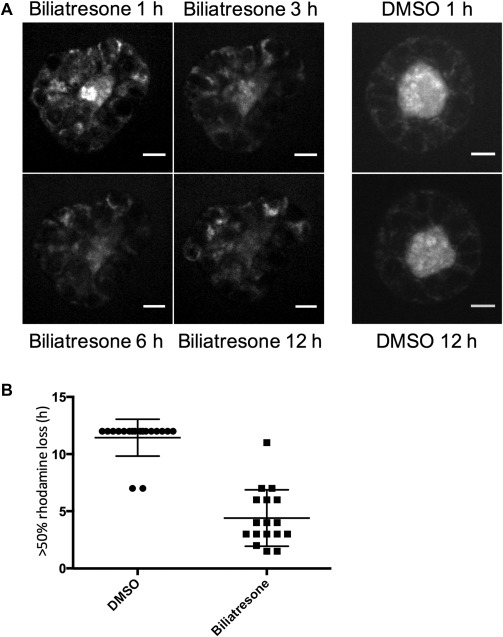
Biliatresone causes epithelial leakiness. Cholangiocyte spheroids were loaded with rhodamine, treated with vehicle (DMSO) or biliatresone for 12 hours, and imaged every 20 minutes. (A) Representative spheroids at 1, 3, 6, and 12 hours. Scale bars: 10 μm. (B) Quantification of rhodamine loss from spheroids over 12 hours (biliatresone, n = 17; DMSO, n = 18) from six independent experiments. The time at which >50% of the luminal signal was lost was scored. Data are presented as the mean ± standard deviation (P < 0.0001).
In the setting of increased epithelial permeability in vivo, cholangiocyte exposure to biliatresone could result in leakage of bile acids or biliatresone itself into the duct submucosa and deeper structures. We adapted methods used for long‐term culture of precision cut liver (and other tissue) slices in a high‐oxygen environment28 to the culture of isolated intact neonatal mouse EHBDs. In this explant culture system, ducts treated with biliatresone for 1 day and stained with antibodies against the cholangiocyte marker K19 demonstrated monolayer disruption with patchy lumen obstruction when compared with vehicle‐treated ducts, which were uniformly completely open. TUNEL staining did not demonstrate increases in apoptosis in biliatresone‐treated ducts (Supporting Fig. 4B). Biliatresone‐treated ducts also demonstrated an increase in immunostaining in periductal regions for the myofibroblast marker α‐SMA, and the fibrosis‐associated matrix proteins fibronectin EIIIA and type I collagen (Fig. 4). These results suggest that exposure of neonatal EHBDs to biliatresone could result in ductal disruption and fibrosis, as is seen in human BA patients.
Figure 4.
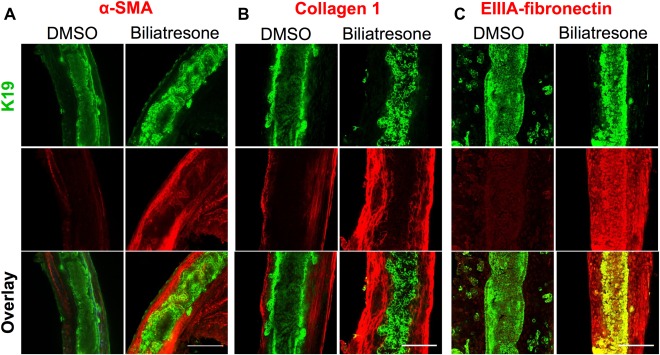
Biliatresone causes monolayer disruption, patchy obstruction, myofibroblast activation, and fibrosis in neonatal EHBD explant cultures. Neonatal mouse EHBDs were incubated for 24 hours with biliatresone or vehicle (DMSO) and immuno stained for the cholangiocyte marker K19 (green) and (A) the myofibroblast marker α‐SMA (red) and the fibrosis markers (B) collagen 1 (red) or (C) EIIIA‐fibronectin (red). K19 staining demonstrates ductal disruption, whereas the other stains demonstrate myofibroblast accumulation and matrix deposition in the submucosal and other periductal regions with biliatresone treatment. Images are representative of (A) seven independent experiments with 13 ducts for each condition, (B) three independent experiment with six ducts for each condition, and (C) three independent experiments with six ducts for each condition. Scale bars: 100 μm.
After demonstrating the effects of biliatresone on cholangiocytes in spheroid and duct explant culture, we studied its mechanism of action. We have shown that biliatresone contains an α‐methylene‐ketone reactive group and binds GSH strongly and rapidly.29 Given that decreases in GSH in some cell types have been reported to result in diminished cellular tubulin staining,14, 15, 16, 17 as we observed in cholangiocytes after biliatresone treatment, we hypothesized that biliatresone acts through GSH. We first measured GSH levels in cholangiocytes treated with biliatresone and observed a 43.6% decrease in GSH after 1 hour of treatment, with recovery over time (Fig. 5A). When we measured GSH in tissue, we found that the EHBDs had significantly less GSH (total and reduced) than the liver in both neonates and adults, suggesting that the EHBDs might be particularly susceptible to biliatresone‐induced decreases in GSH (Supporting Fig. 6), although given the lack of a significant difference in GSH levels between neonates and adults, this would be an unlikely explanation for the susceptibility of neonates to BA‐like diseases.
Figure 5.
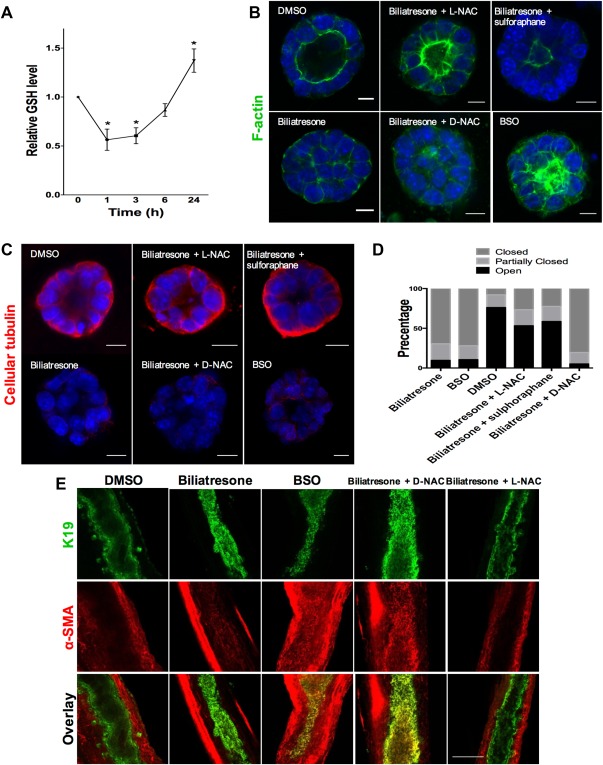
The effects of biliatresone are secondary to decreased GSH. (A) GSH levels were measured in cholangiocytes in two‐dimensional (2D) culture over 24 hours after treatment with biliatresone. Data are presented as the mean ± standard error of the mean. *P < 0.05 (versus 0 time point) and are from three independent experiments, each with three technical replicates. (B) Cholangiocyte spheroids in 3D culture were treated with the compounds indicated for 24 hours and were then immunostained for F‐actin (green) and with the nuclear stain DAPI (blue). Only spheroids that were fully visualized along the z axis were scored. Scale bars: 10 μM. (C) Spheroids treated as described in panel B and, immunostained for cellular tubulin (red). Scale bars: 10 μM. (D) Quantification of the spheroids from panel B. Spheroids (96‐119) in three different experiments for each condition were graded as open (lumen wide open), partially closed (small lumen), or closed (obstructed lumen). (E) Immunofluorescence staining of neonatal mouse EHBDs incubated for 24 hours in a high‐oxygen environment (see Materials and Methods), treated with the compounds indicated and immunostained for K19 (green) and α‐SMA (red). Scale bar: 100 μM. Images are representative of (B,C) three independent experiments, each of which was performed in duplicate, and (E) four independent experiments with at least eight ducts for each condition.
To determine the role of GSH in cholangiocyte damage resulting from biliatresone, we added compounds that increase GSH to biliatresone‐treated cultures. The effects of biliatresone on cholangiocyte spheroids were prevented by cotreatment with L‐NAC or by pretreatment with sulforaphane, but not by cotreatment with D‐NAC (Fig. 5B‐D). Sulforaphane is an Nrf2 activator that increases GSH.30 L‐NAC and D‐NAC are both antioxidants and bind biliatresone equivalently; however, only L‐NAC increases GSH synthesis, suggesting that the effects of L‐NAC are mediated by increasing GSH rather than by sequestering biliatresone.31, 32 We quantified the effects of the different compounds by grading spheroid lumens as open, partially open or closed, and confirmed that lumen patency was influenced by GSH levels (Fig. 5D).
To determine whether decreased GSH is both a necessary and sufficient mediator of biliatresone effects, we treated cholangiocyte spheroids and neonatal mouse EHBD explants with buthionine sulfoximine (BSO), which inhibits γ‐glutamylcysteine synthetase, the enzyme that mediates the first step in GSH synthesis. BSO, like biliatresone, caused a rapid decrease in GSH in cholangiocytes (Fig. 6A). Decreasing GSH with BSO mimicked the effects of biliatresone, with lumen obstruction and disrupted polarity in spheroids, and lumen narrowing and increased periductal α‐SMA, in EHBD explants (Fig. 5A‐E). BSO, similarly to biliatresone, resulted in decreased immunostaining for cellular tubulin, and this change was prevented with L‐NAC and sulforaphane treatment, but not treatment with D‐NAC. The sequence of events observed in cholangiocyte spheroids after BSO treatment (decreased tubulin staining followed by mislocalization of polarity markers and lumen closure) was the same as that observed for biliatresone (Fig. 6B). In addition, BSO treatment for 1 hour followed by incubation with BSO‐free media showed that spheroids appeared damaged at 12 hours, but after a 24‐hour washout, polarity was restored and lumens were reformed (Fig. 6C). These data suggest that the effects of BSO, like those of biliatresone, are reversible in the spheroid model (Fig. 6C and Supporting Fig. 5).
Figure 6.
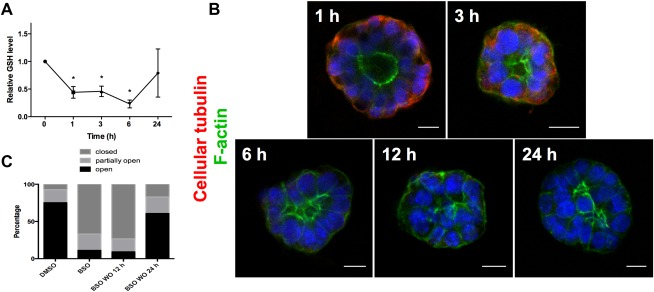
GSH reduction causes transient cholangiocyte spheroid disruption. (A) GSH levels were measured in cholangiocytes in 2D culture over 24 hours after treatment with BSO. Data are presented as the mean ± standard error of the mean. *P < 0.05 (versus 0 time point). Representative of three independent experiments, each of which was performed in duplicate. (B) Cholangiocyte spheroids were treated with BSO for 1‐24 hours as indicated, then stained for F‐actin (green) and β1 integrin (red). Nuclei were stained with DAPI (blue). Cellular tubulin changes, F‐actin redistribution, monolayer disruption, and loss of lumens followed the same sequence and time course as for biliatresone. Scale bars: 10 μm. (C) Spheroids were treated with vehicle (DMSO) or BSO for 24 hours (two left bars) or with BSO for 1 hour followed by a 12‐ or 24‐hour washout period (two right bars). Lumens were scored as open, closed, or partially open as described in Materials and Methods (n = 286‐327 spheroids per condition).
In addition to investigating the role of GSH in biliatresone effects, we studied the role of the transcription factor SOX17. A BA‐like phenotype including loss of integrity of the ductal epithelial monolayer is seen in Sox17 haploinsufficient mice in certain genetic backgrounds.10 To determine whether SOX17 expression is affected by biliatresone, we examined SOX17 levels in cholangiocytes after biliatresone treatment. Sox17 messenger RNA and protein levels were reduced significantly after biliatresone treatment (Fig. 7A,B). To determine whether decreases in SOX17 mimicked the effects of biliatresone in our cholangiocyte model system, we silenced Sox17 by way of siRNA treatment and achieved 95% knockdown, as confirmed by quantitative real‐time PCR and immunohistochemistry (Supporting Fig. 7). Treatment with Sox17 siRNA but not scrambled siRNA resulted in lumen obstruction and apical tight junction disruption, mimicking the effects of biliatresone (Fig. 7 and Supporting Video 1 and Supporting Video 2). Sox17 siRNA treatment resulted in the same sequence of events in spheroids (taking into account approximately 24 hours required for Sox17 knockdown after transfection) as seen for biliatresone and BSO, with decreased staining for cellular tubulin occurring first, followed by loss of apical polarity and lumen obstruction (Fig. 7D). Overexpression of SOX17 by transfection of complementary DNA encoding Sox17 resulted in spheroid disruption in both the presence and absence of biliatresone, suggesting that SOX17 levels are tightly controlled, and preventing us from performing rescue experiments. Silencing of Sox17 had no effect on GSH levels (Fig. 7E), suggesting that decreases in SOX17 were either caused by GSH reductions or in a parallel pathway.
Figure 7.

Biliatresone decreases SOX17, and silencing Sox17 phenocopies the biliatresone effect. (A) Sox17 messenger RNA expression in cholangiocytes in 2D culture treated with biliatresone or vehicle (DMSO) for 24 hours. Data are presented as the mean ± standard deviation. *P < 0.05. (B) Western immunoblotting of cholangiocytes in 2D culture treated with biliatresone or vehicle (DMSO) for 24 hours. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (C) Cholangiocyte spheroids treated with siRNA for Sox17 or with scrambled RNA for 24 hours, then stained for ZO1 and with DAPI. Images are representative of three independent experiments with duplicate wells of spheroids. Scale bars: 10 μm. (D) Cholangiocyte spheroids were treated with siRNA for Sox17 for the times indicated and were then immunostained for F‐actin (green) and cellular tubulin (red). Nuclei were stained with DAPI (blue). Images are representative of three independent experiments with duplicate wells of spheroids. Scale bars: 10 μm. (E) GSH levels in cholangiocytes in 2D culture treated with siRNA for Sox17 or scrambled RNA (P = 0.98). Data are presented as the mean ± standard deviation.
Discussion
The etiology of BA is unknown, although epidemiological studies support an infection or toxin exposure, likely occurring prenatally,5, 6 in the setting of genetic susceptibility.4, 33, 34, 35, 36 Our group recently identified an isoflavonoid called biliatresone that has been implicated in outbreaks of a BA‐like syndrome in Australian livestock and also causes selective EHBD destruction in zebrafish.13 This is important evidence supporting the hypothesis that toxins can cause BA. Here we used cultured cholangiocyte spheroids and a novel bile duct explant culture system to delineate the effects of biliatresone, showing that biliatresone disrupts the cholangiocyte monolayer and, in duct explants, causes periepithelial fibrosis. We identified decreases in GSH and SOX17 as key mediators of these effects and demonstrated that decreased GSH independent of biliatresone causes duct explant obstruction and fibrosis. Our data suggest that environmental agents that reduce GSH and reach the neonatal bile duct might cause BA. Redox stress is also a contributing factor in biliatresone‐induced cholangiocyte injury in zebrafish, although unlike in our mammalian cell systems, decreased GSH potentiates but is not sufficient for injury.37
A decrease in GSH was sufficient for cholangiocyte injury in our model systems. GSH levels rapidly decline after biliatresone treatment, although there is partial restoration over time. BSO similarly causes rapid decreases in cholangiocyte GSH, and the time course of injury follows the same sequence of events as for biliatresone. BSO causes damage in spheroids even after 1 hour of exposure, but spheroids recover after a 24‐hour washout. Bile ducts treated with DMSO and biliatresone demonstrated no difference in apoptosis or proliferation, though it is likely that apoptosis occurs later in the process. This suggests the possibility that a potentially transient insult to the bile ducts could cause significant damage, but also that cholangiocyte recovery could occur in vivo if the injury were stopped. These findings may have important implications for human disease. Although there is no evidence that pregnant women are exposed to biliatresone itself, humans may be exposed to other GSH‐depleting molecules, and it is conceivable that some of these cross the placenta and are excreted in neonatal bile.
One of the first signs of injury we observed in cholangiocyte spheroids was a decrease in cellular tubulin immunoreactivity. Microtubule disruption caused by decreases in GSH has been described in other cell types—including colonic, kidney, and neuronal cells14, 15, 16, 17—and our data suggest that microtubule changes in cholangiocyte spheroids are the result of decreased GSH after biliatresone treatment. Microtubules play an important role in lumen formation and the maintenance of polarity in several types of epithelial cells, including cholangiocytes.18, 26 In biliatresone‐mediated cholangiocyte injury, changes in cellular tubulin occur after and as a result of a decrease in GSH and are accompanied by loss of cell‐cell adhesion and increased epithelial permeability. Polarity abnormalities have been reported in human BA; a recent study found that extracellular matrix proteins and adhesion molecules that mediate cellular polarity and integrity are abnormal in BA patients.38 Additionally, in zebrafish, it was shown that that planar cell polarity plays an important role in vertebrate biliary development.39
The importance of the epithelium in preventing injuries and the injury response has been studied in the lung, where loss of bronchial epithelial cell integrity leads to inflammation and fibrosis.40, 41 We observed increased epithelial permeability after biliatresone treatment. Similar damage to the cholangiocyte monolayer in bile ducts could potentially result in leakage of both bile and biliatresone into the periductular tissues, either of which could potentially cause myofibroblast activation and fibrosis. Our finding that biliatresone exposure causes increased α‐SMA and collagen expression in the EHBD explant culture system is consistent with both indirect and direct effects of biliatresone on myofibroblast precursor cells. In this context, it is interesting that biliatresone‐mediated injuries in cholangiocyte spheroids are reversible. We speculate that cholangiocytes could recover from an initial injury, but that the exposure of the submucosal and deeper structures to bile (or biliatresone or similar toxins) might lead to more permanent damage. Obstruction could potentially be secondary to cholangiocyte injury, associated inflammation, or a secondary fibrotic process.
Another potential cause of bile duct obstruction is epithelial deciduation, which is observed in Sox17 heterozygote mice that develop a BA‐like syndrome.10 In these mice, SOX17 is required to maintain the epithelial architecture of the gallbladder and cystic duct. We find that biliatresone reduces SOX17 in cholangiocytes, and that silencing Sox17 mimics the effects of biliatresone on the biliary epithelium. SOX17 has been implicated as key protein in EHBD development,42 and our model suggests that it is important to duct maintenance as well.
One of the key questions in BA research is why early manifestations in humans are relatively selective for the EHBDs. Using spheroid culture systems, we found that multiple ductular epithelial cells, including mammary ductal, pancreatic ductal, and kidney tubular cells were affected by biliatresone, whereas enterocytes, which are epithelial cells but not ductular, were not affected. Similarly, we found that intrahepatic and extrahepatic cholangiocytes in vitro appeared to be equally susceptible to injury. These findings raise interesting questions about the specificity of biliatresone for extrahepatic cholangiocytes in zebrafish and livestock. It appears likely that biliatresone is toxic to many cells, but that in vivo it is selective for extrahepatic cholangiocytes because of excretion and/or concentration in bile. In support of this hypothesis, we found in zebrafish that intrahepatic bile ducts are required for biliatresone toxicity. We speculate that biliatresone crosses the placenta and enters the fetal liver and the biliary system; maternal hepatic metabolism may prevent cholangiocyte exposure in the mother. The stasis of bile in utero as well as concentration of bile and biliatresone in the gallbladder might explain EHBD toxicity. We hypothesize that biliatresone undergoes metabolism by hepatocytes and that there may be significant differences in metabolism between neonatal and adult livers. Further studies will be needed to answer this question. Low levels of GSH may also predispose the EHBD to injury. We previously reported that hepatocytes are relatively resistant to the effects of biliatresone.13 GSH levels in the liver parenchyma are higher than in the EHBDs, which may protect hepatocytes, although there is no difference in GSH levels between adult and neonatal mouse EHBDs, highlighting the potential importance of late fetal or neonatal versus adult hepatic transport and metabolism of biliatresone. The GSH levels of intrahepatic compared with extrahepatic cholangiocytes are unknown but could potentially account for selective extrahepatic toxicity. Finally, it is possible that both intrahepatic and extrahepatic cholangiocytes are injured by biliatresone, but that the regenerative abilities of the liver and intrahepatic cholangiocytes compared with the EHBDs result in repair after injury rather than fibrosis.
There are a wealth of data implicating both immune dysregulation and genetic factors in human BA. Toxin‐induced BA is not inconsistent with these findings and may represent a primary injury, with immune dysregulation representing a secondary insult. Ongoing studies are focused on determining links between biliatresone treatment and immune dysregulation.
In conclusion, our findings are consistent with a model of biliatresone‐induced injury that includes primary injury to the EHBDs, loss of cell‐cell adhesion, cholangiocyte polarity changes, increased epithelial permeability, and resulting myofibroblast activation and fibrosis. These events are secondary to reductions in GSH and are accompanied by reduced SOX17. Elements of this model—including the role of GSH, the potentially transient nature of the insult, the increase in epithelial permeability, and the reversibility of cholangiocyte damage—may provide important insights into both the pathogenesis and clinical course of human BA and may lead to the development of new preventive measures or novel treatments in the future.
Author names in bold designate shared co‐first authorship
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28599/suppinfo.
Supporting Information
Supporting Information
Supporting Information
Acknowledgment
We thank Basil Bakir, Anil Rustgi, Mauricio Reginato, Katalin Susztak, Mary Ann Crissey, and John Lynch for providing cells and the Cell and Developmental Biology Microscopy core and the NIDDK Center for Molecular Studies in Digestive and Liver Disease Molecular Pathology and Imaging Core (P30 DK050306) at the University of Pennsylvania for imaging support.
Potential conflict of interest: Nothing to report.
Supported by National Institute of Diabetes and Digestive and Kidney Diseases grants R01 DK092111 (to M.P., J.R.P., and R.G.W.) and 5T32 HD043021‐10 and 1T32 DK101371‐01 (to O.W.‐Z.); an AASLD Advanced Hepatology and Liver Transplant Fellowship grant (to O.W.‐Z.); and grants from the Fred and Suzanne Biesecker Pediatric Liver Center (to M.P. and R.G.W.).
REFERENCES
- 1. Haber BA, Russo P. Biliary atresia. Gastroenterol Clin North Am 2003;32:891‐911. [DOI] [PubMed] [Google Scholar]
- 2. Bezerra JA. Potential etiologies of biliary atresia. Pediatr Transplant 2005;9:646‐651. [DOI] [PubMed] [Google Scholar]
- 3. Riepenhoff‐Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res 1993;33:394‐399. [DOI] [PubMed] [Google Scholar]
- 4. Tiao MM, Tsai SS, Kuo HW, Chen CL, Yang CY. Epidemiological features of biliary atresia in Taiwan, a national study 1996‐2003. J Gastroenterol Hepatol 2008;23:62‐66. [DOI] [PubMed] [Google Scholar]
- 5. Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics 2011;128:e1428‐e1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou K, Lin N, Xiao Y, Wang Y, Wen J, Zou GM, et al. Elevated bile acids in newborns with biliary atresia (BA). PLoS One 2012;7:e49270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hertel PM, Estes MK. Rotavirus and biliary atresia: can causation be proven? Curr Opin Gastroenterol 2012;28:10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gibelli NE, Tannuri U, de Mello ES, Rodrigues CJ. Bile duct ligation in neonatal rats: is it a valid experimental model for biliary atresia studies? Pediatr Transplant 2009;13:81‐87. [DOI] [PubMed] [Google Scholar]
- 9. Holder TM, Ashcraft KW. Production of experimental biliary atresia by ligation of the common bile duct in the fetus. Surg Forum 1966;17:356‐357. [PubMed] [Google Scholar]
- 10. Uemura M, Ozawa A, Nagata T, Kurasawa K, Tsunekawa N, Nobuhisa I, et al. Sox17 haploinsufficiency results in perinatal biliary atresia and hepatitis in C57BL/6 background mice. Development 2013;140:639‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harper P, Plant JW, Unger DB. Congenital biliary atresia and jaundice in lambs and calves. Aust Vet J 1990;67:18‐22. [DOI] [PubMed] [Google Scholar]
- 12. Robson S. Congenital biliary atresia and jaundice in lambs New South Wales Animal Health Surveillance April‐June 2007:2. [Google Scholar]
- 13. Lorent K, Gong W, Koo KA, Waisbourd‐Zinman O, Karjoo S, Zhao X, et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci Transl Med 2015;7:286ra267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aquilano K, Baldelli S, Cardaci S, Rotilio G, Ciriolo MR. Nitric oxide is the primary mediator of cytotoxicity induced by GSH depletion in neuronal cells. J Cell Sci 2011;124:1043‐1054. [DOI] [PubMed] [Google Scholar]
- 15. Hosono T, Hosono‐Fukao T, Inada K, Tanaka R, Yamada H, Iitsuka Y, et al. Alkenyl group is responsible for the disruption of microtubule network formation in human colon cancer cell line HT‐29 cells. Carcinogenesis 2008;29:1400‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang X, Chen L, Liu W, Qiao Q, Wu K, Wen J, et al. Involvement of oxidative stress and cytoskeletal disruption in microcystin‐induced apoptosis in CIK cells. Aquat Toxicol 2015;165:41‐50. [DOI] [PubMed] [Google Scholar]
- 17. Zuelke KA, Jones DP, Perreault SD. Glutathione oxidation is associated with altered microtubule function and disrupted fertilization in mature hamster oocytes. Biol Reprod 1997;57:1413‐1419. [DOI] [PubMed] [Google Scholar]
- 18. Akhtar N, Streuli CH. An integrin‐ILK‐microtubule network orients cell polarity and lumen formation in glandular epithelium. Nat Cell Biol 2013;15:17‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueno Y, Alpini G, Yahagi K, Kanno N, Moritoki Y, Fukushima K, et al. Evaluation of differential gene expression by microarray analysis in small and large cholangiocytes isolated from normal mice. Liver Int 2003;23:449‐459. [DOI] [PubMed] [Google Scholar]
- 20. Karjoo S, Wells RG. Isolation of neonatal extrahepatic cholangiocytes. J Vis Exp 2014. doi: 10.3791/51621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reichert M, Takano S, Heeg S, Bakir B, Botta GP, Rustgi AK. Isolation, culture and genetic manipulation of mouse pancreatic ductal cells. Nat Protoc 2013;8:1354‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 2009;459:262‐265. [DOI] [PubMed] [Google Scholar]
- 23. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double‐fluorescent Cre reporter mouse. Genesis 2007;45:593‐605. [DOI] [PubMed] [Google Scholar]
- 24. Koo KA, Lorent K, Gong W, Windsor P, Whittaker SJ, Pack M, et al. Biliatresone, a reactive natural toxin from Dysphania glomulifera and D. littoralis: discovery of the toxic moiety 1,2‐Diaryl‐2‐Propenone. Chem Res Toxicol 2015;28:1519‐1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dipaola F, Shivakumar P, Pfister J, Walters S, Sabla G, Bezerra JA. Identification of intramural epithelial networks linked to peribiliary glands that express progenitor cell markers and proliferate after injury in mice. Hepatology 2013;58:1486‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanimizu N, Miyajima A, Mostov KE. Liver progenitor cells fold up a cell monolayer into a double‐layered structure during tubular morphogenesis. Mol Biol Cell 2009;20:2486‐2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diaz R, Kim JW, Hui JJ, Li Z, Swain GP, Fong KS, et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum Pathol 2008;39:102‐115. [DOI] [PubMed] [Google Scholar]
- 28. Klassen LW, Thiele GM, Duryee MJ, Schaffert CS, DeVeney AL, Hunter CD, et al. An in vitro method of alcoholic liver injury using precision‐cut liver slices from rats. Biochem Pharmacol 2008;76:426‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koo KA, Waisbourd‐Zinman O, Wells RG, Pack M, Porter JR. Reactivity of biliatresone, a natural biliary toxin, with glutathione, histamine and amino acids. Chem Res Toxicol 2016;29:142‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Steele ML, Fuller S, Patel M, Kersaitis C, Ooi L, Munch G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol 2013;1:441‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sarnstrand B, Tunek A, Sjodin K, Hallberg A. Effects of N‐acetylcysteine stereoisomers on oxygen‐induced lung injury in rats. Chem Biol Interact 1995;94:157‐164. [DOI] [PubMed] [Google Scholar]
- 32. Karg E, Tunek A, Brotell H, Rosengren E, Rorsman H. Alteration of glutathione level in human melanoma cells: effect of N‐acetyl‐L‐cysteine and its analogues. Pigment Cell Res 1990;3:11‐15. [DOI] [PubMed] [Google Scholar]
- 33. Garcia‐Barceló M‐M, Yeung M‐Y, Miao X‐P, Tang CS‐M, Chen G, So M‐T, et al. Genome‐wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum Mol Genet 2010;19:2917‐2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leyva‐Vega M, Gerfen J, Thiel BD, Jurkiewicz D, Rand EB, Pawlowska J, et al. Genomic alterations in biliary atresia suggest region of potential disease susceptibility in 2q37.3. Am J Med Genet A 2010;152A:886‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rurarz M, Czubkowski P, Chrzanowska K, Cielecka‐Kuszyk J, Marczak A, Kaminska D, et al. Biliary atresia in children with aberrations involving chromosome 11q. J Pediatr Gastroenterol Nutr 2014;58:e26‐e29. [DOI] [PubMed] [Google Scholar]
- 36. Tsai EA, Grochowski CM, Loomes KM, Bessho K, Hakonarson H, Bezerra JA, et al. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum Genet 2014;133:235‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. X Zhao, K Lorent, B Wilkins, DM Marchione, K Gillespie, O Waisbourd‐Zinman, et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology 2016. doi: 10.1002/hep.28603. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 38. Whitby T, Schroeder D, Kim HS, Petersen C, Dirsch O, Baumann U, et al. Modifications in integrin expression and extracellular matrix composition in children with biliary atresia. Klin Padiatr 2015;227:15‐22. [DOI] [PubMed] [Google Scholar]
- 39. Cui S, Capecci LM, Matthews RP. Disruption of planar cell polarity activity leads to developmental biliary defects. Dev Biol 2011;351:229‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eurlings IM, Reynaert NL, van den Beucken T, Gosker HR, de Theije CC, Verhamme FM, et al. Cigarette smoke extract induces a phenotypic shift in epithelial cells; involvement of HIF1alpha in mesenchymal transition. PLoS One 2014;9:e107757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao W, Li L, Wang Y, Zhang S, Adcock IM, Barnes PJ, et al. Bronchial epithelial cells: The key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology 2015;20:722‐729. [DOI] [PubMed] [Google Scholar]
- 42. Spence JR, Lange AW, Lin SC, Kaestner KH, Lowy AM, Kim I, et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell 2009;17:62‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28599/suppinfo.
Supporting Information
Supporting Information
Supporting Information