

Abstract
Posttraumatic stress disorder (PTSD) is defined as a psychiatric disorder; however, PTSD co-occurs with multiple somatic manifestations. People living with PTSD commonly manifest dysregulations in the systems that regulate the stress response, including the hypothalamic-pituitary-adrenal (HPA) axis, and development of a pro-inflammatory state. Additionally, somatic autoimmune and inflammatory diseases and disorders have a high rate of co-morbidity with PTSD. Recognition and understanding of the compounding effect that these disease states can have on each other, evidenced from poorer treatment outcomes and accelerated disease progression in patients suffering from co-morbid PTSD and/or other autoimmune and inflammatory diseases, has the potential to lead to additional treatment opportunities.
Keywords: Post-traumatic stress disorder, PTSD, Autoimmune disease, Inflammatory diseases, Rheumatoid arthritis
Introduction
Although traditionally considered a type of anxiety disorder, post-traumatic stress disorder (PTSD) is classified as a Trauma and Stress related disorder in DSM-V. PTSD is a chronic psychiatric illness that develops subsequent to experiencing a significant traumatic event. Although exposure to a stressful event is required for PTSD, only a minority (8–18%) [1–3] of trauma exposed individuals go on to develop the disorder. DSM-V criteria for PTSD include delayed onset of behavioral changes that can be grouped into 4 distinct diagnostic clusters: re-experiencing, avoidance, hyper-arousal and negative cognitions and mood.
PTSD is also a somatic condition, such that patients with PTSD have been found to have biological alterations in several primary pathways involving the neuroendocrine [4] and immune systems [5]. Much like physiological stress, chronic psychological stress stimulates the stress response pathways of the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic nervous system leading to downstream release of glucocorticoids (GC) and catecholamines. Cortisol, which is the primary endogenous GC hormone in humans, acts on the central nervous system, metabolic system, and immune system to modulate the stress response. GCs impact physiology and behavior by binding to the intracellular GC receptor (GR) on target tissues leading to downstream effects of immunosuppression, increased energy metabolism, and negative feedback inhibition of the HPA axis. In this way, GC signaling is central to the neuroendocrine modulation of the immune system.
PTSD co-occurs with dysregulation of the HPA axis, impaired GC signaling, and the development of a pro-inflammatory state. Not surprisingly, PTSD is associated with poor self-reported physical health as well as high rates of comorbidities, such as cardiovascular, respiratory, gastrointestinal, inflammatory and autoimmune diseases [6–8]. Recently a large retrospective cohort study of 666,269 Iraq war veterans showed a two-fold increase in the risk of autoimmune diseases in individuals with PTSD compared to those without any psychiatric illness and a 51% increased risk when compared to individuals with other psychiatric illnesses [9]. Dysregulations in immune function as a result of the complex interplay between the neuroendocrine and immune systems in PTSD may unmask a predisposition to, or accelerate the progression of, autoimmune (AI)/inflammatory diseases, thereby compounding the disease burden in these patients. In the following sections we will review the mechanistic links between neuroendocrine and immune dysfunctions in PTSD and outline existing and novel pharmacological treatment options that may be able to address both the psychological and biological disturbances observed in patients living with PTSD.
Candidate Mechanisms Linking PTSD and Immune Dysfunction
Reduced Circulating Cortisol
As mentioned above, neuroendocrine alterations in people living with PTSD may precipitate immune disturbances. The dogmatic physiological signature of chronic stress is simultaneous elevations in concentrations of cortisol and catecholamines [5]. Contrary to this dogmatic view, PTSD is associated with low levels of morning cortisol and elevated levels of norepinephrine (NE). Excessive activation of the HPA axis in response to trauma and sustained increases in corticotrophin-releasing factor (CRF) are collectively proposed to downregulate CRF receptors in the pituitary leading to downstream reduction in GC signaling, decreased secretion of cortisol, and subsequently increased GC sensitivity [10]. These changes, along with enhanced sympathetic activity driven stimulation of β2-adrenergic receptor on immune cells, may lead to increased cytokine production, fostering the hyper-inflammatory state frequently co-morbid with PTSD.
An alternative view is that low cortisol concentrations are a precipitator of PTSD rather than a consequence of PTSD. Low salivary and urinary cortisol immediately following trauma have been reported to be predictors of PTSD suggesting that low cortisol concentrations may in fact be a preexisting vulnerability for developing PTSD rather than a consequence of PTSD [11]. Interestingly, inadequate cortisol secretion in relation to the chronic inflammation observed in rheumatoid arthritis, suggests that the suboptimal production of cortisol may be involved in onset and/or progression of autoimmune disease [12,13]. Hence, overlap may exist between the mechanisms by which low cortisol concentrations may precipitate both psychiatric and somatic diseases and disorders.
However, because of the multifaceted regulation and impact of the GR, information about glucocorticoids alone is not consistently predictive of their function or dysfunction. GRs have extensive variability in their actions depending on the target tissue. Multiple independent promoters present in the GR gene contribute to GR variability by influencing tissue specificity of GR gene expression. Epigenetic modulation of some of these promoters has been demonstrated to change GR gene expression and function in PTSD [14]. Moreover, single nucleotide polymorphisms in the GR gene NR3C1 and the FKBP5 gene (co-chaperone of hsp90 which regulates GR sensitivity) are associated with altered HPA axis sensitivity (GR hypersensitivity or GR resistance) [15]. Hence, alterations in multiple factors including GR expression levels, GR affinity, co-factors, GR heterogeneity and GR density in target tissues are sufficient to affect GC signaling and are candidate mechanisms for enhanced inflammation associated with PTSD.
Chronic Low Grade Inflammation
PTSD is linked to cytokinemia and a recent meta-analysis of 20 studies found increased plasma levels of pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β) & interleukin-6 (IL-6) in individuals with PTSD compared to healthy controls [16]. In addition, there is a prospective association of plasma C-reactive protein (CRP) concentrations with the development of PTSD [17], and higher mitogen induced cytokine production in trauma exposed soldiers correlates with augmentation of PTSD symptoms in response to subsequent stressors [18]. These findings suggest that inflammation may predispose an individual to PTSD, and inflammation may even form the biological basis of stress sensitization [18] that precipitates PTSD after trauma exposure.
The relationship between the HPA axis and cytokines is bidirectional. In addition to the previously discussed effects of the HPA axis on cytokines, cytokines can influence HPA axis signaling and impair cellular processes by stimulating oxidative stress. The sequela that follows cytokine-induced changes in the HPA axis and central nervous system has been proposed to lead to the manifestation of PTSD symptoms [19]. This proposition is consistent with results of a study using the predator exposure animal model of PTSD that demonstrated elevated levels of pro-inflammatory cytokines and reactive oxygen species in the brain (hippocampus, amygdala, pre-frontal cortex) and in the periphery as a consequence of stressor exposure [20]. Conversely, administration of the anti-inflammatory agent minocycline following a laboratory stressor is sufficient to block the development of PTSD-like behaviors in a rodent model [21].
Chronic inflammation is a pathological feature of multiple somatic diseases that are highly co-morbid with PTSD including cardiovascular disease, rheumatoid arthritis, asthma, psoriasis, metabolic syndrome, fibromyalgia, chronic pain syndromes and hypothyroidism. Common cytokines implicated in enhanced inflammation in PTSD and other diseases may therefore serve as a potent therapeutic target in the treatment of both types of conditions.
Alterations in Innate and Adaptive Immunity
A burgeoning area of study is the relationship between PTSD and innate and adaptive immunity. A recent study of U.S. Marines applied weighted gene co-expression network analysis to RNA-Seq and microarray assessment of peripheral blood leukocyte gene expression taken pre- and post-deployment. The authors reported that PTSD risk and PTSD cases groups both had enhanced differential expression of genes associated with innate immune responses mediated by interferon signaling. These findings add to the authors previous work showing that differential expression of CRP and genes involved in antiviral interferon response were associated with the risk of developing PTSD [22] and suggest that innate immunity up-regulation may be both a risk for, and consequence of, PTSD.
A relationship between PTSD and adaptive immunity is also plausible given that cytokines drive differentiation of T cell subsets, and individuals living with PTSD exhibit elevated cytokine production. To this end, a recent study demonstrated an association between PTSD and a T cell phenotype consistent with increased differentiation of T cells and interpreted as early aging of the immune system [23]. Furthermore, T cells may provide a window into the susceptibility of an individual to psychiatric disorders such that responsiveness of T cells to the synthetic GC dexamethasone prior to military deployment predicted both PTSD and depression following deployment [24]. Collectively, the shifts in T cell biology observed in PTSD push towards a preponderance of CD4+ T helper 1 (Th1) cells over the C4+ T helper 2 (Th2) type cells which correlates with increased plasma levels of Interferon-gamma (IFN-γ) in PTSD [25]. Although mechanisms have not been elucidated to date, Marpe Bam et al., has shown that epigenetic modifications and miRNAs were associated with elevated gene expression of the pro-inflammatory cytokine interleukin-12 (IL-12) in peripheral blood mononuclear cells (PBMCs) of PTSD patients [26]. In addition to PTSD, several autoimmune diseases are associated with alterations of the Th1 versus Th2 cytokine balance including rheumatoid arthritis, multiple sclerosis, type 1 diabetes, and autoimmune thyroid disease. In these somatic conditions, the balance is skewed towards Th1 and an excess of IL-12 and TNF-α production, whereas Th2 and production of anti-inflammatory interleukin-10 (IL-10) appear to be deficient [12].
In addition to shifts in T helper cells, Jergovic et al., found a ~50% decrease in the number of regulatory T cells (Treg) in PTSD patients compared to healthy controls [27]. Treg are essential for controlling immune responses and maintaining self-tolerance by inhibiting auto reactive T cells. A decrease in number and function of peripheral Treg has been associated with the development of multiple autoimmune diseases that are highly co-morbid with PTSD [28].
Telomere Shortening and Premature Immunosenescence
In addition to elevated levels of terminally differentiated T cells and an altered Th1/Th2 balance, PTSD has been associated with the age-related phenomenon of telomere shortening. Chronic inflammation has been shown to accelerate telomere shortening leading to cellular aging and premature senescence that have been implicated in loss of control of the immune system [29]. Senescent cells are terminally differentiated and no longer fully functional, but instead of undergoing cell death, they exist in a zombie-like state spewing cytokines into the cellular milieu. Telomere shortening has been identified in many autoimmune diseases [30] and is associated with acceleration of the manifestation of age-related diseases. Telomere shortening of leukocytes/PBMCs has emerged as a biomarker of PTSD and a recent literature review of 32 studies between 2001 and 2014 found reduced leukocyte telomere length and increased pro-inflammatory markers in PTSD patients suggesting early immunosenescence [23]. Additionally, PTSD is associated with earlier onset of age-related conditions linked to telomere shortening and increased mortality [31].
Sex Differences and PTSD
Similar to other neuropsychiatric and somatic disorders, sex differences have been reported in the context of PTSD. There are well-known differences in PTSD risk between men and women with women exhibiting a higher frequency of PTSD than men (2:1)[33], not explained solely on the basis of exposure type and/or severity alone. Dias et. al demonstrated that female-specific elevation of pituitary adenylate cyclase-activating peptide (PACAP) and differential methylation of a single nucleotide polymorphism (rs2267735) on the PACAP gene (Adcyap1r1) was associated with a PTSD diagnosis in females, but not in males [32]. Differences in the neuroendocrine response to stress in males and females can be attributed to genomic (as above) or hormonal differences to the neuroendocrine response to stress between the sexes [34].
Additionally, autoimmune diseases disproportionally impact females over males, reflected in the study conducted by O’Donovan et al., showing that women with PTSD were three times more likely to be diagnosed with an autoimmune condition [9]. Interestingly however, the magnitude of PTSD-related increased risk was similar between the sexes and the authors therefore did not find a sex difference in the relative risk of autoimmune diseases in PTSD patients. They did however find that a history of Military Sexual Trauma (MST) and PTSD were associated with the highest risk of autoimmune diseases in both men and women and thus MST was an independent risk factor in the development of autoimmune disease. Notably, the patient populations in a large majority of studies referenced in this paper are composed of combat veterans exposed to the trauma of war. But the finding of MST as an independent risk factor for the development of PTSD points to the possibility that the type of trauma may correlate with severity and/or risk of autoimmune or somatic illness, and warrants further work in this area.
PTSD Treatment Opportunities: Immune System Intervention
The literature summarized here establishes that in addition to the commonly appreciated psychiatric manifestations of PTSD, marked alterations in the neuroendocrine and immune systems exist in individuals living with PTSD. As such, intervention strategies that target neuroendocrine and immune dysfunction may prove beneficial to the treatment of PTSD. A similar angle has been assessed in the context of depression such that a meta-analysis illustrates that elevations in CRP and IL-6 precede development of depression and that patients with increased inflammation are less likely to respond to conventional anti-depressants and more likely to respond to adjunctive anti-inflammatory treatment [35].
Although mechanistically interventions that target function of the HPA axis and/or GR should prove effective in the treatment of both PTSD and immune dysfunction, these neuroendocrine interventions have had mixed utility which may be due to the pervasive nature of the GR on multiple organ systems. Mifepristone, a GR antagonist, has been reported to effectively improve metrics of PTSD symptoms [36], but a more recent report from the same research group demonstrates improvements in cognition but not in symptoms of PTSD or metrics of physical health [37]. More targeted treatment of GR function, through manipulation of GR co-chaperones such as FKBP5, may be a more advantageous route of intervention given that this type of intervention should leave non-pathological GRs intact [38]. To this end, studies of rapamycin, a drug which, among other things, can alter function of GR co-chaperones, has shown promise in rodent models of PTSD [39] and is already FDA approved and in clinical trials unrelated to PTSD.
Given the potential limitations to interventions at the level of GR and the HPA axis, attention to immune-centric interventions is also warranted. Several pro-inflammatory cytokines elevated in PTSD are also implicated in autoimmune diseases and therefore are uniquely positioned to function as biomarkers for diagnosis and treatment of both conditions. For instance, plasma levels of IL-1β and IL-6 have been shown to positively correlate with PTSD symptom duration and severity respectively [16], and can therefore be used to monitor treatment response in PTSD. Drugs aimed at decreasing concentrations of pro-inflammatory cytokines in the circulation might have dual benefits and help ease disease burden in PTSD patients. Canakinumab, a monoclonal antibody against IL-1β, and anakinra, an IL-1 receptor antagonist, are two such medications that target IL-1β. These drugs have been used in the treatment of rheumatoid arthritis and other inflammatory conditions with positive results [40]. Clazakizumab, a monoclonal antibody against IL-6, is in phase 2 clinical trials to treat rheumatoid arthritis with promising results [41]. Furthermore, in rheumatoid arthritis patients, long term treatment with anti-TNF agents has been shown to raise cortisol levels (inadequate cortisol in relation to inflammation implicated in chronic low-grade inflammation) and normalize the HPA axis leading to rapid clinical improvement [42].
In addition, targeting senescent cells may be an advantageous point of intervention. Senolytics are a new intervention strategy in aging research and in diseases of aging which show particular promise. These treatments target and remove the senescent cells, many of which are believed to contribute to cytokinemia, without damaging healthy cells. This exfoliation of the immune system may confer benefits for both traditional immune disorders and neuropsychiatric disorders with an immune component. Although initial studies used methods for clearance of senescent cells that lacked translational potential, recent work demonstrates successful administration of a pharmacological senolytic agent in a mouse model [43].
In addition to these novel immune-driven interventions, it is important to recognize that some of the existing treatments for PTSD confer immune benefits. SSRIs are first line treatment of PTSD, and have been shown to exert anti-inflammatory effects on T-lymphocytes, dendritic cells, and neutrophils [44]. Specifically fluoxetine and citalopram were found to exhibit an anti-arthritic effect on murine collagen-induced arthritis and in a human ex vivo disease model of rheumatoid arthritis [45]. The anti-inflammatory effect of SSRIs in human rheumatoid arthritis tissue was due to reduction of spontaneous cytokine production from macrophages (IL-6, INF-γ and IL-10) through toll-like receptors. Previous studies have found SSRIs to improve symptoms in encephalomyelitis, a multiple sclerosis model through reduction in pro-inflammatory cytokines [46–48]. Other drugs used in the treatment of PTSD that have been found to have anti-inflammatory effects include prazosin (alpha-1-adrenoreceptor blocker) and ketamine. Prazosin has been shown to be effective in treatment resistant cases of PTSD in which recurrent nightmares are problematic [49]. Previous studies have found prazosin and doxazosin, also an alpha-1 blocker, to exhibit anti-inflammatory effects in rodent models of inflammation by inhibiting the production of lipopolysaccharide induced pro-inflammatory cytokines TNF-α and IL-1β [50,51]. Ketamine infusion has been shown to have a rapid reduction in symptom severity in patients with chronic PTSD [52] and ketamine possesses anti-inflammatory actions which have been attributed to inhibition of transcription factors activator protein-1 and nuclear factor (NF)-κB, as well as lowering of serum levels of IL-6, TNF-α, inducible nitric oxide synthase and CRP [53].
Conclusions
Although once believed to be an immune-privileged site, the bidirectional communication between the brain and periphery is now commonly appreciated. The growing recognition that neuropsychiatric disorders are also somatic disorders will improve understanding of disease pathogenesis and lead to advances in treatment options. In the case of PTSD, the relationship with the immune system appears to be multi-tiered and bidirectional. Continued monitoring of developments in immunological interventions and efforts to apply these interventions to PTSD is essential to advancing biological psychiatry. Furthermore, given the bidirectional nature of the relationship between PTSD and immune system function, recognition and treatment of PTSD may improve immunological outcomes for individuals living with primary disorders of the immune system.
Figure 1. A Psychoneuroimmunological Model of PTSD.
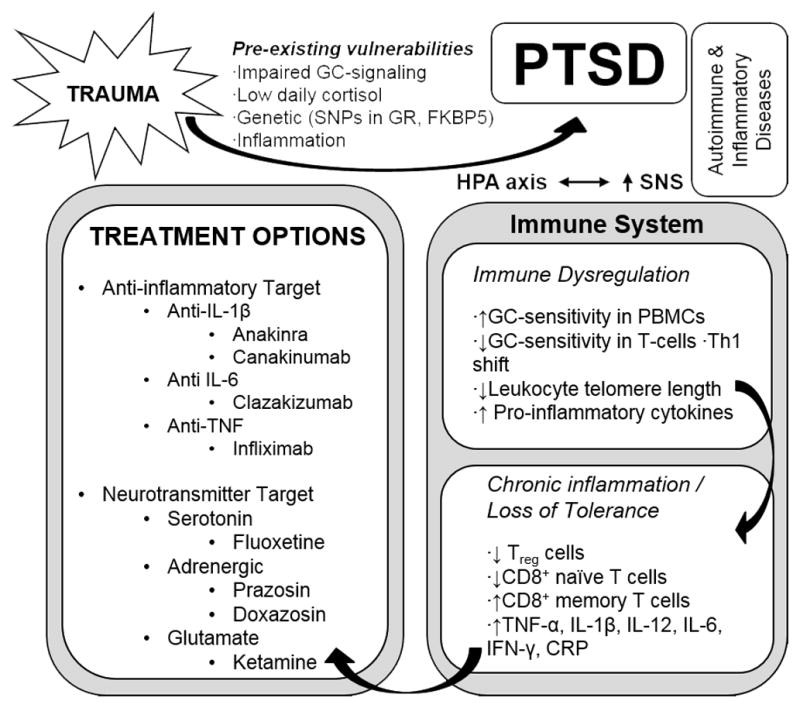
Exposure to severe psychological trauma in the presence of pre-existing risk factors leads to PTSD. Immune system changes in PTSD include altered glucocorticoid (GC) sensitivity in target immune cells, shifts in immune cell distribution, immunosenescence, elevated pro-inflammatory cytokines and a decrease in regulatory T cells. A complex interplay of the biological alterations in the stress response known to exist in PTSD, along with immune alterations, are hypothesized to increase the risk for co-morbid somatic autoimmune and inflammatory disorders. Immune interventions may improve both primary PTSD symptoms and co-morbid somatic disorders related to the immune system.
Highlights.
PTSD co-occurs with somatic diseases.
PTSD is commonly associated with neuroendocrine and immune dysfunction
Targeting neuroendocrine and immune dysfunction may improve PTSD symptoms.
Targeting PTSD may improve somatic co-morbidities.
Translational reciprocity between biological psychiatry and immunology may advance treatment options.
Acknowledgments
G.N. Neigh’s effort on this work was supported by startup funds from the Department of Anatomy & Neurobiology of Virginia Commonwealth University’s School of Medicine. Research in G.N. Neigh’s laboratory is currently supported by R01NR014886 from the National Institute of Nursing Research and K18MH105098 from the National Institute of Mental Health.
Footnotes
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. [Internet] Arch Gen Psychiatry. 1995;52:1048–60. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 2.Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis GC, Andreski P. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. [Internet] Arch Gen Psychiatry. 1998;55:626–32. doi: 10.1001/archpsyc.55.7.626. [DOI] [PubMed] [Google Scholar]
- 3.White J, Pearce J, Morrison S, Dunstan F, Bisson JI, Fone DL. Risk of post-traumatic stress disorder following traumatic events in a community sample. [Internet] Epidemiol Psychiatr Sci. 2015;24:249–57. doi: 10.1017/S2045796014000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heim C, Nemeroff CB. Neurobiology of posttraumatic stress disorder. [Internet] CNS Spectr. 2009;14:13–24. [PubMed] [Google Scholar]
- 5.Pace TWW, Heim CM. A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. [Internet] Brain Behav Immun. 2011;25:6–13. doi: 10.1016/j.bbi.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Boscarino JA. Posttraumatic stress disorder and physical illness: results from clinical and epidemiologic studies. [Internet] Ann N Y Acad Sci. 2004;1032:141–53. doi: 10.1196/annals.1314.011. [DOI] [PubMed] [Google Scholar]
- 7.Kubzansky LD, Koenen KC. Is posttraumatic stress disorder related to development of heart disease? An update. [Internet] Cleve Clin J Med. 2009;76(Suppl 2):S60–5. doi: 10.3949/ccjm.76.s2.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalcanti-Ribeiro P, Andrade-Nascimento M, Morais-de-Jesus M, de Medeiros GM, Daltro-Oliveira R, Conceição JO, Rocha MF, Miranda-Scippa Â, Koenen KC, Quarantini LC. Post-traumatic stress disorder as a comorbidity: impact on disease outcomes. [Internet] Expert Rev Neurother. 2012;12:1023–37. doi: 10.1586/ern.12.77. [DOI] [PubMed] [Google Scholar]
- 9.O’Donovan A, Cohen BE, Seal KH, Bertenthal D, Margaretten M, Nishimi K, Neylan TC. Elevated risk for autoimmune disorders in iraq and afghanistan veterans with posttraumatic stress disorder. [Internet] Biol Psychiatry. 2015;77:365–74. doi: 10.1016/j.biopsych.2014.06.015. The authors conducted a retrospective cohort study of 666,269 Iraq and Afghanistan veterans under the age of 55 who were enrolled in the VA healthcare system between October, 2001 and March 2011. Using generalized linear models, they found that veterans diagnosed with PTSD had a significantly higher adjusted relative risk for diagnosis with an autoimmune disease compared to veterans with no psychiatric illness and compared to veterans diagnosed with a psychiatric illness other than PTSD. Although previous studies of Vietnam veterans found higher prevalence of self reported auto-immune disorders and physician dianosed RA associated with PTSD, this is the first large scale study to examine if PTSD increased the risk all autoimmune diseases as diagnosed by a physician using definitive diagnostic criteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wieck A, Grassi-Oliveira R, Hartmann Do Prado C, Teixeira AL, Bauer ME. Neuroimmunoendocrine interactions in post-traumatic stress disorder: Focus on long-term implications of childhood maltreatment. Neuroimmunomodulation. 2014;21:145–151. doi: 10.1159/000356552. [DOI] [PubMed] [Google Scholar]
- 11.van Zuiden M, Kavelaars A, Geuze E, Olff M, Heijnen CJ. Predicting PTSD: Pre-existing vulnerabilities in glucocorticoid-signaling and implications for preventive interventions [Internet] Brain Behav Immun. 2013;30:12–21. doi: 10.1016/j.bbi.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Elenkov IJ. Neurohormonal-cytokine interactions: Implications for inflammation, common human diseases and well-being. Neurochem Int. 2008;52:40–51. doi: 10.1016/j.neuint.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 13.Spies CM, Straub RH, Cutolo M, Buttgereit F. Circadian rhythms in rheumatology--a glucocorticoid perspective. [Internet] Arthritis Res Ther. 2014;16(Suppl 2):S3. doi: 10.1186/ar4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Labonté B, Azoulay N, Yerko V, Turecki G, Brunet A. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. [Internet] Transl Psychiatry. 2014;4:e368. doi: 10.1038/tp.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro-Vale I, van Rossum EFC, Machado JC, Mota-Cardoso R, Carvalho D. Genetics of glucocorticoid regulation and posttraumatic stress disorder—What do we know? [Internet] Neurosci Biobehav Rev. 2016;63:143–157. doi: 10.1016/j.neubiorev.2016.02.005. The authors outline a comprehensive review of the role the GR and GC signaling plays in the pathophysiolgy of PTSD from studies that have been published thus-far. Changes in levels of cortisol, GR polymorphisms, GR sensitivity and epigenetic changes to stress cam all impact the functioning of the GR in the HPA axis response to stress and in the development of PTSD. The authors cite consensus findings and outliers regarding the regulation of GR in PTSD and point out the potential targets in GC signaling that can be used in the treatment and prevention of PTSD. [DOI] [PubMed] [Google Scholar]
- 16.Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, Salum G, Magalhães PV, Kapczinski F, Kauer-Sant’Anna M. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. [Internet] The lancet Psychiatry. 2015;2:1002–12. doi: 10.1016/S2215-0366(15)00309-0. The authors conducted a meta-analysis and meta-regression of studies comparing inflammatory markers between patients with PTSD and healthy controls. The meta-analysis was based on 20 published studies and found that levels of IL-1β, IL-6 and IFN-γ were elevated in individuals with PTSD compared to healthy controls, with standardised mean differences ranging from .49 to 1.42. [DOI] [PubMed] [Google Scholar]
- 17.Eraly SA, Nievergelt CM, Maihofer AX, Barkauskas DA, Biswas N, Agorastos A, O’Connor DT, Baker DG. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. [Internet] JAMA psychiatry. 2014;71:423–31. doi: 10.1001/jamapsychiatry.2013.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smid GE, van Zuiden M, Geuze E, Kavelaars A, Heijnen CJ, Vermetten E. Cytokine production as a putative biological mechanism underlying stress sensitization in high combat exposed soldiers [Internet] Psychoneuroendocrinology. 2015;51:534–546. doi: 10.1016/j.psyneuen.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Furtado M, Katzman MA. Neuroinflammatory pathways in anxiety, posttraumatic stress, and obsessive compulsive disorders. [Internet] Psychiatry Res. 2015;229:37–48. doi: 10.1016/j.psychres.2015.05.036. [DOI] [PubMed] [Google Scholar]
- 20.Wilson CB, McLaughlin LD, Nair A, Ebenezer PJ, Dange R, Francis J. Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post-traumatic stress disorder in a predator exposure animal model. [Internet] PLoS One. 2013;8:e76146. doi: 10.1371/journal.pone.0076146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levkovitz Y, Fenchel D, Kaplan Z, Zohar J, Cohen H. Early post-stressor intervention with minocycline, a second-generation tetracycline, attenuates post-traumatic stress response in an animal model of PTSD. [Internet] Eur Neuropsychopharmacol. 2015;25:124–32. doi: 10.1016/j.euroneuro.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Breen MS, Maihofer AX, Glatt SJ, Tylee DS, Chandler SD, Tsuang MT, Risbrough VB, Baker DG, O’Connor DT, Nievergelt CM, et al. Gene networks specific for innate immunity define post-traumatic stress disorder. [Internet] Mol Psychiatry. 2015;20:1538–45. doi: 10.1038/mp.2015.9. By applying weighted gene co-expression network analysis (WGCNA) to RNA-Seq and microarray peripheral blood leukocyte gene expression taken from U.S. Marines pre- and post-deployment, the authors identified groups of enriched genes that were differentially expressed between PTSD cases and controls. The PTSD risk and PTSD cases groups both had enhanced differential expression of genes associated with innate immune responses mediated by interferon signaling. These findings add to the authors previous work showing differential expression of CRP and genes involved in antiviral interferon response to be associated with risk of developing PTSD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aiello AE, Dowd JB, Jayabalasingham B, Feinstein L, Uddin M, Simanek AM, Cheng CK, Galea S, Wildman DE, Koenen K, et al. PTSD is associated with an increase in aged T cell phenotypes in adults living in Detroit. [Internet] Psychoneuroendocrinology. 2016;67:133–141. doi: 10.1016/j.psyneuen.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Zuiden M, Kavelaars A, Vermetten E, Olff M, Geuze E, Heijnen C. Pre-deployment differences in glucocorticoid sensitivity of leukocytes in soldiers developing symptoms of PTSD, depression or fatigue persist after return from military deployment. [Internet] Psychoneuroendocrinology. 2015;51:513–24. doi: 10.1016/j.psyneuen.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Zhou J, Nagarkatti P, Zhong Y, Ginsberg JP, Singh NP, Zhang J, Nagarkatti M. Dysregulation in microRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. [Internet] PLoS One. 2014;9:e94075. doi: 10.1371/journal.pone.0094075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bam M, Yang X, Zhou J, Ginsberg JP, Leyden Q, Nagarkatti PS, Nagarkatti M. Evidence for Epigenetic Regulation of Pro-Inflammatory Cytokines, Interleukin-12 and Interferon Gamma, in Peripheral Blood Mononuclear Cells from PTSD Patients. [Internet] J Neuroimmune Pharmacol. 2015;11:168–81. doi: 10.1007/s11481-015-9643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jergović M, Bendelja K, Vidović A, Savić A, Vojvoda V, Aberle N, Rabatić S, Jovanovic T, Sabioncello A. Patients with posttraumatic stress disorder exhibit an altered phenotype of regulatory T cells. [Internet] Allergy Asthma Clin Immunol. 2014;10:43. doi: 10.1186/1710-1492-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS. Regulatory T-cells in autoimmune diseases: challenges, controversies and--yet--unanswered questions. [Internet] Autoimmun Rev. 2015;14:105–16. doi: 10.1016/j.autrev.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Rane G, Dai X, Shanmugam MK, Arfuso F, Samy RP, Lai MKP, Kappei D, Kumar AP, Sethi G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res Rev. 2016;25:55–69. doi: 10.1016/j.arr.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Georgin-Lavialle S, Aouba A, Mouthon L, Londono-Vallejo JA, Lepelletier Y, Gabet AS, Hermine O. The telomere/telomerase system in autoimmune and systemic immune-mediated diseases [Internet] Autoimmun Rev. 2010;9:646–651. doi: 10.1016/j.autrev.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 31.Lohr JB, Palmer BW, Eidt CA, Aailaboyina S, Mausbach BT, Wolkowitz OM, Thorp SR, Jeste DV. Is Post-Traumatic Stress Disorder Associated with Premature Senescence? A Review of the Literature. [Internet] Am J Geriatr Psychiatry. 2015;23:709–25. doi: 10.1016/j.jagp.2015.04.001. The authors reviewed 22 articles published between 2000 and 2014 from a total of 66 studies that analyzed targeted outcomes in order to identify the link, if any, between PTSD and early aging. They found PTSD to be associated with senescent-like changes including shorter leukocyte telomere length, increased pro-inflammatory cytoines, higher incidence of age associated illnesses like CVD, dementia and an overall increased mortality. This literature review highlights premature senescence as the possible mechanism that leads to a high prevelance of adverse outcomes in PTSD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dias BG, Ressler KJ. PACAP and the PAC1 receptor in post-traumatic stress disorder. [Internet] Neuropsychopharmacology. 2013;38:245–6. doi: 10.1038/npp.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.NB. The epidemiology of posttraumatic stress disorder: what is the extent of the problem? J Clin Psychiatry. 2001;62:16–22. [PubMed] [Google Scholar]
- 34.Sherin JE, Nemeroff CB. Post-traumatic stress disorder: the neurobiological impact of psychological trauma. [Internet] Dialogues Clin Neurosci. 2011;13:263–78. doi: 10.31887/DCNS.2011.13.2/jsherin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cattaneo A, Gennarelli M, Uher R, Breen G, Farmer A, Aitchison KJ, Craig IW, Anacker C, Zunsztain PA, McGuffin P, et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline “predictors” and longitudinal “targets”. [Internet] Neuropsychopharmacology. 2013;38:377–85. doi: 10.1038/npp.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golier JA, Caramanica K, Demaria R, Yehuda R. A Pilot Study of Mifepristone in Combat-Related PTSD. [Internet] Depress Res Treat. 2012;2012:393251. doi: 10.1155/2012/393251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golier JA, Caramanica K, Michaelides AC, Makotkine I, Schmeidler J, Harvey PD, Yehuda R. A randomized, double-blind, placebo-controlled, crossover trial of mifepristone in Gulf War veterans with chronic multisymptom illness. [Internet] Psychoneuroendocrinology. 2016;64:22–30. doi: 10.1016/j.psyneuen.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Gaali S, Kirschner A, Cuboni S, Hartmann J, Kozany C, Balsevich G, Namendorf C, Fernandez-Vizarra P, Sippel C, Zannas AS, et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. [Internet] Nat Chem Biol. 2015;11:33–7. doi: 10.1038/nchembio.1699. [DOI] [PubMed] [Google Scholar]
- 39.Mac Callum PE, Hebert M, Adamec RE, Blundell J. Systemic inhibition of mTOR kinase via rapamycin disrupts consolidation and reconsolidation of auditory fear memory. [Internet] Neurobiol Learn Mem. 2014;112:176–85. doi: 10.1016/j.nlm.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Cavalli G, Dinarello CA. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. [Internet] Rheumatology (Oxford) 2015;54:2134–44. doi: 10.1093/rheumatology/kev269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mease P, Gottlieb AB, Berman A, Drescher E, Xing J, Wong R, Banerjee S. The Efficacy and Safety of Clazakizumab, an Anti-Interleukin-6 Monoclonal Antibody, in a Phase 2b Study of Adults with Active Psoriatic Arthritis. [Internet] Arthritis Rheumatol (Hoboken, NJ) 2016 doi: 10.1002/art.39700. [DOI] [PubMed] [Google Scholar]
- 42.Atzeni F, Straub RH, Cutolo M, Sarzi-Puttini P. Anti-TNF therapy restores the hypothalamic-pituitary-adrenal axis. [Internet] Ann N Y Acad Sci. 2010;1193:179–81. doi: 10.1111/j.1749-6632.2009.05366.x. [DOI] [PubMed] [Google Scholar]
- 43.Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. [Internet] Nat Med. 2015;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walker FR. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? [Internet] Neuropharmacology. 2013;67:304–17. doi: 10.1016/j.neuropharm.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Sacre S, Medghalchi M, Gregory B, Brennan F, Williams R. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit toll-like receptors. [Internet] Arthritis Rheum. 2010;62:683–93. doi: 10.1002/art.27304. [DOI] [PubMed] [Google Scholar]
- 46.Cambron M, Mostert J, Haentjens P, D’Hooghe M, Nagels G, Willekens B, Heersema D, Debruyne J, Van Hecke W, Algoed L, et al. Fluoxetine in progressive multiple sclerosis (FLUOX-PMS): study protocol for a randomized controlled trial. [Internet] Trials. 2014;15:37. doi: 10.1186/1745-6215-15-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taler M, Gil-Ad I, Korob I, Weizman A. The immunomodulatory effect of the antidepressant sertraline in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. [Internet] Neuroimmunomodulation. 2011;18:117–22. doi: 10.1159/000321634. [DOI] [PubMed] [Google Scholar]
- 48.Mostert JP, Admiraal-Behloul F, Hoogduin JM, Luyendijk J, Heersema DJ, van Buchem MA, De Keyser J. Effects of fluoxetine on disease activity in relapsing multiple sclerosis: a double-blind, placebo-controlled, exploratory study. [Internet] J Neurol Neurosurg Psychiatry. 2008;79:1027–31. doi: 10.1136/jnnp.2007.139345. [DOI] [PubMed] [Google Scholar]
- 49.Green B. Prazosin in the treatment of PTSD. [Internet] J Psychiatr Pract. 2014;20:253–9. doi: 10.1097/01.pra.0000452561.98286.1e. [DOI] [PubMed] [Google Scholar]
- 50.Dong J, Mrabet O, Moze E, Li K, Neveu PJ. Lateralization and catecholaminergic neuroimmunomodulation: prazosin, an alpha1/alpha2-adrenergic receptor antagonist, suppresses interleukin-1 and increases interleukin-10 production induced by lipopolysaccharides. [Internet] Neuroimmunomodulation. 10:163–8. doi: 10.1159/000067178. [DOI] [PubMed] [Google Scholar]
- 51.Tung D, Ciallella J, Cheung PH, Saha S. Novel anti-inflammatory effects of doxazosin in rodent models of inflammation. [Internet] Pharmacology. 2013;91:29–34. doi: 10.1159/000343762. [DOI] [PubMed] [Google Scholar]
- 52.Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, Kirkwood K, Aan Het Rot M, Lapidus KAB, Wan L-B, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. [Internet] JAMA psychiatry. 2014;71:681–8. doi: 10.1001/jamapsychiatry.2014.62. [DOI] [PubMed] [Google Scholar]
- 53.Potter DE, Choudhury M. Ketamine: repurposing and redefining a multifaceted drug. [Internet] Drug Discov Today. 2014;19:1848–54. doi: 10.1016/j.drudis.2014.08.017. [DOI] [PubMed] [Google Scholar]