

Abstract
In Alzheimer's disease (AD), tau undergoes numerous modifications, including increased phosphorylation at serine-422 (pS422). In the human brain, pS422 tau protein is found in prodromal AD, correlates well with cognitive decline and neuropil thread pathology, and appears associated with increased oligomer formation and exposure of the N-terminal phosphatase-activating domain (PAD). However, whether S422E phosphorylation contributes to toxic mechanisms associated with disease-related forms of tau remains unknown. Here, we report that S422-pseudophosphorylated tau (S422E) lengthens the nucleation phase of aggregation without altering the extent of aggregation or the types of aggregates formed. When compared to unmodified tau aggregates, the S422E modification significantly increased the amount of SDS-stable tau dimers, despite similar levels of immunoreactivity with an oligomer-selective antibody (TOC1) and another antibody that reports PAD exposure (TNT1). Vesicle motility assays in isolated squid axoplasm further revealed that S422E tau monomers inhibited anterograde, kinesin-1 dependent fast axonal transport (FAT). Unexpectedly, and unlike unmodified tau aggregates, which selectively inhibit anterograde FAT, aggregates composed of S422E tau were found to inhibit both anterograde and retrograde FAT. Highlighting the relevance of these findings to human disease, pS422 tau was found to colocalize with tau oligomers and with a fraction of tau showing increased PAD exposure in the human AD brain. This study identifies novel effects of pS422 on tau biochemical properties, including prolonged nucleation and enhanced dimer formation, which correlate with a distinct inhibitory effect on FAT. Taken together, these findings identify a novel mechanistic basis by which pS422 confers upon tau a toxic effect that may directly contribute to axonal dysfunction in AD and other tauopathies.
Keywords: Alzheimer's disease, tauopathy, oligomer, protein phosphorylation, axonal transport
Introduction
Tau is a major microtubule-associated protein component of the neuronal cytoskeleton that plays an important role on the regulation of signaling pathways within axon (Kanaan et al., 2013). Under pathological conditions, tau transitions from a soluble monomeric state to oligomeric and filamentous aggregates, which accumulate as neurofibrillary tangles (NFTs), neuropil threads, and neuritic plaques (Grundke-Iqbal et al., 1986a; 1986b; Kosik et al., 1986; Wood et al., 1986a). The accumulation of tau aggregates in various morphological states is characteristic of a number of neurodegenerative diseases referred to as tauopathies, including Alzheimer's disease (AD) (Spillantini and Goedert, 1998). Pathological tau inclusions in these diseases display numerous abnormal alterations, some of which can facilitate aggregation and attenuate microtubule binding and stabilization (Bierer et al., 1995; Biernat et al., 1993; Drewes et al., 1995). At least 25 threonine/serine sites are phosphorylated in filamentous tau (Morishima-Kawashima et al., 1995); however, many of these phosphorylation sites are shared with normal tau protein derived from human brain biopsies (Matsuo et al., 1994). Additionally, NFT formation follows a prescribed sequence of events during disease progression; some sites are phosphorylated early in the pathogenic process, whereas other sites are not phosphorylated until after NFTs have already formed (Kimura et al., 1996; Stoothoff and Johnson, 2005). Hence, specific tau phosphorylation events contributing to disease-causing processes remain unclear. From this, it is essential to understand the effect of individual phosphorylation sites on tau aggregation and neurodegenerative mechanisms
C-terminal phosphorylation of tau at serine-422 (pS422) is a disease-related modification that occurs early in the progression of AD (Guillozet-Bongaarts et al., 2006; Kimura et al., 1996; Vana et al., 2011). In AD, pS422 tau is found in pre-tangle neurons (Guillozet-Bongaarts et al., 2006), and appearance of the pS422 epitope is a stronger correlate of disease neuropathology and cognitive decline than mature NFT deposition (Vana et al., 2011). Moreover, the lack of pS422 tau in cognitively intact control brain samples further supports the disease-specificity of phosphorylation at S422 (Bussière et al., 1999; Hasegawa et al., 1996). Evidence from animal models of AD supports this conclusion. For example, appearance of the pS422 epitope is dependent upon β-amyloid-induced NFT formation in transgenic mice expressing mutant P301L human tau protein (Deters et al., 2009; Gotz et al., 2001; Grueninger et al., 2010).
Recent studies from our group and others have implicated two pathogenic events associated with disease-related forms of tau. First, conformational changes in tau that aberrantly expose the phosphatase-activating domain (PAD, amino acids 2-18) activate a PP1/GSK3β signaling pathways and promote inhibition of anterograde fast axonal transport (FAT) (Kanaan et al., 2011; LaPointe et al., 2009b). The tau N-terminal 1 (TNT1) antibody, a monoclonal antibody raised against amino acids 2-18, demonstrates that PAD exposure is a disease-related event (Kanaan et al., 2011; 2012). The pattern of TNT1-immunoreactivity indicates diffuse, granular cytoplasmic staining in pretangle neurons preceding the appearance of the early phospho-epitope AT8 in postmortem human tissue (Kanaan et al., 2011). Second, prefibrillar oligomeric species of tau are more potent inducers of neurodegeneration than fibrillar tau aggregates (Berger et al., 2007; Kopeikina et al., 2011; Maeda et al., 2007; Patterson et al., 2011a; Sahara et al., 2013). In this regard, a monoclonal antibody that preferentially labels tau oligomers (tau oligomeric complex 1; TOC1) (Grundke-Iqbal et al., 1986b; 1986a; Kosik et al., 1986; Patterson et al., 2011a; Ward et al., 2013; Wood et al., 1986a) displays elevated immunoreactivity in prodromal AD brains (mild cognitive impairment) (Mufson et al., 2014; Spillantini and Goedert, 1998), and does not colocalize with later events in tangle formation, such as truncation at residue 391 or formation of β-sheet structures (Bierer et al., 1995; Biernat et al., 1993; Drewes et al., 1995; Patterson et al., 2011a). Interestingly, pS422 accompanies PAD exposure and tau oligomer formation in early tau pathology observed in several brain regions (e.g. medial temporal lobe, temporal cortex and cholinergic basal forebrain) of Braak I-VI cases (Kanaan et al., 2016; Morishima-Kawashima et al., 1995; Mufson et al., 2014; Patterson et al., 2011a). However, whether S422 phosphorylation modulates PAD exposure or oligomer formation remains unknown.
The present study sought to determine whether S422 phosphorylation modulates tau aggregation, PAD exposure, oligomer formation, and the toxic effect of aggregated tau on FAT. We present evidence indicating that pseudophosphorylation at S422 (S422E) lengthens the nucleation phase of tau aggregation kinetics, without altering the overall extent of aggregation or the types of aggregates formed. We also demonstrate that, compared to unmodified tau aggregates, the S422E modification enhances the formation of SDS-resistant dimers without altering exposure of the TNT1 or TOC1 epitopes. Using vesicle motility assays in isolated squid axoplasm, we further report that S422E monomers and aggregates inhibit FAT. Lastly, we establish that the tau species isolated from human AD brain containing the pS422 modification also have PAD exposed (TNT1 reactive) and are in oligomeric conformations (TOC1 reactive). Collectively, these results provide evidence of a functional role for the pS422 disease-related modification in mechanisms of tau toxicity relevant to AD and other tauopathies.
Materials & Methods
Recombinant tau proteins
Full-length tau protein (hT40) corresponds to the longest isoform in the adult human brain, consisting of 441 amino acids and four microtubule-binding repeats. Pseudophosphorylation (i.e., switching serine to a glutamate) was used to mimic phosphorylation at position 422 in tau. Pseudophosphorylation is a routine approach to examine both single and multiple phosphorylation residues on the tau protein (Guillozet-Bongaarts et al., 2006; Jeganathan et al., 2008; Kanaan et al., 2012; 2011; LaPointe et al., 2009a; Matsuo et al., 1994; Necula and Kuret, 2005). Pseudophosphorylated S422E tau was generated using mutagenesis of the hT40 construct per the manufacturer's instructions (QuikChange Lightning Multi Site-Directed Mutagenesis Kit, Agilent Technologies, 210515). The forward primer used in this reaction was: CAGCATCGACATGGTAGACGAGCCCCAGCTCGCCACGCTAG. pT7c plasmid DNAs containing either hT40 or S422E were amplified in XL10-Gold ultracompetent Escherichia coli, purified using the PureLink Quick Plasmid Miniprep Kit (Invitrogen, K2100), and verified by DNA sequencing. Proteins were expressed in T7 Express supercompetent Escherichia coli, and purified through C-terminal His-tag metal affinity chromatography (Talon resin, Clontech, 635502) followed by size exclusion chromatography using an S200 26/60 column (GE Healthcare, 17-1195-01) as previously described (Carmel et al., 1996; Kimura et al., 1996; Stoothoff and Johnson, 2005).
In vitro tau polymerization reaction
Tau polymerization was induced with arachidonic acid (ARA; Cayman Chemical, 90010) as previously described (Gamblin et al., 2000; Guillozet-Bongaarts et al., 2006; Kimura et al., 1996; LaPointe et al., 2009a; Vana et al., 2011). Briefly, recombinant tau (2μM) was incubated in polymerization buffer (5mM dithiothreitol, 100mM NaCl, 0.1mM EDTA, 10mM HEPES, pH 7.6) with 75μM ARA (final concentration) at room temperature for 6 h. ARA was stored at −20°C, and working solutions were prepared in 100% ethanol immediately prior to use. Control (monomer) samples were prepared in polymerization buffer with ARA vehicle (i.e., 100% ethanol). A combination of right angle laser light scatter, thioflavin S (ThS), and transmission electron microscopy was used to validate tau aggregation.
Right angle laser light scatter (LLS) assay
Tau polymerization was monitored in real-time by measuring the intensity of right angle light scatter (iS; (Gamblin et al., 2000; Guillozet-Bongaarts et al., 2006)). A 475 nm laser (B&W Tek, Inc., BWI-475-20-E) was used to illuminate polymerization reactions. Images of scattered light were captured throughout the 6h polymerization reaction using a high sensitivity CMOS digital camera (Thor Labs, DCC1240M) and imaging software uc480 Viewer version 4.2. Images were collected using an exposure time of 175.15 ms, a frame rate of 4.01 fps, and a pixel clock of 11 MHz. Individual images were imported into Photoshop CS5 Version 12.1 (Adobe Systems, Inc.) where the marquee tool was used to select a region of interest (75 × 10 pixels) within the scattered light near the middle of the cuvette. The intensity of scattered light was obtained using the histogram feature. The background intensity was subtracted by measuring scattering in the cuvette prior to the addition of ARA (Gamblin et al., 2000; Vana et al., 2011). Each experiment was repeated five independent times, and background-corrected light scatter data were plotted as a function of time. The polymerization kinetics curve was best fit with the sigmoidal Gompertz equation (Bussière et al., 1999; Hasegawa et al., 1996; Necula and Kuret, 2004a; Winsor, 1932).
Thioflavin S
A 0.0175% solution of ThS (Sigma, #T1892) was prepared on the day of each experiment and filtered using a 0.2μm syringe filter before use. To measure ThS fluorescence, 6 μL of 0.0175% ThS was added to 150 μL of 2μM aggregated tau (hT40 or S422E) in a black 96-well plate (Fisher Scientific, 06-443-2), such that the final concentration of ThS was 0.0007%. Fluorescence measurements were performed at 25°C in a GloMax Multi Detection System (Promega) with an excitation wavelength of 490 nm and an emission wavelength of 510-570 nm. Each experiment was repeated three independent times. To compare measurements from independent experiments, background fluorescence was measured in wells containing tau (hT40 or S422E) + vehicle (ethanol), and subtracted from each respective treatment condition.
Transmission electron microscopy (TEM)
Aliquots of polymerized tau samples were fixed with 2% glutaraldehyde, spotted onto 300 mesh Formvar carbon coated copper grids (Electron Microscopy Sciences, 215-412-8400), and negatively stained with 2% uranyl acetate. Grids were examined with a JEOL JEM-1400 Plus electron microscope at 80 kV and ×10,000 magnification. Images were captured with an AMT XR81 digital camera and AMT software version 602.6 (Advanced Microscopy Techniques). Tau samples were diluted to a final concentration of 1.6 μM to capture representative images of each polymerization reaction, or diluted to a final concentration of 0.2μM for quantitative TEM analysis. A higher dilution was used for the quantitative analysis to obtain samples with discrete, non-overlapping aggregates.
Images were processed using Image J to measure total aggregate mass, individual aggregate area, aggregate number, and individual aggregate lengths (Fig. S1). For the individual aggregate measures (i.e., number and length) objects were separated into three categories based on area; 1) oligomers were <1000nm2, 2) short filaments were 1000 – 5000 nm2, or 3) long filaments were >5000 nm2. All images were captured at ×10,000 magnification with a standard beam intensity and gain for consistency across samples. A macro was used in Image J to measure total aggregate mass, individual aggregate area and aggregate number. The scale was set at 373.5 pixels equal to 400 nm. Images were smoothed twice to eliminate small variations in estimations of object area, and auto threshold was used to distinguish aggregates over background. Total mass of aggregated tau was determined by summating the area of individual aggregates (nm2) covering the field. Aggregate counts were performed for three populations of aggregates sorted by area: oligomers = < 1000 nm2, short filaments = 1000 – 5000 nm2, long filaments = > 5000 nm2. Aggregate length was not an output of the Image J macro, thus these measurements were made manually in Image J by measuring the distance along the longest axis of individual oligomers (100 per sample), short filaments (average of 24 per sample) and long filaments (average of 50 per sample). Each experiment was repeated three independent times, and five fields from each sample were randomly captured during each experiment and used for analysis.
Soluble tau extraction from fresh frozen human tissue
Soluble tau was prepared from frontal cortex tissue of non-demented control (n=6; age: mean=83yrs, range=75-95 yrs; sex: 4 female, 2 male; Braak stage II-III) and AD cases (n=6; age: mean=84 yrs, range=69-93 yrs; sex: 0 female, 6 male; Braak stage VI) as described previously (Deters et al., 2009; Gotz et al., 2001; Grueninger et al., 2010; Kanaan et al., 2016). Briefly, tissue pieces from cortex (0.5-1 grams) were homogenized on ice in ten volumes (1g=10ml) of brain homogenization buffer (50mM Tris pH 7.4, 274mM NaCl, 5mM KCl, 1mM PMSF, and 10 μg/ml each of pepstatin, leupeptin, bestatin, and aprotinin). The soluble tau fraction was collected in the supernatant following centrifugation at 27,000 × g for 20 minutes at 4°C. The samples were assayed for total protein concentration using the Lowry protein assay and stored at -80°C until used in sandwich ELISAs for analysis (see below).
Tau sandwich enzyme-linked immunosorbent assays (ELISAs)
A novel sandwich ELISA was developed to measure the relative levels of oligomer formation and PAD exposure in recombinant tau proteins and pS422-containing tau species in the frontal cortex from human brains samples. All steps were performed at room temperature. Washing was performed between each step using 200μL/well of ELISA wash buffer (100mM borate acid, 25mM sodium borate, 75mM NaCl, 0.25mM thimerosal, 0.4% (w/v) bovine serum albumin, 0.05% (v/v) Tween-20), and all other steps were performed using 50μL solution/well. For recombinant tau protein sandwich ELISAs, the following capture antibodies were used: Tau5, TNT1, or TOC1. Tau5 is a monoclonal mouse IgG1 raised against bovine tau, with a continuous epitope between amino acids 215-225. Tau5 is a pan-tau antibody that labels monomeric and aggregated tau equally well (Carmel et al., 1996; Kanaan et al., 2011; LaPointe et al., 2009b; LoPresti et al., 1995; Porzig et al., 2007). TNT1 is a monoclonal mouse IgG1 directed against N-terminal amino acids 2-18 (AEPRQEFEVMEDHAGTY), the PAD (Kanaan et al., 2012; 2011). In native assays, reactivity of TNT1 is dependent upon conformational display of PAD, which is a disease-specific event (Kanaan et al., 2012; 2011). TOC1 is a monoclonal mouse IgM raised against tau dimers (Berger et al., 2007; Kopeikina et al., 2011; Maeda et al., 2007; Patterson et al., 2011a; Sahara et al., 2013). The TOC1 epitope (amino acids 209-224) is conformation dependent as it is revealed upon dimerization and oligomerization of tau (Ward et al., 2013). High binding 96-well microplates (Fisher Scientific, 07-200-35) were coated with Tau5 (1μg/ml), TNT1 (2μg/ml) or TOC1 (2μg/ml) capture antibody diluted in borate saline (100mM borate acid, 25mM sodium borate, 75mM NaCl, 0.25mM thimerosal) for 60 min. Plates were washed twice with ELISA wash buffer, and then blocked for 60 min with ELISA wash containing 5% non-fat dry milk (blocking reagent). Each well was washed twice and then recombinant tau samples were added to each well for 90 min. Recombinant hT40 and S422E tau monomers or aggregates from the in vitro polymerization assays were diluted with polymerization buffer to a final concentration of 5nM (Tau5 and TNT1 assays) or 20nM (TOC1 assay). Titer assays were performed for each capture antibody sandwich ELISA to ensure recombinant tau protein concentrations produced signal within the linear range. Wells were rinsed 3 times with ELISA wash and then incubated for 90 min with the rabbit polyclonal pan-tau antibody R1 (Berry et al., 2004) diluted 1:20,000 in blocking reagent, as the detection antibody. Wells were washed 3 times, and then incubated for 60 min with goat anti-rabbit IgG conjugated to horseradish peroxidase (1:5000; Vector Labs, PI-1000) diluted in blocking reagent. Wells were washed 3 times, and assays were developed using 3,3′,5,5′-tetramethylbenzidine (TMB; Sigma) for 15 min (Tau5, TNT1) or 30 min (TOC1). Development reactions were stopped with 3.5% sulfuric acid and absorbance was read at 450nm on a SpectraMax Plus 384 microplate reader (Molecular Devices). The amount of total tau applied in the assay was known and equivalent between groups since these are recombinant protein samples, but absorbance (A) is not a linear measure (i.e., A=Log10 (1/transmittance) and not useful for comparing across samples (e.g. A=0.3 is 50% light absorbed, A=1 is 90% light absorbed and A=2 is 99% light absorbed). Thus, the absorbance data were converted to percent light absorbed (a linear scale) using the following equation %A=(1–10-x)*100, where x is absorbance. The percent light absorbed data were used for statistical comparisons across groups.
A similar protocol was used to measure the levels of PAD exposure and tau oligomers in pS422-containing tau species in the soluble tau fractions of the frontal cortex from control and AD brains (Kanaan et al., 2016). Tau species containing pS422 protein were captured by coating ELISA microplates for 60 min with rabbit anti-pS422 antibody (Abcam, ab79415, diluted to 2μg/ml in borate saline). Plates were rinsed twice, blocked for 60 min with blocking reagent, and rinsed twice before samples were added to the well for 90 min. Human brain extracts were diluted to a final total protein concentration of 0.4 μg/μL (i.e., 20 μg/well). The optimal protein amount for the soluble tau fraction was determined by titering to ensure the ELISAs were performed within the linear rage of reactivity. Wells were rinsed twice, and then incubated for 90 min with Tau5 (0.005μg/ml), TNT1 (0.01μg/ml) or TOC1 (1μg/ml) as the detection antibodies (diluted in blocking reagent). Wells were rinsed 3 times and incubated for 90 min with horse anti-mouse IgG conjugated to horseradish peroxidase (Vector Labs, PI-2000) or goat anti-mouse IgM-specific conjugated to horseradish peroxidase (Jackson, 115-035-020) diluted (0.2μg/ml) in blocking reagent. The wells were rinsed 3 times, signal was detected by developing TMB for 6 min (TOC1) or 30 min (Tau5 and TNT1), and then the reaction was stopped using 3.5% sulfuric acid. Tau standard ELISAs were performed with human sample sandwich ELISAs to estimate the amount of tau captured by anti-pS422 and detected with Tau 5, TNT1 or TOC1. For the tau standards, a serial dilution of recombinant full-length human tau protein ranging from 100-3.13 ng was bound to the ELISA plate for 60 min, blocked as above, and detected using either Tau5, TNT1 or TOC1 antibodies and HRP-secondary antibodies exactly as in the recombinant protein sandwich ELISAs above. Each standard was run in duplicate and developed simultaneously with the sandwich ELISAs to ensure accurate interpolation of unknown tau amounts. The standard curve data were log10 transformed and best fit to a sigmoidal curve (r2=0.99 for all three). This provided a standard curve of absorbance values that were derived from Tau5, TNT1 or TOC1 reactivity with known amounts of tau protein. The quantity of tau (ng) in each human sample was interpolated from the tau standard curves and then converted to a concentration of ng/μL by dividing the interpolated quantity by the volume of the sample used (i.e., 50μL). Finally, the data (i.e., concentrations of tau) were normalized to reduce skewness using logarithmic transformations and then used for statistical comparisons.
Immunoblotting
Monomeric and aggregated tau samples were prepared in Laemmli sample buffer (20mM Tris pH 6.8, 6% glycerol (v/v). 1.6% sodium dodecyl sulfate (v/v), 0.85% 2-mercaptoethanol (v/v), 0.002% bromophenol blue (v/v)), incubated at 90°C for 5 min, separated by SDS-PAGE using 4-20% Criterion TGX precast gels (Bio-Rad Laboratories, Inc., 5671094) and transferred to 0.22 μm nitrocellulose membranes (Pall Corporation, 66485). Membranes were blocked with 2% non-fat dry milk in Tris-buffered saline (TBS; 50mM Tris, 150mM NaCl, pH 7.4), and incubated overnight at 4°C in Tau13 antibody (1:50,000), a mouse monoclonal IgG1 antibody against the N-terminus of tau (García-Sierra et al., 2003). Membranes were rinsed four times in TBS + 0.1% Tween 20, and developed with the affinity-purified secondary antibody conjugated to IRDye 680RD goat anti-mouse IgG (H+L) (LiCor, 925-68070). Image acquisition and intensity measurements were performed using a LiCor Odyssey system.
Squid axoplasm motility assay
FAT was evaluated in freshly extruded squid axoplasm from giant squid axon (Loligo pealii; Marine Biological Laboratory, Woods Hole, MA) as previously described (Brady et al., 1993; Morfini et al., 2007; Song et al., 2016). Tau protein (monomer or aggregate) was diluted in X/2 buffer (175 mM potassium aspartate, 65 mM taurine, 35 mM betaine, 25 mM glycine, 10 mM HEPES, 6.5 mM MgCl2, 5 mM EGTA, 1.5 mM CaCl2, 0.5mM glucose, 10mM adenosine triphosphate, pH 7.2) and perfused into isolated axoplasm at a final concentration of 2 μM. The tau protein concentration of 2 μM was chosen specifically because it is within the physiological range of tau in the brain (Alonso et al., 1996; King et al., 1999). Motility was analyzed using a Zeiss Axiomat microscope equipped with a 100× (1.3 numerical aperture) objective and differential interference contrast optics. Images were obtained using a Hammamatsu C2400 CCD with an Argus 20 for image adjustment and the video was further processed using a Hamamatsu Photonics Microscopy C2117 video manipulator for generation of calibrated cursors and scale bars (Song et al., 2016). Anterograde and retrograde FAT were measured by matching calibrated cursor movements to the speed of vesicles moving in the axoplasm preparation. Vesicle motility was measured for 50 min after perfusion and data were plotted as a function of time. Full-length hT40 monomer was used as the control group since no changes are observed for this experimental group when compared to buffer control-perfused axoplasms (LaPointe et al., 2009b; Morfini et al., 2007). Two comparisons were made between groups: i) the average velocity of transport over the last 20 min of the assay was compared to measure the extent of inhibition across treatment groups, and ii) the rate of change in transport velocity over the entire 50 min assay was evaluated by comparing the slope of the linear regression curve fit for each group.
Triple-Label Immunofluorescence (IF) for Confocal Microscopy
Triple-label IF was used to characterize co-localization between tau phosphorylated at S422, PAD exposed tau and oligomeric tau. Tissue sections from age-matched, non-demented controls (n=6; age: mean=81 yrs, range=73-91 yrs; sex: 1 female, 5 males; Braak stage I-III) and severe AD cases (n=6; age: mean=83, range=60-92 yrs; sex: 4 female, 2 males; Braak stage V-VI) were processed for triple-label IF using the TNT1 (mouse IgG1), TOC1 (mouse IgM) and pS422 (rabbit) antibodies according to published methods (Kanaan et al., 2016). The sections were incubated overnight at 4° C in a primary antibody solution containing TNT1 (1:30,000), TOC1 (1:2,000) and pS422 (1:1,000) antibodies followed by incubation in a secondary antibody solution of Alexa Fluor 488 goat anti-mouse IgG1-specific (Invitrogen, A-21121), Alexa Fluor 568 goat anti-mouse IgM-specific (Invitrogen, A-21043), and Alexa Fluor 405 goat anti-rabbit specific (Invitrogen, A-31556) antibodies (all diluted 1:500) for 2 hours. Following the staining procedure, sections were mounted on microscope slides, autofluorescence was blocked using sudan black as described (Kanaan et al., 2012; 2011; 2007), and coverslipped using hardset Vectashield mounting media. Control sections with one of the three primary antibodies omitted confirmed that each secondary label was specific to the appropriate primary antibody (i.e., no staining was observed with the fluorophore for the omitted antibody; data not shown). A Nikon A1+ laser scanning confocal microscope system equipped with solid-state lasers (488, 561, and 640) and Nikon Elements AR software were used to acquire image z-stacks (0.5μm step size), and the images (maximum intensity projections) were prepared for publication using Adobe Photoshop and Illustrator.
Statistics
All experiments were repeated at least three times (see figure legends). All experiments were analyzed by Student's t-test or two-way analysis of variance as indicated in figure legends. When overall significance was achieved, the Holm-Sidak post-hoc test was used to make all possible comparisons. Data were expressed as mean ± SEM. All tests were two-tailed, and significance was set at p ≤ 0.05. GraphPad Prism 6 software (GraphPad Software, Inc.) was used for all statistical tests.
Results
S422E lengthens the nucleation phase of aggregate formation in vitro, but overall extent of aggregation and the types of aggregate species formed are similar to wild-type tau
Recombinant tau protein (2μM) was induced to aggregate with ARA (75μM) to determine whether pseudophosphorylation at S422 influences tau polymerization characteristics. Aggregation was monitored in real-time for 360 min by right-angle LLS (Fig. 1). The data were best fit to the Gompertz sigmoidal function to evaluate polymerization kinetics (Table 1). The time to initiate polymerization (i.e., lag time or nucleation time) for S422E tau (8.5 ± 1.7 min) was 1.85 times longer than hT40 tau (4.6 ± 0.6 min). However, the rate of polymerization (kapparent) did not differ between hT40 tau and S422E tau. At 360 min, a similar intensity of light scattering was observed for hT40 (122.5 ± 1.2) and S422E tau (134.5 ± 1.7) indicating that the extent of aggregate formation was comparable between these two proteins. ThS fluorescence, measured at 360 min, also was comparable between hT40 aggregates (946.4 ± 73.8) and S422E aggregates (927.9 ± 63.7).
Figure 1. Pseudophosphorylation of S422 slows the initiation of tau polymerization but does not alter the elongation rate or overall extent of aggregation.
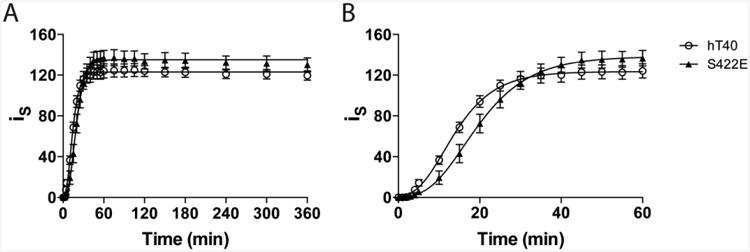
Polymerization kinetics of hT40 (open circles; n=4) and S422E (closed triangles; n=4) during the entire 360 min reaction (A). The first 60 min of the aggregation reaction shown in Panel A are displayed in (B) to illustrate the differences in lag time. Polymerization was measured as intensity of scattered light (iS) and polymerization curves were fit to Gompertz sigmoid function (curve fit data shown in Table 1).
Table 1.
Polymerization kinetics of hT40 and S422E fit to the Gompertz sigmoidal function.
| Protein | Lag Time (min) | kapparent (time-1) | Maximum (pixel intensity) | r2 | n |
|---|---|---|---|---|---|
| hT40 | 4.6 ± 0.6 | 0.154 ± 0.005 | 122.5 ± 1.2 | 0.9658 | 4 |
| S422E | 8.5 ± 1.7 | 0.135 ± 0.005 | 134.5 ± 1.7 | 0.9452 | 4 |
Lag time: indicates the time until detectable polymerization signal is present
kapparent (arbitrary units): proportional to the maximum rate of polymerization
Maximum: LLS signal after 6hr
Values are expressed as mean ± SE
Quantitative analysis of polymerized hT40 and S422E samples was performed on TEM micrographs to verify the results of LLS assays and evaluate whether the S422E modification modulates the types of aggregates formed by hT40 (Fig. 2A-E). Total aggregate mass (Fig. 2C; t4=1.73, p=0.16) and mean aggregate length (Fig. 2D; t4=0.82, p=0.46) were not significantly different between hT40 and S422E tau constructs. Aggregates were sorted by area into oligomers, short filaments and long filaments for comparisons between hT40 and S422E samples. Area measurements aligned well with previously defined criteria for oligomers using length (i.e., <50nm) (Patterson et al., 2011a)). Previously, filaments were defined as aggregates >50 nm in length (Patterson et al., 2011a), but here we further segregated filaments into short and long categories using area (short: 1000-5000 nm2 corresponding to ∼35-275 nm lengths and long: >5000nm2 corresponding to ∼200-1800 nm lengths) to better characterize these two populations of filaments (Fig. S1, Table S1). For both tau constructs, there were significantly more oligomer-type aggregates than short or long filaments formed, but no significant difference between the numbers of short or long filament (Fig. 2H; F(2,12)=86.64, p<0.0001). S422E did not differ significantly from hT40 in the number of oligomers, short filaments, or long filaments formed (F(1,12)=0.05, p=0.83).
Figure 2. Pseudophosphorylation at S422 did not alter the type or extent of aggregate formation.
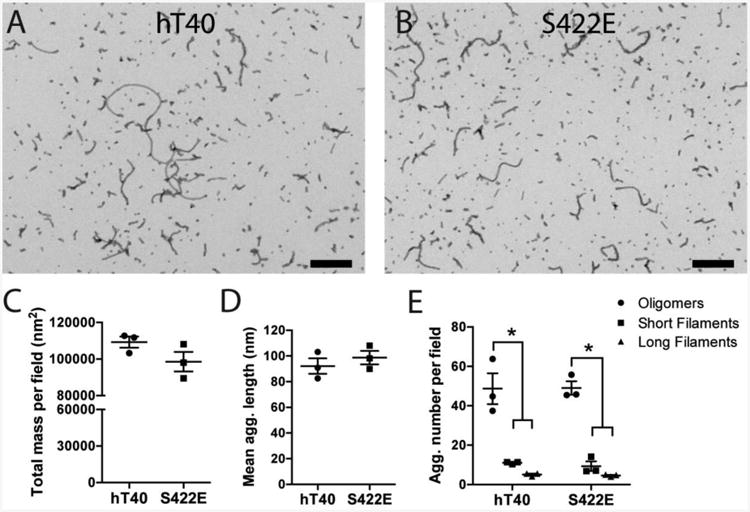
A-B) Electron micrographs of hT40 (A) and S422E (B) diluted to 1.6 μM provide representative images of the aggregate population. Scale bar = 400 nm. C-E) Quantitative EM analysis of total aggregate mass per field (C), mean aggregate length (D), and aggregate number per field (E) revealed no statistical differences between hT40 and S422E. Within each construct, there were significantly more oligomeric aggregates (circles) than short filaments (squares) or long filaments (triangles), but no difference between the numbers of short and long filaments. Values are expressed as mean ± SEM. n=3 samples/group, 5 replicate images used per sample. Data was analyzed using a 2-way analysis of variance with the Holm-Sidak post-hoc test. *p ≤ 0.05 vs. oligomers of the same construct.
S422 pseudophosphorylation does not alter the extent of PAD exposure and oligomer formation in vitro
We used a sandwich ELISA to determine whether monomer and aggregated hT40 and S422E tau differ in the extent of PAD exposure and oligomer formation (Fig. 3). Total tau was assessed using Tau5 sandwich ELISAs, and as expected the monomer and aggregate samples were equivalent between all groups (Fig. 3A; hT40 vs. S422E, F(1,12)=3.09, p=0.10; monomer vs. aggregate, F(1,12) =1.80, p=0.20). Compared to monomers, aggregation significantly increased PAD exposure for both hT40 and S422E samples (Fig. 3B; F(1,12)=685.8, p<0.0001), as indicated by increased TNT1 reactivity. Aggregation also significantly increased oligomer formation (TOC1 reactivity) compared to monomers in both hT40 and S422E samples (Fig. 3C; F(1,12)=109.3, p<0.0001). However, there were no differences in either TNT1 (F(1,12)=0.17, p=0.69) or TOC1 (F(1,12)=0.02, p=0.89) immunoreactivity between hT40 and S422E monomers or aggregates. The robust differences between monomers and aggregates, but lack of difference between hT40 and S422E constructs, were consistent across a range of sample concentrations (1.56-200 nM; data not shown).
Figure 3. Aggregation of both hT40 and S422E tau produces oligomers and exposure of the N-terminal phosphatase-activating domain.
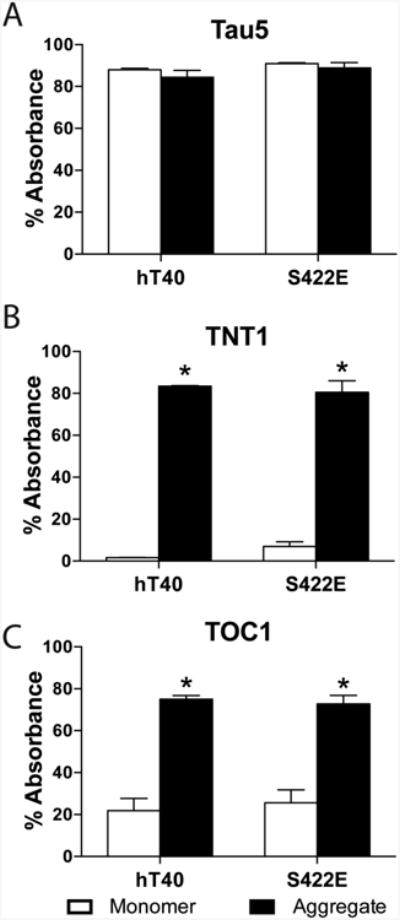
A-C) Sandwich ELISAs were used to evaluate (A) total tau using the monoclonal Tau5 antibody, (B) PAD exposure using the monoclonal TNT1 antibody, or (C) oligomer formation using the monoclonal TOC1 antibody as the capture antibodies. Tau aggregates (black bars) exhibited significantly more reactivity for both TOC1 and TNT1 as compared to monomer (white bars), however, there were no significant differences between hT40 and S422E aggregates or hT40 and S422E monomers. Values are expressed as mean ± SEM. n=4/group. Data were analyzed using a 2-way analysis of variance with the Holm-Sidak post-hoc test. *p ≤ 0.05 vs. monomer of the same construct.
Pseudophosphorylation at S422 enhances the formation of SDS-stable tau dimers
Next, we sought to investigate whether pseudophosphorylation at S422 altered the formation of SDS-stable tau dimers following aggregation in vitro (Patterson et al., 2011a). Immunoblotting with the high affinity total tau antibody, Tau13, detected tau monomers (∼64 kDa) and apparent 180 kDa tau dimers ((Patterson et al., 2011a); Fig. 4A). The 64 kDa molecular weight band was observed in hT40 and S422E monomer and aggregated samples as expected, and signal intensity was not significantly different between either tau construct (hT40 vs. S422E, F(1,12)=0.03, p=0.86) or tau species (monomer vs. aggregate, F(1,12)=0.02, p=0.88) (Fig. 4B). In contrast, aggregation of either hT40 or S422E significantly increased the amount of SDS-stable dimers at 180 kDa (Fig. 4C; F(1,12)=110.0; p<0.0001), as compared to the levels of these species in the respective monomer samples. Furthermore, S422E aggregation significantly increased the amount of SDS-stable dimers compared to hT40 aggregation (p=0.03).
Figure 4. Pseudophosphorylation of S422 increases SDS-stable dimer formation.
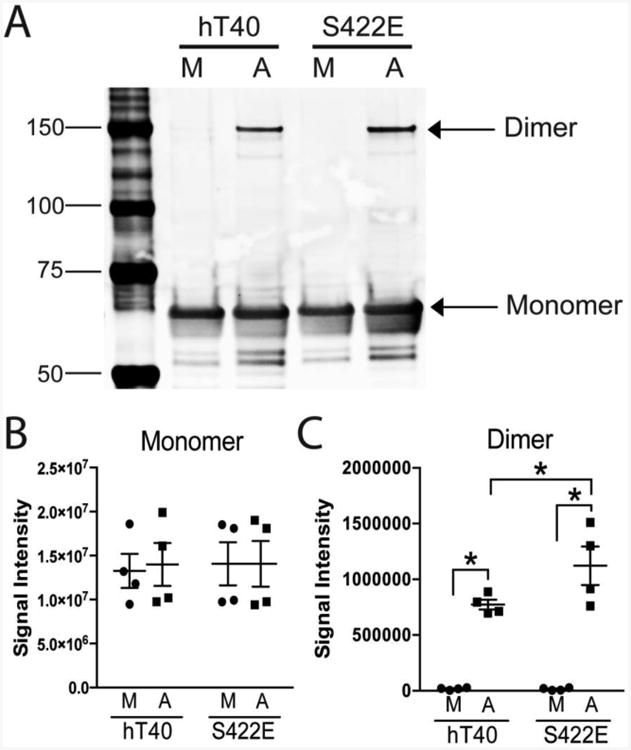
A) A representative immunoblot of monomeric (M, i.e., unaggregated) and aggregated (A) hT40 and S422E tau proteins probed with the monoclonal tau antibody Tau13 (total tau). Monomers (∼64 kDa) are similar across all samples and dimers (∼180 kDa) are present in aggregated tau samples. B-C) Quantitative analysis of immunoblot signal intensity. There were equivalent levels of tau in the monomer band (B) for both monomeric (M, i.e., unaggregated) and aggregated (A) hT40 and S422E samples. Aggregation of tau protein increased the intensity of the dimer band (C) in both hT40 and S422E, and there were significantly more tau dimers in the aggregated S422E samples when compared to aggregated hT40 (p=0.03). Values are expressed as mean ± SEM. n=4/group. Data were analyzed using a 2-way analysis of variance with a Holm-Sidak post-hoc test. *p ≤ 0.05 vs. monomer of the same construct, and **p ≤ 0.05 vs. hT40 aggregates.
Inhibition of fast axonal transport by S422-pseudophosphorylated tau monomers and aggregates
Vesicle motility assays in isolated squid axoplasm were used to evaluate whether S422 pseudophosphorylation modulates the toxic effect of tau aggregates on FAT (Brady et al., 1993; Morfini et al., 2007). Vesicle motility was monitored for 50 min following perfusion of 2μM hT40 monomer (Fig. 5A), hT40 aggregates (Fig. 5B), S422E monomer (Fig. 5C), or S422E aggregates (Fig. 5D). Differences between groups were compared using average FAT rate (μm/sec) over the last 20 min of the assay (Fig. 6A,B). As previously reported, perfusion of hT40 monomer had no effect on the rate of anterograde FAT in the squid axoplasm (Fig. 5A), whereas perfusion of hT40 aggregates significantly inhibited anterograde FAT as compared to hT40 monomer (Fig. 5B; Fig. 6A; p=0.003) (LaPointe et al., 2009b). Neither hT40 monomers nor hT40 aggregates altered the rate of retrograde FAT (Fig. 5A,B; Fig. 6B). In contrast to hT40 monomer, perfusion of S422E monomer selectively inhibited anterograde transport (Fig. 5C; Fig. 6A; p=0.028), but not retrograde FAT. Surprisingly, aggregated S422E significantly inhibited both anterograde and retrograde FAT rates (Fig. 5D; Fig. 6A,B) compared to S422E monomer (anterograde, p=0.012; retrograde, p=0.002) and hT40 aggregates (retrograde only, p=0.019).
Figure 5. Vesicle motility assays in isolated squid axoplasm.
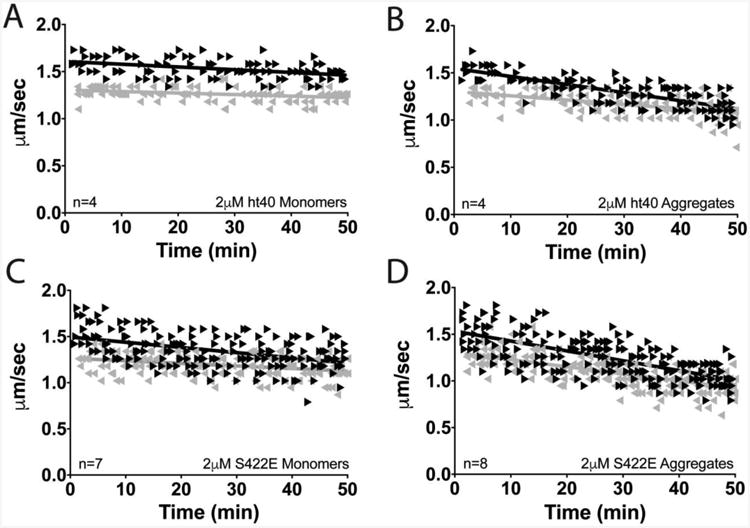
Individual velocity (μm/sec) measurements (arrowheads) are plotted as a function of time (min). (A) Perfusion of hT40 monomer did not alter anterograde (▸) or retrograde (◂) FAT. (B) Perfusion of hT40 aggregates selectively inhibited anterograde but not retrograde FAT. (C) Perfusion of S422E monomer selectively inhibited anterograde FAT. (D) Aggregates of S422E inhibited both anterograde and retrograde FAT.
Figure 6. Pseudophosphorylation of S422 inhibits anterograde and retrograde fast axonal transport (FAT).
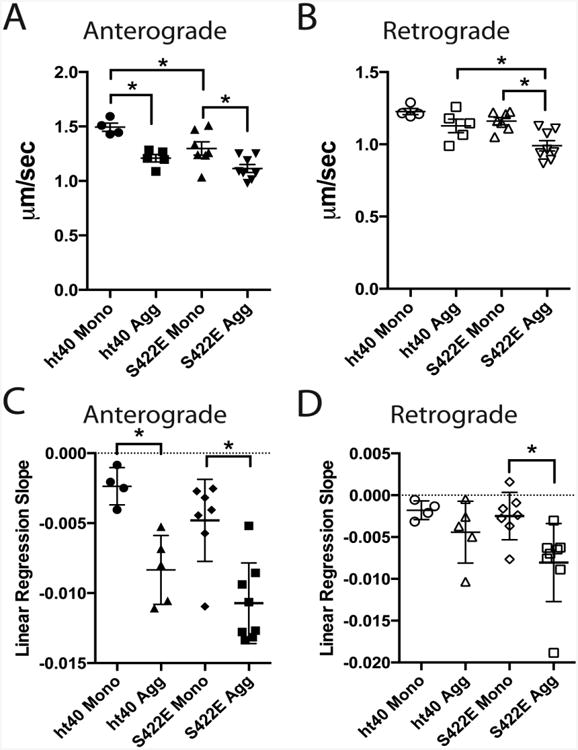
A-B) Quantification of average anterograde and retrograde FAT rates during the last 20 minutes of Fig. 5A-D indicates that hT40 aggregates, S422E monomers, and S422E aggregates all significantly inhibit anterograde FAT (A) compared to ht40 monomers, whereas only S422E aggregates significantly inhibit retrograde FAT (B). C-D) Data in Fig. 5A-D were fit using linear regressions and the slopes were compared to assess the rate of change in transport velocity (all average slopes were negative). The rate of decline in anterograde transport (C) in hT40 and S422E aggregate treated axoplasms is significantly different compared to axoplasms treated with their respective monomer samples. Anterograde rates of decline did not differ between axoplasms treated with hT40 or S422E aggregates. The rate of change in retrograde transport (D) in S422E aggregates is different from S422E monomers. All differences between groups were compared using a 2-way analysis of variance with a Holm-Sidak post-hoc test. *p ≤ 0.05.
We also performed a linear regression analysis of FAT rates over the 50 min assay and compared the slope of fitted lines to evaluate whether the rate of change in transport velocity was different among the treatment groups (all average slopes were negative; Fig. 6C,D). The slope of hT40 aggregates (m=-0.008) and S422E aggregates (m=-0.011) were significantly different from their respective monomer samples (Fig. 6A; hT40 monomer, m=-0.002, r2=0.17, p=0.003; S422E monomer, m=-0.005, r2=0.20, p=0.001), but hT40 and S422E aggregates were not significantly different when compared (p=0.243). The slopes for anterograde FAT in the S422E and hT40 monomer samples were statistically similar (Fig. 6C; p=0.243). The slope for retrograde transport in S422E aggregate-treated axoplasms (m=-0.008) was significantly different from S422E monomer-treated samples (Fig. 6D; m=-0.002, r2=0.12, p=0.015), while the slope for retrograde hT40 monomer and aggregates were not different (p=0.289). The slopes for retrograde transport in the hT40 aggregate (m=-0.004) and S422E aggregate groups (Fig. 6D; p=0.176) were not statistically distinguished from each other.
pS422 tau colocalizes with TOC1 and TNT1 tau in AD
Triple-label IF confocal microscopy (Fig. 7A-D) and sandwich ELISAs (Fig. 7E-G) were used to evaluate colocalization of pS422 with TOC1 and TNT1 in post-mortem AD brains. Staining in fixed tissue sections confirmed several previous reports (Kanaan et al., 2016; Mufson et al., 2014; Patterson et al., 2011a) that pS422 (Fig. 7A) extensively colocalizes with TNT1 (Fig. 7B) and TOC1 (Fig. 7C) in both control and AD brains, despite the relatively sparse appearance of tau pathology in the temporal cortices of control cases (data not shown). Sandwich ELISAs were developed to examine the co-occurrence of pS422 with either TNT1 or TOC1 in the same tau species isolated from the soluble fraction of control and AD frontal cortices. The amount of total tau captured with pS422 (detected with the pan-tau antibody, Tau5) was significantly higher in AD compared to control (Fig. 7E; t10=6.07, p=0.0001). The level of pS422 tau that also contained PAD exposed tau (i.e., TNT1 reactive) was significantly higher in AD compared to control (Fig. 7F; t10=2.31, p=0.0435). Similarly, the level of pS422 tau that also contained an oligomeric conformation (i.e., TOC1 reactive) was significantly higher in AD compared to control (Fig. 7G; t10=1.51, p=0.0029).
Figure 7. Phosphorylation at S422 occurs with PAD exposure and oligomer formation in Alzheimer's disease.
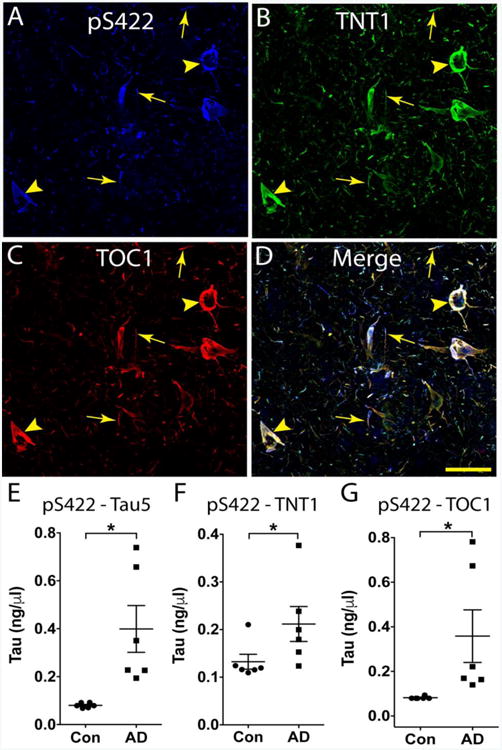
A-D) Triple label immunofluorescence confirmed that pS422 (blue, A), TNT1 (green, B) and TOC1 (red, C) are highly colocalized in AD temporal lobe tissue (merge, D). The coincidence of pS422, TNT1 and TOC1 was evident in both neuronal (arrowheads) and thread (arrows) inclusions. Scale bar in D is 50μm and applies to A-D. E-G) Novel sandwich ELISAs were developed to capture pS422 positive tau species from the soluble fraction of control (i.e., age-matched non-demented) and AD frontal cortex (n=6/group). E) The total amount of tau captured with pS422 antibody was significantly higher in AD compared to controls (detected with Tau5, a pan-tau antibody). F) The amount of pS422 tau with PAD exposed tau was significantly higher in AD compared to control (detected with TNT1, a marker of PAD exposure). G) The amount of pS422 that was oligomeric was significantly greater in AD compared to controls (detected with TOC1, a tau oligomer selective antibody). Data were compared using an unpaired t-test. *p ≤ 0.05.
Discussion
Phosphorylation of tau protein at residue S422 is a disease-related modification (Bussière et al., 1999; Hasegawa et al., 1996) that occurs very early in AD, prior to NFT formation (Guillozet-Bongaarts et al., 2006; Kimura et al., 1996). Despite an appreciation for the presence of pS422 in disease, a mechanistic link between pS422 and neuronal dysfunction, or degeneration, was not yet established. Previously, we demonstrated that appearance of the pS422 epitope is concomitant with early markers of tau toxicity, including oligomer formation (Mufson et al., 2014; Patterson et al., 2011a) and exposure of the N-terminal PAD (Kanaan et al., 2016). Here, we extended these previous immunohistochemical reports by demonstrating colocalization of pS422 with both TOC1 and TNT1 in situ and in tau species isolated from human brain tissue. Additionally, we have characterized the aggregation kinetics of S422E tau, and demonstrated that this modification stabilizes the formation of multimeric tau species. Further, S422E tau aggregates inhibited both anterograde and retrograde FAT in squid axoplasms. Collectively, these findings identify potential mechanisms by which phosphorylation of tau at S422 may contribute to toxicity in AD.
The effect of S422 pseudophosphorylation on tau aggregation
Purified recombinant tau protein does not detectably aggregate over days of incubation under physiological conditions (King et al., 1999). However, aggregation can be accelerated in vitro by incubation with an anionic inducer, such as ARA (Chirita et al., 2003; Wilson and Binder, 1997). When induced, tau polymerization typically follows nucleation-dependent kinetics, characterized by a nucleation phase (i.e., lag time), followed by a more extended phase of exponential growth, before reaching a steady-state equilibrium (Chirita and Kuret, 2004). Nucleation follows a ligand-mediated mechanism, whereby inducer interaction stabilizes an altered conformation of tau that then undergoes self-aggregation (King et al., 1999; Necula and Kuret, 2004b). Results from the present analysis demonstrated that pseudophosphorylation of S422 increased the lag time of tau polymerization without altering the rate of elongation or maximum polymerization signal. Additionally, aggregates formed by S422E tau and hT40 tau had similar morphology, and showed comparable levels of β-sheet structures (i.e., ThS signal). Collectively, these observations suggest that S422E may prolong aggregate nucleation, but once elongation is initiated the aggregation reaction proceeds similarly to wild-type tau protein.
Previously, pseudophosphorylation in the C-terminal domain of tau, including S422E and S409E, was found to have little effect on tau polymerization (Necula and Kuret, 2004a). For example, aggregated S422E or S409E accumulated a similar amount of filamentous tau relative to full-length wild-type tau, whereas mutations clustered near the proline-rich domain, (i.e., S212E/S214E, S199E/S202E/T205E) enhanced filament mass by 2-3-fold (Necula and Kuret, 2004a). Our results are consistent with these observations, and support the hypothesis that modifications in various locations along the polypeptide chain differentially affect tau aggregation (Necula and Kuret, 2005; 2004a). The observed increase in lag time could indicate that pseudophosphorylation at S422 impedes the ability of ARA to interact with tau (King et al., 1999), or decreases the ability of tau to adopt a specific filament assembly-competent conformation (Necula and Kuret, 2004b). The stabilization of dimers further supports the notion that S422 modification has an impact on the structures formed during tau aggregation in vitro, but ultimately the final aggregated sample is comparable to wild-type tau by several measures. Since pS422 is known to colocalize with pretangle pathology (Guillozet-Bongaarts et al., 2006; Kimura et al., 1996; Mufson et al., 2014; Patterson et al., 2011a), these in vitro results suggest this modification may contribute more to the stabilization of pre-fibrillar conformations rather than directly facilitating filament elongation.
A novel role for S422 pseudophosphorylation in tau dimer stabilization
Accumulating evidence suggests that multimeric species of tau that precede NFTs are responsible for neuronal dysfunction and degeneration. For instance, reducing tau overexpression in the rTg4510 transgenic mouse decreases neuronal cell loss even though filamentous inclusions continue to form (Santacruz et al., 2005), and multimeric tau species correlate with spatial memory deficits (Berger et al., 2007). Similarly, in human brain tissue, levels of granular tau oligomers are elevated, even in Braak stage I-II control cases (Maeda et al., 2007; 2006; Patterson et al., 2011a) with limited NFT pathology (H. Braak and E. Braak, 1997).
In the present study, aggregation of hT40 or S422E produced detectable SDS-stable tau multimers migrating as a major band with an apparent molecular weight of 180 kDa. The 180 kDa band has been previously identified as a tau dimer by SELDI-TOF mass spectrometry (Patterson et al., 2011a; Sahara et al., 2007). S422E enhanced formation of the 180 kDa dimer as compared to hT40 suggesting that phosphorylation at this residue stabilizes a dimeric tau conformation. These dimers also corresponds to the SDS-stable species (∼140 and 170 kDa) described in the rTg4510 transgenic mouse, although the sizes are slightly different since our recombinant tau construct contains a histidine tag (Berger et al., 2007; Sahara et al., 2013; 2007). Berger et al., (2007) demonstrated that these dimeric forms of tau are abnormally phosphorylated, including phosphorylation at S422, and they were negatively correlated with spatial memory performance in the Morris water maze. Despite S422E-mediated stabilization of tau multimer formation, TOC1 reactivity was not increased in comparison to hT40. One possible explanation for this is that TOC1 appears to label only a subpopulation of oligomeric aggregates (Patterson et al., 2011a). Hence, more than one tau oligomer conformation is likely to exist, and S422E may stabilize one or more multimer conformations not detected by the TOC1 antibody. The lack of differences in levels of PAD exposure between hT40 and S422E aggregate samples is consistent with the formation of similar levels and morphologies of tau polymers by the end of the in vitro polymerization assay. Taken together, these results indicate that S422 phosphorylation facilitates the formation of SDS-stable tau dimers.
Coincidence of pS422, PAD exposure, and oligomer formation in AD
Co-localization between pS422, TNT1 and TOC1 was extensive in the temporal cortex of AD brains, consistent with previous reports (Guillozet-Bongaarts et al., 2006; Kanaan et al., 2016; Mufson et al., 2014; Patterson et al., 2011a). The sandwich ELISAs captured pS422-reactive tau species and then used TNT1 and TOC1 to detect whether the pool of pS422 tau also contained PAD exposed and oligomer tau species, respectively. These are the first data to confirm that these modifications coexist on the same tau species. Interestingly, low total levels of tau with pS422 were captured from age-matched control brains in the ELISAs, and the data suggest that the majority of the pS422 tau in controls (∼0.1ng/ul) is reactive with TNT1 and TOC1, further supporting a close association between these modifications even in control brains. Together, these data confirm that pS422 tau species also exhibit increased PAD exposure and oligomerization in AD, all of which are linked to tau-mediated transport toxicity (Kanaan et al., 2016; 2012; 2011; LaPointe et al., 2009b; Mufson et al., 2014; Patterson et al., 2011a; 2011b; Vana et al., 2011).
Inhibition of axonal transport by S422 pseudophosphorylation
Abnormalities in kinase activity and deficits in FAT, a cellular process critical for the maintenance of appropriate neural connectivity, are increasingly implicated in the pathogenesis of various neurodegenerative diseases, including AD and tauopathies (Morfini et al., 2002a; 2009a). Dystrophic neurites, synaptic loss, and mislocalization of proteins are early pathogenic AD features consistent with this hypothesis (Bell and Claudio Cuello, 2006; Dessi et al., 1997; Scheff et al., 1990), but relationships among these critical pathogenic events remained unclear. Recently, our group described a mechanism by which physiological levels of aggregated tau inhibits kinesin-dependent anterograde FAT (Kanaan et al., 2012; 2011; LaPointe et al., 2009b). This effect is mediated by N-terminal amino acids 2-18 (Kanaan et al., 2011), comprising the PAD. Increased PAD exposure activates a protein phosphatase-1/glycogen kinase-3 (PP1/GSK3) signaling cascade that, in turn, promotes phosphorylation of conventional kinesin and detachment from its transported cargoes (Kanaan et al., 2012; 2011; LaPointe et al., 2009b). Collectively, these findings provided a mechanistic basis linking abnormal kinase activation, aberrant tau phosphorylation, and neuritic atrophy in AD (Kanaan et al., 2013; Morfini et al., 2002a).
Our results indicate that both monomeric and aggregated S422E inhibit anterograde FAT in the squid axoplasm model. These findings are consistent with other disease-related forms of tau where increased PAD exposure was predicted in monomers. For example, mimicking the phosphoepitope of AT8 (i.e., S199E/S202E/T205E) impairs folding of the amino terminus in the paperclip conformation (Deters et al., 2009; Gotz et al., 2001; Grueninger et al., 2010; Grundke-Iqbal et al., 1986a; 1986c; Jeganathan et al., 2006; Kosik et al., 1986; Wood et al., 1986b) and this protein inhibited anterograde FAT as a monomer (Kanaan et al., 2011; LaPointe et al., 2009b; Spillantini and Goedert, 1998). Moreover, a mutant form of tau associated with inherited frontotemporal dementia that lacks exons 6-9 (including the proline-rich region) also inhibited anterograde FAT as a monomer. This protein is unlikely to form the paperclip conformation because the hinge region is eliminated, ultimately increasing PAD exposure. The effects of S422E aggregates and monomers on anterograde FAT are likely due to the PAD-dependent activation of PP1/GSK3 pathway previously identified (Bierer et al., 1995; Biernat et al., 1993; Drewes et al., 1995; Kanaan et al., 2012; 2011; LaPointe et al., 2009b), but future studies are necessary to directly confirm this conclusion.
Unexpectedly, we observed a toxic effect of S422E aggregates on retrograde FAT, a unique effect among all tau constructs tested to date in the isolated squid axoplasm preparation (Kanaan et al., 2012; 2011; LaPointe et al., 2009b; Morfini et al., 2007; Morishima-Kawashima et al., 1995; Patterson et al., 2011b). Based on results from biochemical analyses here, S422E-mediated inhibition of retrograde FAT may result from activation of kinases other than GSK3, which selectively inhibits only anterograde FAT (Berger et al., 2007; Kopeikina et al., 2011; Maeda et al., 2007; Matsuo et al., 1994; Morfini et al., 2002b; Patterson et al., 2011a; Sahara et al., 2013). Accordingly, several phosphotransferases inhibit both anterograde and retrograde FAT through multiple mechanisms in the axoplasm preparation (Kanaan et al., 2013; Kimura et al., 1996; Morfini et al., 2006; 2009b; Patterson et al., 2011a; Pigino et al., 2009; Stoothoff and Johnson, 2005; Ward et al., 2013). Given these precedents, it is conceivable that one or more of the phosphotransferases responsible for phosphorylation of S422 in situ (i.e., JNK and p38) (Guillozet-Bongaarts et al., 2006; Kimura et al., 1996; Mufson et al., 2014; Reynolds et al., 1997a; 1997b; Vana et al., 2011; Yoshida et al., 2004) could indirectly promote FAT deficits, but the precise mechanism of this effect remains to be identified.
The squid axoplasm model system used here provides an opportunity to study axon-autonomous effects because the axoplasm is isolated from the somatodendritic compartment and synapses. In addition to the mechanism described above, various kinase-independent mechanisms have been proposed to inhibit FAT that could be alternate explanations for S422E effects. Because phosphorylation at S422 is unlikely to negatively affect microtubule affinity, monomers of this modified form of tau would be in position to impair axonal transport through more direct interactions with motor proteins on microtubules. Some studies have proposed that tau can regulate motor protein binding to microtubules or physically block motor proteins (Mandelkow, 2003), but our previous studies found that application of normal tau monomers up to approximately 20-fold higher than physiological levels does not negatively affect transport in either direction in the squid axoplasm assay used here (Morfini et al., 2007). Moreover, aggregates of S422E, which would not be expected to bind microtubules, are more potent in affecting FAT. Another proposed mechanism involves alterations of transcription or translation but these are also unlikely to contribute to these effects because the axoplasm model is isolated from the somatodendritic compartment and synapses.
In addition to its effects on FAT, these studies do not rule out the possibility that pS422 tau could exert toxicity through alternate pathways. Other proposed mechanisms for neurotoxicity include enhanced formation of degradation-resistant multimers that could overwhelm protein degradation machinery (Myeku et al., 2015; Tai et al., 2012; Wang et al., 2009) and/or compromise chaperone activity (Patterson et al., 2011a), impairment of neurophysiological functions (Fá et al., 2016), or promotion of mitochondria dysfunction (Kopeikina et al., 2011). However, the relatively low concentrations of S422E tau and the short incubation time used presently would argue against these mechanisms as an explanation for the effects on FAT. Finally, PAD-dependent effects on synaptic function (Moreno et al., 2016) could be enhanced by pS422. Additional studies are required to establish whether S422E tau plays a role in one or more of these alternative pathways of toxicity.
Conclusions
In summary, the present work using S422E tau identifies three potential effects of pS422-modified tau relevant to AD. First, S422E slows the nucleation phase of tau aggregation. Delayed filament formation may stabilize pre-fibrillar tau conformations that contribute to toxicity. Second, S422E facilitates the formation of stable tau dimers. Whether these two mechanisms are related is currently unknown; however the observation that phosphorylation at a single residue can stabilize tau multimer conformations known to contribute to disease (Berger et al., 2007; Bussière et al., 1999; Guillozet-Bongaarts et al., 2006; Hasegawa et al., 1996; Jeganathan et al., 2008; Kanaan et al., 2012; 2011; LaPointe et al., 2009a; Necula and Kuret, 2005) is novel. Finally, we identify a unique effect of aggregated S422-phosphorylated tau on FAT. Not only is S422E tau capable of inhibiting anterograde FAT as a monomer and aggregate, but it also is capable of inhibiting retrograde FAT when aggregated. Interestingly, the mechanism of this retrograde inhibition remains to be identified and is likely PAD-independent, as S422E does not appear to expose PAD to a greater extent than wild-type tau. In combination with our data from human AD brains, these results reveal a novel mechanistic link between tau phosphorylation at S422 and toxicity in AD.
Supplementary Material
Highlights.
SDS-stable dimer formation is enhanced by pseudophosphorylated S422 tau (S422E).
S422E tau monomers inhibit anterograde fast axonal transport in squid axoplasm.
S422E tau aggregates inhibit both anterograde and retrograde fast axonal transport.
pS422 colocalizes with pathological tau conformations in Alzheimer's disease.
Acknowledgments
We thank our late colleague, mentor, and friend Lester “Skip” I. Binder for his dedication and contributions to the field of tau biology. We also acknowledge Tessa Grabinski and Chelsey Hamel for their technical assistance on this work, and Hunter college students Alison Klein, Zach Gershon, Brenda Abdelmesih and Jennifer Purks for their assistance with the squid axoplasm experiments in Woods Hole, MA. We are grateful to Dr. Thomas Beach and the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human biological materials (i.e., fixed brain tissue sections). We gratefully acknowledge the assistance of this Neuropathology Core in the Alzheimer Disease Core Center at Northwestern University, Chicago, IL for providing the fresh frozen human tissue samples.
Funding: This work was supported by the National Institutes of Health [Grant numbers P01 AG14449 (SEC, NMK), R01 AG044372 (NMK) and R01 NS082730 (NMK, STB), R01 NS23868, NS23320, and NS41170 (STB), NS066942A (GM)] and the Secchia Family Foundation (NMK).
Abbreviations
- AD
Alzheimer's disease
- ARA
arachidonic acid
- E
glutamic acid
- ELISA
enzyme-linked immunosorbent assay
- FAT
fast axonal transport
- hT40
full-length wild-type tau protein
- is
intensity of scattered light
- LLS
laser light scatter
- NFT
neurofibrillary tangle
- PAD
phosphatase-activating domain
- pS422
phosphorylation of tau at serine 422
- S
serine
- TEM
transmission electron microscopy
- ThS
Thioflavin S
- TNT1
tau N-terminal 1
- TOC1
tau oligomeric complex 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Bell KFS, Claudio Cuello A. Altered synaptic function in Alzheimer's disease. Eur J Pharmacol. 2006;545:11–21. doi: 10.1016/j.ejphar.2006.06.045. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C. Accumulation of Pathological Tau Species and Memory Loss in a Conditional Model of Tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry RW, Sweet AP, Clark FA, Lagalwar S, Lapin BR, Wang T, Topgi S, Guillozet-Bongaarts AL, Cochran EJ, Bigio EH, Binder LI. Tau epitope display in progressive supranuclear palsy and corticobasal degeneration. J Neurocytol. 2004;33:287–295. doi: 10.1023/B:NEUR.0000044190.96426.b9. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer's disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64:749–760. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow E. Phosphorylation of Ser 262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. NBA. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Brady ST, Richards BW, Leopold PL. Assay of vesicle motility in squid axoplasm. Methods Cell Biol. 1993;39:191–202. doi: 10.1016/s0091-679x(08)60171-5. [DOI] [PubMed] [Google Scholar]
- Bussière T, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Buée L, Delacourte A. Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol. 1999;97:221–230. doi: 10.1007/s004010050978. [DOI] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50“s selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Chirita CN, Kuret J. Evidence for an Intermediate in Tau Filament Formation. Biochemistry. 2004;43:1704–1714. doi: 10.1021/bi036034b. [DOI] [PubMed] [Google Scholar]
- Chirita CN, Necula M, Kuret J. Anionic Micelles and Vesicles Induce Tau Fibrillization in Vitro. J Biol Chem. 2003;278:25644–25650. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- Dessi F, Colle MA, Hauw JJ, Duyckaerts C. Accumulation of SNAP- 25 immunoreactive material in axons of Alzheimer's disease. Neuroreport. 1997;8:3685–3689. doi: 10.1097/00001756-199712010-00006. [DOI] [PubMed] [Google Scholar]
- Deters N, Ittner LM, Götz J. Substrate-specific reduction of PP2A activity exaggerates tau pathology. Biochem Biophys Res Commun. 2009;379:400–405. doi: 10.1016/j.bbrc.2008.12.140. [DOI] [PubMed] [Google Scholar]
- Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, Mandelkow EM, Mandelkow E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–7688. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fá M, Puzzo D, Piacentini R, Staniszewski A, Zhang H, Baltrons MA, Puma DDL, Chatterjee I, Li J, Saeed F, Berman HL, Ripoli C, Gulisano W, Gonzalez J, Tian H, Costa JA, Lopez P, Davidowitz E, Yu WH, Haroutunian V, Brown LM, Palmeri A, Sigurdsson EM, Duff KE, Teich AF, Honig LS, Sierks M, Moe JG, D'Adamio L, Grassi C, Kanaan NM, Fraser PE, Arancio O. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci Rep. 2016;6:1–15. doi: 10.1038/srep19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, Berry RW, Binder LI. In Vitro Polymerization of Tau Protein Monitored by Laser Light Scattering: Method and Application to the Study of FTDP-17 Mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- García-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by A beta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Grueninger F, Bohrmann B, Czech C, Ballard TM, Frey JR, Weidensteiner C, von Kienlin M, Ozmen L. Phosphorylation of tau at S422 is enhanced by Aβ in TauPS2APP triple transgenic mice. Neurobiol Dis. 2010;37:294–306. doi: 10.1016/j.nbd.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986a;261:6084–6089. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. PNAS. 1986b;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. PNAS. 1986c;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Cahill ME, Cryns VL, Reynolds MR, Berry RW, Binder LI. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem. 2006;97:1005–1014. doi: 10.1111/j.1471-4159.2006.03784.x. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Jakes R, Crowther RA, Lee VM, Ihara Y, Goedert M. Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Letters. 1996;384:25–30. doi: 10.1016/0014-5793(96)00271-2. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global Hairpin Folding of Tau in Solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of early pathological tau conformations and phosphoryaltion in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2016;75:19–34. doi: 10.1093/jnen/nlv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Kordower JH, Collier TJ. Age-related accumulation of Marinesco bodies and lipofuscin in rhesus monkey midbrain dopamine neurons: Relevance to selective neuronal vulnerability. J Comp Neurol. 2007;502:683–700. doi: 10.1002/cne.21333. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, Leitman E, Binder LI, Brady ST. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. NBA. 2012;33:826.e15–826.e30. doi: 10.1016/j.neurobiolaging.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J Neurosci. 2011;31:9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Pigino GF, Brady ST, Lazarov O, Binder LI, Morfini GA. Axonal degeneration in Alzheimer's disease: When signalling abnormalities meet the axonal transport system. Exp Neurol. 2013;246:44–53. doi: 10.1016/j.expneurol.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Ono T, Takamatsu J, Yamamoto H, Ikegami K, Kondo A, Hasegawa M, Ihara Y, Miyamoto E, Miyakawa T. Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia. 1996;7:177–181. doi: 10.1159/000106875. [DOI] [PubMed] [Google Scholar]
- King ME, Ahuja V, Binder LI, Kuret J. Ligand-Dependent Tau Filament Formation: Implications for Alzheimer's Disease Progression. Biochemistry. 1999;38:14851–14859. doi: 10.1021/bi9911839. [DOI] [PubMed] [Google Scholar]
- Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL. Tau Accumulation Causes Mitochondrial Distribution Deficits in Neurons in a Mouse Model of Tauopathy and in Human Alzheimer's Disease Brain. AJPA. 2011;179:2071–2082. doi: 10.1016/j.ajpath.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. PNAS. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe NE, Horowitz PM, Guillozet-Bongaarts AL, Silva A, Andreadis A, Binder LI. Tau 6D and 6P Isoforms Inhibit Polymerization of Full-Length Tau in Vitro. Biochemistry. 2009a;48:12290–12297. doi: 10.1021/bi901304u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J Neurosci Res. 2009b;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. PNAS. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A. Granular Tau Oligomers as Intermediates of Tau Filaments. Biochemistry. 2007;46:3856–3861. doi: 10.1021/bi061359o. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiology of Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O'Connor M, Trojanowski JQ, Lee VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- Moreno H, Morfini G, Buitrago L, Ujlaki G, Choi S, Yu E, Moreira JE, Avila J, Brady ST, Pant H, Sugimori M, Llinas RR. Tau Pathology-mediated presynaptic dysfunction. Neuroscience. 2016;325:30–38. doi: 10.1016/j.neuroscience.2016.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST. Fast axonal transport misregulation and Alzheimer's disease. Neuromolecular Med. 2002a;2:89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Mizuno N, Kikkawa M, Brady ST. Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J Neurosci Res. 2007;85:2620–2630. doi: 10.1002/jnr.21154. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002b;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal Transport Defects in Neurodegenerative Diseases. J Neurosci. 2009a;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, You YM, Pollema SL, Kaminska A, Liu K, Yoshioka K, Björkblom B, Coffey ET, Bagnato C, Han D, Huang CF, Banker G, Pigino G, Brady ST. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009b;12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–829. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ward S, Binder L. Prefibrillar Tau Oligomers in Mild Cognitive Impairment and Alzheimer's Disease. Neurodegener Dis. 2014;13:151–153. doi: 10.1159/000353687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, Duff KE. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. 2015;22:46–53. doi: 10.1038/nm.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necula M, Kuret J. Site-specific pseudophosphorylation modulates the rate of tau filament dissociation. FEBS Letters. 2005;579:1453–1457. doi: 10.1016/j.febslet.2005.01.047. [DOI] [PubMed] [Google Scholar]
- Necula M, Kuret J. Pseudophosphorylation and Glycation of Tau Protein Enhance but Do Not Trigger Fibrillization in Vitro. J Biol Chem. 2004a;279:49694–49703. doi: 10.1074/jbc.M405527200. [DOI] [PubMed] [Google Scholar]
- Necula M, Kuret J. A static laser light scattering assay for surfactant-induced tau fibrillization. Anal Biochem. 2004b;333:205–215. doi: 10.1016/j.ab.2004.05.044. [DOI] [PubMed] [Google Scholar]
- Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of Prefibrillar Tau Oligomers in Vitro and in Alzheimer Disease. J Biol Chem. 2011a;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Ward SM, Combs B, Voss K, Kanaan NM, Morfini G, Brady ST, Gamblin TC, Binder LI. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry. 2011b;50:10300–10310. doi: 10.1021/bi2009147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. PNAS. 2009;106:5907–5912. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porzig R, Singer D, Hoffmann R. Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem Biophys Res Commun. 2007;358:644–649. doi: 10.1016/j.bbrc.2007.04.187. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Nebreda AR, Gibb GM, Utton MA, Anderton BH. Reactivating kinase/p38 phosphorylates tau protein in vitro. J Neurochem. 1997a;69:191–198. doi: 10.1046/j.1471-4159.1997.69010191.x. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Utton MA, Gibb GM, Yates A, Anderton BH. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem. 1997b;68:1736–1744. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- Sahara N, DeTure M, Ren Y, Ebrahim AS, Kang D, Knight J, Volbracht C, Pedersen JT, Dickson DW, Yen SH, Lewis J. Characteristics of TBS-extractable hyperphosphorylated tau species: aggregation intermediates in rTg4510 mouse brain. J Alzheimers Dis. 2013;33:249–263. doi: 10.3233/JAD-2012-121093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007;25:3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. NBA. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002;21:4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Kang M, Morfini G, Brady ST. Fast axonal transport in isolated axoplasm from the squid giant axon. Methods Cell Biol. 2016;131:331–348. doi: 10.1016/bs.mcb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GVW. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. AJPA. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112:2355–2367. doi: 10.1242/jcs.112.14.2355. [DOI] [PubMed] [Google Scholar]
- Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, Binder LI. Progression of Tau Pathology in Cholinergic Basal Forebrain Neurons in Mild Cognitive Impairment and Alzheimer's Disease. AJPA. 2011;179:2533–2550. doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Gen. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis. 2013;37:593–602. doi: 10.3233/JAD-131235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. AJPA. 1997;150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- Winsor CP. The Gompertz Curve as a Growth Curve. PNAS. 1932;18:1–8. doi: 10.1073/pnas.18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) PNAS. 1986a;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) PNAS. 1986b;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.