

Abstract
Objective
Aging is a main risk factor for the development of osteoarthritis (OA) and the molecular mechanisms underlying the aging-related changes in articular cartilage include increased mammalian target of rapamycin (mTOR) signaling and defective autophagy. REDD1 is an endogenous inhibitor of mTOR that regulates cellular stress responses. In this study we measured REDD1 expression in normal, aged and OA cartilage and assessed REDD1 function in human and mouse articular chondrocytes.
Methods
REDD1 expression was analyzed in human and mouse articular cartilage by qPCR, western blotting, and immunohistochemistry. For functional studies, REDD1 and TXNIP knockdown or overexpression was performed in chondrocytes in the presence or absence of rapamycin and chloroquine, and mTOR signaling and autophagy were measured by western blotting. REDD1/TXNIP protein interaction was assessed by co-immunoprecipitation experiments.
Results
Human and mouse cartilage from normal knee joints expressed high levels of REDD1. REDD1 expression was significantly reduced in aged and OA cartilage. In cultured chondrocytes, REDD1 knockdown increased whereas REDD1 overexpression decreased mTOR signaling. In addition, REDD1 activated autophagy by an mTOR independent mechanism that involved protein/protein interaction with TXNIP. The REDD1/TXNIP complex was required for autophagy activation in chondrocytes.
Conclusion
The present study shows that REDD1 is highly expressed in normal human articular cartilage and reduced during aging and OA. REDD1 in human chondrocytes negatively regulates mTOR activity and is essential for autophagy activation. Reduced REDD1 expression thus represents a novel mechanism for the increased mTOR activation and defective autophagy observed in OA.
Keywords: REDD1, mTOR, cartilage, osteoarthritis, autophagy
Introduction
Osteoarthritis (OA) is the most prevalent musculoskeletal pathology and a leading cause of pain and disability (1). OA is predominantly characterized by the progressive degradation of articular cartilage due to an imbalance between anabolic and catabolic processes that eventually involves other tissues within the joint and results in joint dysfunction (2). Aging is the main risk factor for OA (2). Age-related changes occur in articular cartilage and other joint tissues and they are thought to represent a major risk factor for OA development (3). Articular cartilage changes during aging include reduced thickness, reduced cell density, cellular senescence associated with aberrant secretory activity, and impaired cellular defense mechanisms (3). Understanding the molecular mechanisms that drive cartilage aging has become a main focus of OA research.
The PI3K/AKT/mTOR pathway has emerged as a central axis in the control of lifespan and healthspan in model organisms (4, 5). It transduces growth factor signals to regulate cell growth, transcription, protein translation as well as cellular stress responses (6). mTOR is a serine/threonine protein kinase that interacts with several proteins to form two distinct complexes named mTOR complex 1 (mTORC1) and 2 (mTORC2) (7). mTORC1 is rapamycin sensitive and regulates different aspects of cellular growth and metabolism whereas mTORC2 regulates cell survival and cytoskeletal organization (7). A major function of mTORC1 is the suppression of autophagy, a protective mechanism that involves degradation and recycling of dysfunctional cellular organelles (8). There is increasing evidence that mTOR signaling is dysregulated in OA articular cartilage (9). A recent study showed elevated mTOR expression in human OA cartilage, as well as in mouse and dog models of experimental OA (10). In addition, reduced expression of key autophagic markers has been reported in aged and OA articular cartilage (11). Pharmacological or genetic inhibition of mTOR is protective against experimental OA in mice and it is accompanied by an increase in autophagic activity (10, 12). Taken together, these findings suggest that increased mTOR signaling may compromise autophagic activity in chondrocytes and contribute to the development of OA.
The molecular mechanisms underlying mTOR dysregulation in OA cartilage remain poorly understood. A possible mechanism could be increased upstream signaling via PI3K/AKT activation. Several ligands can induce PI3K/AKT signaling in chondrocytes in a context specific fashion (13). However, there is no conclusive evidence for the status of this pathway in articular cartilage, either during normal aging or OA pathogenesis. On the other hand, a decrease in endogenous mTORC1 negative regulators could account for the increased mTOR activity in OA cartilage. Recent reports showed that mTOR inhibitors, such as FOXO, AMPK and Sirt1 have reduced expression in OA cartilage (14-17). Moreover, this reduced expression was associated with defective cellular defenses and a catabolic chondrocyte phenotype (14-17). Whether these effects are mediated by mTOR or autophagy still remains to be elucidated.
REDD1 is an evolutionary conserved protein encoded by the ddit4 gene in mammals. REDD1 expression is induced by hypoxia and other stresses (18, 19) and acts primarily as a canonical mTORC1 inhibitor (20-22). Interestingly, REDD1 is ubiquitously expressed in adult tissues and it has a dual role as a pro-survival or pro-apoptotic factor depending on context and cell type (19, 23, 24). In the present study we examined REDD1 expression in articular cartilage and its function in articular chondrocytes.
Methods
Human cartilage samples
Macroscopically normal articular cartilage was harvested at the time of autopsy from the knees of young (38±13 years) and old donors (77±12 years) with no history of joint disease. OA cartilage was obtained from patients (70±11 years) undergoing total knee arthroplasty. Immediately after full thickness cartilage resection from the subchondral bone, samples were stored at -20°C in Allprotect (Qiagen) for RNA and protein isolation, fixed in Z-Fix (Anatech) for 24 hours and embedded in paraffin for histological analysis, or used for chondrocyte isolation as described below.
Mouse cartilage samples
All animal experiments were approved by the Institutional Animal Care and Use Committee at The Scripps Research Institute. For aging studies, C57BL/6J mice were kept under standard conditions and sacrificed at 6 and 27 months of age. For the surgical OA model, 4-month-old mice were anesthetized and transection of the medial meniscotibial ligament (DMM) and the medial collateral ligament was performed (12, 25). Animals were then euthanized 8 weeks after surgery and knee joints were processed for immunohistochemical analysis as described previously (15).
Human chondrocyte culture
Human primary chondrocytes were isolated as described previously (14) and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% calf serum (CS) at 37°C in 5% CO2. First passage chondrocytes were used in all experiments. The immortalized human chondrocyte cell line T-C/28 (26) was cultured in DMEM containing 10% CS and only cells that had been maintained for fewer than 20 passages were used in all experiments.
For REDD1 knock down experiments, chondrocytes were grown to confluence and transfected with small interfering RNA (siRNA) for DDIT4 or TXNIP (Life Technologies) using lipofectamine RNAiMAX (Life Technologies) in media containing 1% CS for 42 hours. Then, DMSO (0.1%, Sigma), rapamycin (100 nM, LC Labs) or chloroquine (25 μM, Sigma) was added and cells were harvested after 6 hours.
For REDD1 overexpression, confluent chondrocytes were transduced with adenovirus expressing GFP (Ad-GFP) or REDD1 (Ad-REDD1) in media containing 1% CS for 6 hours in the presence of DMSO (0.1%) or rapamycin (100 nM) as indicated.
Mouse chondrocyte culture
Immature murine articular chondrocytes were isolated from knees and hips of 6-days-old wild-type and REDD1 deficient mice (27) following the protocol described by Gosset et al (28). Absence of REDD1 expression in the joints of Redd1-/- mice was confirmed by immunohistochemistry staining with REDD1 specific antibody (Supplemental Figure 1). Isolated cells were cultured in media containing 10% CS and second passage cells were used in all experiments.
RNA isolation and quantitative polymerase chain reaction
Cartilage was pulverized in a freezer mill and homogenized in Qiazol Lysis Reagent (Qiagen). RNA was isolated using the RNeasy Mini kit (Qiagen) followed by removal of proteoglycans using RNAmate (BioChain Institute). In cultured chondrocytes, RNA was collected using Direct-Zol RNA miniprep kit (Zymo Research).
Gene expression was measured using pre-designed TaqMan gene expression assays for the following genes: REDD1, TXNIP, BECN1, BNIP3, FOXO3, GABARAP, MAP1LC3, ULK1, and GAPDH as a reference gene.
Immunohistochemistry
Paraffin-embedded human and mouse samples were deparaffinized and rehydrated as described elsewhere (15). 5-μm thick sections were blocked with 10% goat serum and incubated overnight at 4°C with rabbit anti-REDD1 antibody (1:100 dilution; Proteintech). After washing with phosphate buffered saline (PBS), sections were incubated for 30 minutes with ImmPRESS-AP anti-rabbit IgG polymer detection reagent (Vector Laboratories), dehydrated and mounted. Positive cells were counted in 2 different fields per section and the results are reported as percentage of REDD1-positive cells.
Protein isolation and western blotting
Cultured chondrocytes were washed twice in PBS and lysed in ice-cold RIPA buffer (Pierce) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific). Twenty micrograms of protein were resolved in 4-12% acrylamide gels and transferred to nitrocellulose membranes using the Bolt system (Life Technologies). Membranes were washed in Tris-buffered saline (TBS) and blocked with Odyssey blocking buffer for 1 hour at room temperature. Then, blots were incubated overnight at 4°C with the following antibodies: REDD1 (1:500, Proteintech), p-S6 (1:2000, Cell Signaling), S6 (1:1000, Cell Signaling), p-4EBP (1:1000, Cell Signaling), 4EBP (1:1000, Cell Signaling), LC3 (1:1000, Cell Signaling), TXNIP (1:1000, Cell Signaling), and GAPDH (1:5000, Abcam). After washing three times with TBS with 0.05% Tween 20 (TBS-T), membranes were incubated with secondary antibodies goat anti-rabbit IRDye 800 (1:5000 dilution) and goat anti-mouse IRDye 680 (1:10000 dilution) for 1 hour at room temperature. Blots were washed three times in TBS-T and visualized using the Odyssey Infrared Imaging System (LI-COR). Intensity values were analyzed with the ImageStudioLite software and normalized to those of GAPDH.
Co-immunoprecipitation
For co-immunoprecipitation experiments, T-C/28 cells were transfected with plasmid expressing FLAG-REDD1 protein for 24 hours using lipofectamine 3000 (Life Technologies). Total protein extracts were prepared using RIPA buffer (Pierce) containing a protease inhibitor cocktail (Roche). After removal of insoluble debris by centrifugation, lysates were pre-cleared with 20 μl protein-A magnetic beads (Cell Signaling) for 1 hour at 4°C, followed by overnight incubation with 20 μl anti-FLAG M2 magnetic beads (Sigma). Next, beads were washed four times with RIPA buffer and proteins were eluted with 100 μl of elution buffer (10mM Tris pH 8.0, 2mM EDTA, 2% SDS). 20 μg of proteins were resolved by SDS-PAGE and analyzed by western blotting as described above.
Statistical analysis
Data are reported as the mean ± 95CI (95% confidence interval). Differences between groups were assessed by one-way analysis of variance (ANOVA) followed by a post-hoc Tukey test. Comparisons between two groups were assessed by an unpaired, two-tailed T-test after testing for equal variance using an F-test. All statistical analyses were performed using Prism 6 software (GraphPad Software). P-values less than 0.05 were considered significant.
Results
REDD1 expression is reduced in aged and OA cartilage
REDD1 expression was measured by qPCR and western blotting in human knee cartilage samples from normal and OA donors. REDD1 mRNA and protein levels were significantly decreased in OA samples (Figure 1A,B). Immunohistochemical analysis revealed that REDD1 is highly expressed in normal cartilage where more than 80% of chondrocytes in all cartilage zones showed strong staining (Figure 1C). On the other hand, aged and OA cartilage samples exhibited significantly lower REDD1 expression than normal controls (Figure 1C). REDD1 positive cells were significantly reduced in OA samples when compared with normal aged cartilage. The REDD1 reduction in OA samples was evident in intact regions of the cartilage as well as in fibrillated areas with the chondrocyte clusters also being negative.
Figure 1.
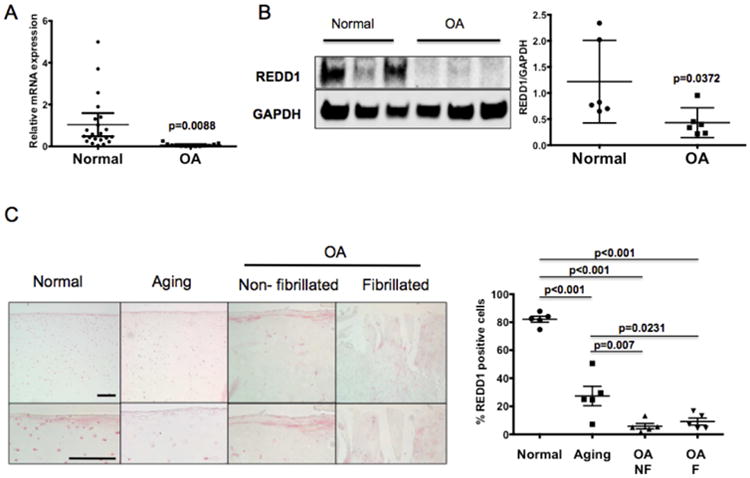
REDD1 expression is reduced in human articular cartilage during aging and OA. A) REDD1 RNA expression analyzed by quantitative PCR in articular cartilage from normal (N=23) and OA donors (N=14). B) Western blot analysis of REDD1 expression in articular cartilage from normal (N=6) and OA donors (N=6). C) REDD1 immunohistochemistry on articular cartilage from normal, aged and OA donors. Six donors were analyzed per group and representative images are shown at two different magnifications. On the right, REDD1 quantitation expressed as percentage of REDD1 immunopositive cells. All values are mean±95%CI. Magnification bar = 100μm.
Altered REDD1 expression in articular cartilage during aging and OA was also observed in murine joints. The number of REDD1 positive cells was significantly higher in the articular cartilage from 6-month-old mice as compared with 27-month-old mice (Figure 2). In addition, mice with surgically induced OA showed less REDD1 expression 8 weeks after surgery as compared with age-matched controls (Figure 2).
Figure 2.
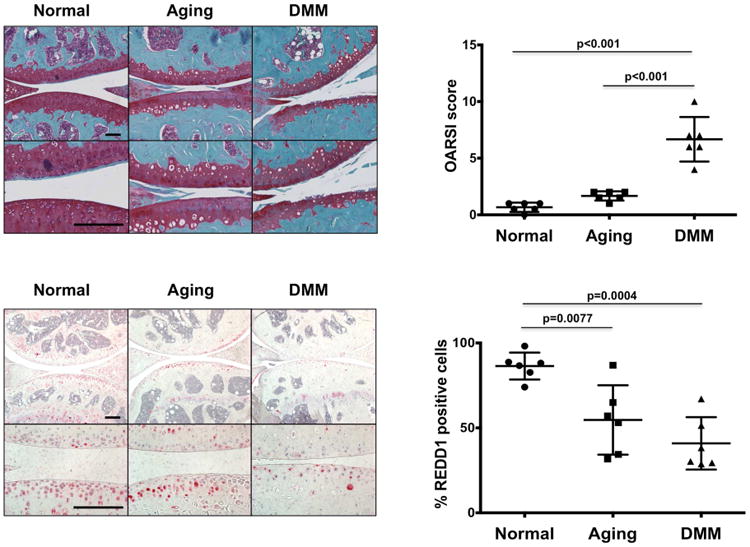
REDD1 expression is reduced in mouse articular cartilage during aging and OA. REDD1 immunohistochemistry on articular cartilage of normal (6-month-old), old (27-month-old), and 6-month-old mice subjected to surgical destabilization of medial meniscus (DMM). A total of 6 mice per group were analyzed and representative images are shown at two different magnifications. On the right, REDD1 quantitation expressed as percentage of REDD1 immunopositive cells. Values are mean±95%CI. Magnification bar = 100μm.
REDD1 is an endogenous mTOR inhibitor in articular chondrocytes
To evaluate the role of REDD1 in mTOR signaling in chondrocytes, we analyzed the phosphorylation status of two downstream effectors of mTORC1 (S6 and 4EBP) upon REDD1 knockdown. REDD1 specific siRNA significantly increased phosphorylation of S6 and 4EBP proteins in human primary chondrocytes (Figure 3A) and T-C/28 cells (Figure 3B). A more marked effect on mTORC1 signaling was observed in chondrocytes from Redd1-/- mice compared to wild-type cells (Figure 3C) as evidenced by a significant increase in S6 and 4EBP phosphorylation. These data collectively suggest that REDD1 is an endogenous mTOR inhibitor and that REDD1 depletion is sufficient to increase mTOR signaling in articular chondrocytes.
Figure 3.
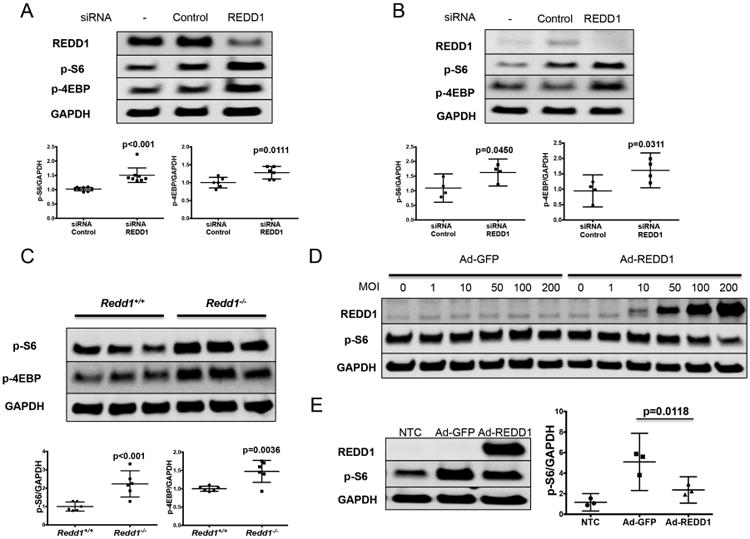
REDD1 inhibits mTORC1 activity in human and mouse articular chondrocytes. A) Western blot analysis of REDD1, phospho-S6 (p-S6) and phospho-4EBP (p-4EBP) in human primary chondrocytes transfected with siRNA control or specific against REDD1. Values are mean±95%CI of eight different human donors. B) Western blot analysis of REDD1, p-S6 and p-4EBP in T-C/28 chondrocytes transfected with siRNA control or specific against REDD1. Values are mean±95%CI of four different experiments. C) Western blot analysis of p-S6 and p-4EBP in immature murine articular chondrocytes isolated from Redd1+/+ and Redd1-/- mice. Values are mean±95%CI of three independent experiments. D) Western blot of REDD1 and p-S6 levels in human primary chondrocytes transduced with adenovirus expressing GFP (Ad-GFP) or REDD1 (Ad-REDD1) at different multiplicities of infection (MOI). E) Western blot analysis of REDD1 and p-S6 levels in T-C/28 chondrocytes transduced with Ad-GFP or Ad-REDD1 at 10 MOI. Values are mean±95%CI of three independent experiments.
Next, we determined whether ectopic REDD1 expression would inhibit mTOR signaling. As shown in Figure 3D, REDD1 overexpression using adenoviral vectors resulted in a dose-response reduction in S6 phosphorylation in human chondrocytes, especially at high multiplicity of infections (MOI). Likewise, a significant reduction in S6 phosphorylation was observed in T-C/28 cells transduced with Ad-REDD1 (Figure 3E).
REDD1 modulates autophagy in articular chondrocytes
Since mTORC1 is a potent inhibitor of autophagy (7), we tested whether REDD1 regulates autophagy in articular chondrocytes using gain- and loss-of-function approaches. To monitor autophagic activity, we detected the conversion of LC3, a key factor required for autophagosome elongation and maturation, from the cytosolic form (LC3-I) to the autophagosome-bound form (LC3-II) (29). As shown in Figures 4A-B, REDD1 depletion resulted in a significant reduction in LC3-II levels in human chondrocytes treated with chloroquine, an inhibitor of autophagosome degradation, as compared with cells transfected with control siRNA. In mouse articular chondrocytes from Redd1-/- mice, autophagic activity was severely impaired when compared with cells from wild-type control mice (Figure 4C). Conversely, REDD1 overexpression using Ad-REDD1 was sufficient to induce autophagy in human chondrocytes as evidenced by a significant increase in LC3-II accumulation (Figures 4D-E).
Figure 4.
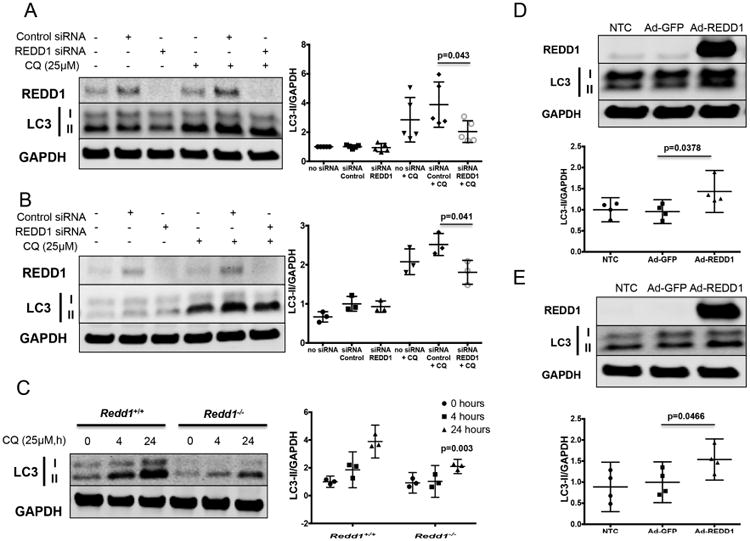
REDD1 regulates autophagy in articular chondrocytes. A) Western blot analysis of REDD1 and LC3 in human primary chondrocytes transfected with siRNA control or specific against REDD1 in the presence of absence of chloroquine (CQ, 25μM). Values are mean±95%CI of five different donors. B) Western blot analysis of REDD1 and LC3 in T-C/28 chondrocytes transfected with siRNA control or specific against REDD1 in the presence of absence of CQ. Values are mean±95%CI of three different experiments. C) Western blot analysis of REDD1 and LC3 in immature murine articular chondrocytes isolated from Redd1+/+ and Redd1-/- mice. Values are mean±95%CI of three independent experiments. Statistical comparisons are Redd1-/- vs Redd1+/+. D) Western blot analysis of REDD1 and LC3 in human primary chondrocytes transduced with adenovirus expressing GFP (Ad-GFP) or REDD1 (Ad-REDD1) at 10 multiplicity of infection. Values are mean±95%CI of four different donors. E) Western blot analysis of REDD1 and p-S6 levels in T-C/28 chondrocytes transduced with Ad-GFP or Ad-REDD1 at 10 MOI. Values are mean±95%CI of four independent experiments.
To investigate whether REDD1 modulates autophagy though mTORC1 signaling, gain- and loss-of-function experiments were carried out in the presence or absence of rapamycin, a specific mTOR inhibitor. Surprisingly, rapamycin treatment did not abolish the effect on autophagy of REDD1 depletion (Figure 5A) or overexpression (Figure 5B) in human chondrocytes. Likewise, rapamycin did not restore the defective autophagy in chondrocytes from Redd1-/- mice (Figure 5C), indicating that REDD1 modulates autophagy in an mTOR independent manner.
Figure 5.
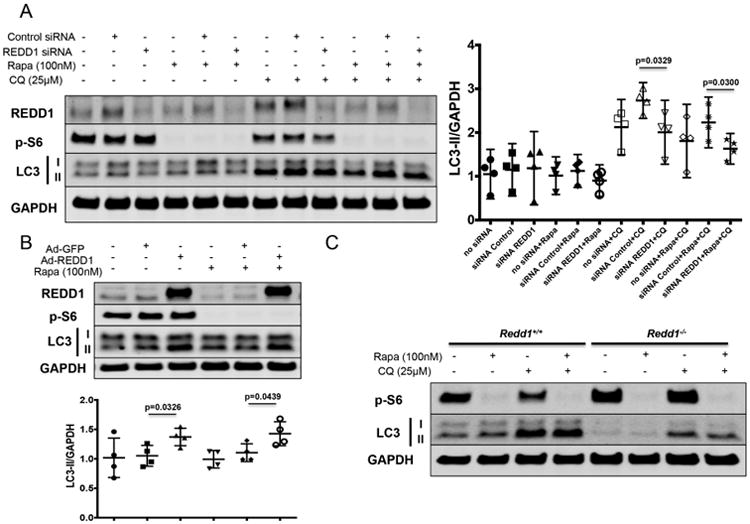
REDD1 induces autophagy in articular chondrocytes through an mTOR independent mechanism. A) Western blot analysis of REDD1, phospho-S6 (p-S6), and LC3 in human primary chondrocytes transfected with siRNA control or specific against REDD1 in the presence of absence of chloroquine (CQ, 25μM) and rapamycin (Rapa, 1μM). Values are mean±95%CI of four different donors. B) Western blot analysis of REDD1, p-S6 and LC3 levels in human primary chondrocytes transduced with adenovirus expressing GFP (Ad-GFP) or REDD1 (Ad-REDD1) at 10 multiplicity of infection, in the presence of absence of CQ and Rapa. Values are mean±95%CI of four different donors. C) Western blot analysis of REDD1, p-S6 and LC3 in immature murine articular chondrocytes isolated from Redd1+/+ and Redd1-/- mice in the presence of absence of CQ and Rapa.
REDD1 interacts with TXNIP to induce autophagy in chondrocytes
Since our results indicated that REDD1 regulation of autophagy is largely mTOR independent in chondrocytes, we wanted to identify the molecular mechanism underlying this function of REDD1. A recent study reported that REDD1 forms a complex with the pro-oxidant protein TXNIP to trigger endogenous ROS production under cellular stress conditions (30). In addition, ROS generated by the REDD1/TXNIP complex were sufficient to induce autophagy in HEK293T cells (30). Based on these findings, we asked whether REDD1 interacts with TXNIP in chondrocytes and whether this protein complex is able to regulate autophagy.
Since TXNIP has not been previously studied in cartilage, we first measured TXNIP expression in articular cartilage of normal and OA donors. As shown in Figures 6A,B, TXNIP mRNA and protein were detected in articular cartilage of normal donors and their expression was significantly reduced in OA.
Figure 6.
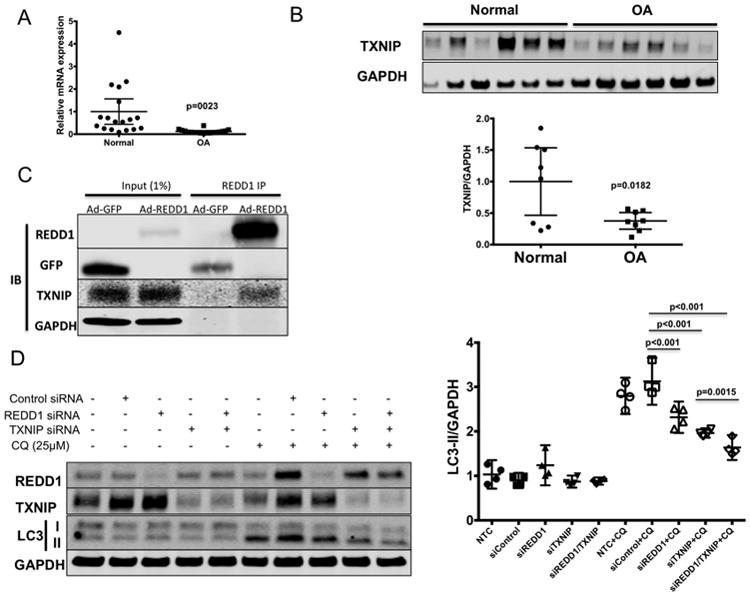
REDD1 forms a complex with TXNIP to induce autophagy in articular chondrocytes. A) TXNIP RNA expression analyzed by quantitative PCR in articular cartilage from normal (N=18) and OA donors (N=20). Values are mean±95%CI. B) TXNIP protein levels in articular cartilage from normal and OA donors assessed by western blot. Eight donors were analyzed per group. C) Co-immunoprecipitation experiment in T-C/28 chondrocytes transduced with adenovirus expressing GFP (Ad-GFP) or FLAG-REDD1 (Ad-REDD1) at 10 multiplicity of infection. Western blot shows REDD1, GFP and TXNIP levels after immunoprecipitation with anti-FLAG antibodies. D) Western blot analysis of REDD1, TXNIP, phospho-S6 (p-S6) and LC3 in human primary chondrocytes transfected with siRNA control or specific against REDD1 or TXNIP as indicated in the presence or absence of chloroquine (CQ, 25μM). Values are mean±95%CI of four different donors.
To determine if REDD1 interacts with TXNIP in chondrocytes, we overexpressed REDD1 in T-C/28 cells and performed co-immunoprecipitation. As shown in Figure 6C, endogenous TXNIP was detected in REDD1 immunoprecipitates, indicating that these two proteins interact in chondrocytes. Next, we determined whether TXNIP is able to regulate autophagy in chondrocytes. In a similar fashion as REDD1 depletion, we found that TXNIP knockdown in human primary chondrocytes resulted in reduced autophagic activity as evidenced by reduced LC3 processing in the presence of chloroquine (Figure 6D). Moreover, combined knockdown of REDD1 and TXNIP resulted in a further reduction of autophagy (Figure 6D) indicating that the REDD1/TXNIP complex is required for autophagy induction in chondrocytes.
Discussion
In the present study we report for the first time that REDD1, an endogenous mTOR inhibitor (20), is highly expressed in normal articular cartilage and that OA and aging are associated with decreased REDD1 mRNA and protein expression. Moreover, our data indicate that REDD1 forms a complex with TXNIP that is required for autophagy induction in articular chondrocytes.
REDD1 is a ubiquitously expressed protein that is tightly regulated during development and was initially identified as transcriptionally induced by hypoxia and DNA damage (18, 19). REDD1 primarily acts as an endogenous mTOR inhibitor under cellular stress conditions such as hypoxia, endoplasmic reticulum stress, energy stress, and oxidative stress (20-22). Mice with global REDD1 deletion are viable, fertile, and do not exhibit any growth or physical abnormalities (27). However, REDD1 deficiency under stress conditions results in tissue specific effects that can be protective or deleterious in a context dependent manner (23, 31-34). mTOR is a central kinase that regulates protein synthesis, autophagy, metabolism and cell survival (7). As part of the normal cellular response to stress, diverse evolutionarily conserved factors such as FOXO or AMPK inhibit mTOR signaling to stop cell growth and proliferation while redirecting energy metabolism to support stress resistance and cell survival (4, 7, 35). Age-related declines of FOXO and AMPK activities have been suggested to potentially lead to age-associated diseases (4) and elevated mTOR signaling has been reported in several age-related pathologies (35), including in OA cartilage (9). While the molecular mechanisms underlying mTOR hyperactivation in OA pathogenesis remain incompletely understood, reduced FOXO (15) and AMPK (16) expression have been found in OA cartilage. The data presented here expands the current knowledge about mTOR regulation in articular cartilage and suggests that decreased REDD1 expression during aging and OA can contribute to exacerbated mTOR signaling.
Recent evidence supports a central role of autophagy in cartilage homeostasis and OA development (36). Reduced expression of key autophagy elements such as ULK1, BECN1 and MAP1LC3 has been reported in aging and OA cartilage (11). Inhibition of mTOR signaling by rapamycin increases autophagy in articular cartilage and is associated with a significant reduction in the severity of cartilage degeneration and synovitis associated with decrease in the expression of ADAMTS-5 and IL-1 in articular cartilage (12). Furthermore, mice with cartilage-specific mTOR deletion showed increased expression of autophagic markers and significant protection from surgically-induced OA (10). Our study shows that REDD1 is an endogenous regulator of autophagy in articular chondrocytes and supports a model where decreased REDD1 expression during aging and OA leads to a deficiency in autophagic activity. Restoring REDD1 expression during aging is a potential therapeutic strategy to prevent the onset of OA.
An important finding of the present study is that REDD1 regulation of autophagy is largely mTOR independent. Instead, REDD1 associates with the pro-oxidant protein TXNIP to form a protein complex that is essential for autophagy induction in chondrocytes. TXNIP is a negative modulator of thioredoxins and it is induced by various types of cellular stress such as oxidative stress, UV radiation, heat shock and apoptotic signaling (37). Recent studies showed that TXNIP interacts with REDD1 under energy stress conditions, resulting in REDD1 protein stabilization and inhibition of mTOR signaling (38). In addition, this REDD1/TXNIP complex has been shown to positively regulate autophagy in 293T cells through production of endogenous radical oxygen species (30). An interesting finding of the latter study is that REDD1 deficiency in vivo also results in mitochondrial dysfunction and accumulation of damaged mitochondria (30), two events that have been described in articular cartilage during aging and OA (39). According to our data, it could be hypothesized that REDD1 deficiency during aging and OA could contribute to defective mitochondrial function in articular chondrocytes. Following this rationale, restoration of REDD1 levels not only would result in autophagy activation but also improve mitochondrial function of articular chondrocytes.
We show that REDD1 expression is significantly reduced in aged and OA cartilage. However, we did not explore the molecular mechanisms responsible for this reduction. REDD1 transcriptional activation can be mediated by different transcription factors such as p63 (18), HIF-1α (20), ATF4 (40), Che-1 (41), or PLZF (42) among others. On the other hand, there is less understanding of REDD1 inhibitory mechanisms. To the best of our knowledge, only one study reported a REDD1 repression by a transcription factor in human hematopoietic stem cells (43). Our data indicates that REDD1 is expressed at high levels in normal articular cartilage. This high basal expression could be due to a sustained transcriptional activation by the above-mentioned transcription factors. REDD1 decreased expression during aging and OA could thus be the consequence of reduced transcriptional activation as opposed to an active transcriptional inhibition. We analyzed REDD1 expression in cultured articular chondrocytes stimulated with interleukin-1β to induce inflammatory stress, or tert-Butyl hydroperoxide to induce oxidative stress, and did not find a reduction in REDD1 protein levels (data not shown). Future studies should be aimed at deeper understanding the REDD1 regulation in articular chondrocytes.
In summary, we report for the first time that REDD1 is an endogenous regulator of mTOR signaling and autophagy in articular chondrocytes. Moreover, REDD1 expression is reduced during aging and OA in articular cartilage, contributing to mTOR hyperactivation and autophagy deficiency that is characteristic of OA pathophysiology.
Supplementary Material
Supplemental figure 1. REDD1 protein expression measured by IHC in articular cartilage from 2-month-old Redd1+/+ and Redd1-/- mice. Magnification bar = 100μm.
{kind=link}
Acknowledgments
We gratefully acknowledge Shantanu Patil MD, Stuart Duffy, and Lilo Creighton for their technical assistance. We would also thank Dr. Irina Budunova and Dr. Rubin Tuder for kindly providing the REDD1-/- mice.
This study was supported by NIH grants AG007996, AG049617 and UL1 TR001114.
Footnotes
The authors have no conflicts of interest.
Author contributions: All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Lotz had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Alvarez-Garcia, Lotz.
Acquisition of data. Alvarez-Garcia, Olmer, Akagi, Akasaki, Fisch, Shen.
Analysis and interpretation of data. Alvarez-Garcia, Su, Lotz.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol. 2014;10(7):437–41. doi: 10.1038/nrrheum.2014.44. [DOI] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51(2):241–8. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328(5976):321–6. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kloet DE, Burgering BM. The PKB/FOXO switch in aging and cancer. Biochimica et biophysica acta. 2011;1813(11):1926–37. doi: 10.1016/j.bbamcr.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(19):1845–6. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 9.Pal B, Endisha H, Zhang Y, Kapoor M. mTOR: a potential therapeutic target in osteoarthritis? Drugs R D. 2015;15(1):27–36. doi: 10.1007/s40268-015-0082-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Vasheghani F, Li YH, Blati M, Simeone K, Fahmi H, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204599. [DOI] [PubMed] [Google Scholar]
- 11.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71(4):575–81. doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beier F, Loeser RF. Biology and pathology of Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in chondrocytes. Journal of cellular biochemistry. 2010;110(3):573–80. doi: 10.1002/jcb.22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akasaki Y, Alvarez-Garcia O, Saito M, Carames B, Iwamoto Y, Lotz MK. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 2014;66(12):3349–58. doi: 10.1002/art.38868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akasaki Y, Hasegawa A, Saito M, Asahara H, Iwamoto Y, Lotz MK. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthritis Cartilage. 2014;22(1):162–70. doi: 10.1016/j.joca.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 2011;63(7):1928–37. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dvir-Ginzberg M, Steinmeyer J. Towards elucidating the role of SirT1 in osteoarthritis. Front Biosci (Landmark Ed) 2013;18:343–55. doi: 10.2741/4105. [DOI] [PubMed] [Google Scholar]
- 18.Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Molecular cell. 2002;10(5):995–1005. doi: 10.1016/s1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- 19.Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Molecular and cellular biology. 2002;22(7):2283–93. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes & development. 2004;18(23):2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corradetti MN, Inoki K, Guan KL. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. The Journal of biological chemistry. 2005;280(11):9769–72. doi: 10.1074/jbc.C400557200. [DOI] [PubMed] [Google Scholar]
- 22.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes & development. 2008;22(2):239–51. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nature medicine. 2010;16(7):767–73. doi: 10.1038/nm.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarzer R, Tondera D, Arnold W, Giese K, Klippel A, Kaufmann J. REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene. 2005;24(7):1138–49. doi: 10.1038/sj.onc.1208236. [DOI] [PubMed] [Google Scholar]
- 25.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15(9):1061–9. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S, Glowacki J, et al. Interleukin-1 beta-modulated gene expression in immortalized human chondrocytes. The Journal of clinical investigation. 1994;94(6):2307–16. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brafman A, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, et al. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Investigative ophthalmology & visual science. 2004;45(10):3796–805. doi: 10.1167/iovs.04-0052. [DOI] [PubMed] [Google Scholar]
- 28.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3(8):1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- 29.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D, Nash Z, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014. doi: 10.1038/ncomms8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolff NC, Vega-Rubin-de-Celis S, Xie XJ, Castrillon DH, Kabbani W, Brugarolas J. Cell-type-dependent regulation of mTORC1 by REDD1 and the tumor suppressors TSC1/TSC2 and LKB1 in response to hypoxia. Molecular and cellular biology. 2011;31(9):1870–84. doi: 10.1128/MCB.01393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ota KT, Liu RJ, Voleti B, Maldonado-Aviles JG, Duric V, Iwata M, et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nature medicine. 2014;20(5):531–5. doi: 10.1038/nm.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li XH, Ha CT, Fu D, Xiao M. REDD1 protects osteoblast cells from gamma radiation-induced premature senescence. PloS one. 2012;7(5):e36604. doi: 10.1371/journal.pone.0036604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dennis MD, McGhee NK, Jefferson LS, Kimball SR. Regulated in DNA damage and development 1 (REDD1) promotes cell survival during serum deprivation by sustaining repression of signaling through the mechanistic target of rapamycin in complex 1 (mTORC1) Cellular signalling. 2013;25(12):2709–16. doi: 10.1016/j.cellsig.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lotz MK, Carames B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7(10):579–87. doi: 10.1038/nrrheum.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fidler IJ, Radinsky R. Search for genes that suppress cancer metastasis. J Natl Cancer Inst. 1996;88(23):1700–3. doi: 10.1093/jnci/88.23.1700. [DOI] [PubMed] [Google Scholar]
- 38.Jin HO, Seo SK, Kim YS, Woo SH, Lee KH, Yi JY, et al. TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene. 2011;30(35):3792–801. doi: 10.1038/onc.2011.102. [DOI] [PubMed] [Google Scholar]
- 39.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7(3):161–9. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 40.Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-beta negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free radical biology & medicine. 2009;46(8):1158–67. doi: 10.1016/j.freeradbiomed.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Desantis A, Bruno T, Catena V, De Nicola F, Goeman F, Iezzi S, et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J. 2015 doi: 10.15252/embj.201489920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hobbs RM, Seandel M, Falciatori I, Rafii S, Pandolfi PP. Plzf regulates germline progenitor self-renewal by opposing mTORC1. Cell. 2010;142(3):468–79. doi: 10.1016/j.cell.2010.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benyoucef A, Calvo J, Renou L, Arcangeli ML, van den Heuvel A, Amsellem S, et al. The SCL/TAL1 Transcription Factor Represses the Stress Protein DDiT4/REDD1 in Human Hematopoietic Stem/Progenitor Cells. Stem Cells. 2015;33(7):2268–79. doi: 10.1002/stem.2028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. REDD1 protein expression measured by IHC in articular cartilage from 2-month-old Redd1+/+ and Redd1-/- mice. Magnification bar = 100μm.