

Abstract
Patients diagnosed with glioblastoma, an aggressive brain tumour, have a poor prognosis, with a median overall survival of less than 15 months. Vasculature within these tumours is typically abnormal, with increased tortuosity, dilation and disorganization, and they typically exhibit a disrupted blood–brain barrier (BBB). Although it has been hypothesized that the ‘normalization’ of the vasculature resulting from anti-angiogenic therapies could improve drug delivery through improved blood flow, there is also evidence that suggests that the restoration of BBB integrity might limit the delivery of therapeutic agents and hence their effectiveness. In this paper, we apply mathematical models of blood flow, vascular permeability and diffusion within the tumour microenvironment to investigate the effect of these competing factors on drug delivery. Preliminary results from the modelling indicate that all three physiological parameters investigated—flow rate, vessel permeability and tissue diffusion coefficient—interact nonlinearly to produce the observed average drug concentration in the microenvironment.
Keywords: multimodality imaging, glioblastoma, drug delivery, perfusion, computational modelling and simulation
1. Background
With a median overall survival of less than 15 months [1], glioblastomas (GBMs) are challenging tumours with unsatisfactory responses to chemotherapy [2–5]. While it is known that many factors, from genetic variants [6] to microscopic tissue oxygenation [7], influence drug effectiveness, the drug delivery within the tumour is a fundamental consideration. Tumours often have abnormal, disorganized vasculature with increased tortuosity, shunting, poor perfusion and permeability, resulting in impaired blood flow. These abnormalities can result in elevated interstitial fluid pressure and hinder the delivery of therapeutic agents to tumours [8–11].
Poor or uneven regional distribution of therapeutic agents in the tumoural tissues of brain tumours, well known to be highly heterogeneous, is believed to contribute to disappointing clinical trial results. Further, complementary drugs such as anti-angiogenic agents, when used along with cytotoxic agents, modulate drug delivery in complex ways. Antiangiogenics have been hypothesized to improve drug delivery through ‘normalization’ of the vasculature [7,12,13]. However, it has also been suggested that the restoration of blood–brain barrier (BBB) integrity might limit the delivery of concomitant cytotoxic agents and hence their effectiveness [14,15].
Regional measurement of tissue drug delivery is thus a foundational measurement that has profound implications for the timing and dosing of drug combinations, yet one that remains elusive in clinical practice. The challenge lies in the complexity of the tumour microenvironment and the spatial and temporal heterogeneity of the tumours. Structural and functional changes in tumour vasculature as well as changes in the cellularity in the interstitial space can impair drug delivery [16]. Determining regional drug distribution in tumours in individual patients non-invasively is a crucial tool needed for improving our understanding of how best to utilize our current drug therapies and evaluate new treatment regimens in individual patients.
Therefore, in order to improve our understanding of how best to use drugs to target these (and other) aggressive tumours, the clinical community needs a tool to measure drug penetration and delivery to brain tumours in individual patients and longitudinally over time [17]. In vivo magnetic resonance imaging (MRI) is a non-invasive tool that has been demonstrated to interrogate microscopic tissue features sensitive to the microvascular space, tissue perfusion and interstitial delivery [7,12,18–21] but methods to use such MRI data to precisely predict therapeutic drug delivery distribution and kinetics directly do not currently exist. For example, figure 1 shows a post-contrast magnetic resonance (MR) image as well as a positron emission tomography (PET) image for the radiolabelled [11C] temozolomide for two patients before and after anti-angiogenic therapy. The post-contrast MR image is a map of the vascular permeability as the gadolinium agent extravasates into the interstitial areas leading to corresponding areas of enhancement. The PET image is similarly a spatial map of the distribution of the temozolomide, the chemotherapy agent. These images suggest that the drug delivery, at least in these patients, appears to be higher in areas of increased vascular permeability and then decreases with decreased permeability. However, it is difficult to test this further from the data available from the images alone. Nonetheless, we believe that MRI can indeed be the basis for such a predictive tool, with the advantages of its ubiquitous clinical availability and non-invasiveness. Further, we believe that MRI methods sensitive to the physical properties of the tumour microenvironment can be used to provide the needed inputs into a detailed, realistic mathematical model of the tumour microenvironment, designed and validated to accurately predict drug delivery. We refer to this approach as ‘Model + MRI’.
Figure 1.

Post-contrast magnetic resonance (MR) and positron emission tomography (PET) images showing the distribution of radiolabelled temozolomide for two patients before (baseline, left-hand side) and after (follow-up, right-hand side) anti-angiogenic therapy.
In this paper, we aim to provide a ‘proof of concept’ of our Model + MRI approach by formulating and validating a mathematical model of perfusion in a vascular network, the actual structure of which is empirically derived from high-resolution three-dimensional optical microscopic features of the vascular network in mouse models, both morphological and physiological. This will allow us to have highly realistic computational models of the essential features that govern local drug delivery and tissue distribution including the permeability of the drug to regions of disrupted and intact BBB, and the complexities of the interstitial space. The empirical data for these model inputs will come from advanced imaging methods that have been developed over the last several years [18,22–28].
2. Material and methods
We have previously developed a hybrid discrete-continuum mathematical model of flow through vascular networks allowing for dynamic adaptation of the vessels, with applications to tumour-induced angiogenesis and drug delivery, wound healing and retinal development [29–37]. Recent extensions to the model have incorporated permeability of the vasculature and diffusion of the extravasated agent through heterogeneous three-dimensional tissue.
Advances in optical imaging technology have enabled us to develop vascular anatomical network (VAN) models based on two-photon microscopy in mouse models [26,38]. These ‘virtual voxels’, representing realistic vascular networks, were used as input to the mathematical models to calculate the flow and distribution of agents within the ‘virtual voxel’.
We have preliminary imaging data for patients with glioblastoma enrolled in an ongoing clinical trial receiving an anti-angiogenic agent. Patients received MR as well as PET imaging before and after treatment with the anti-angiogenic therapy. In addition to standard anatomical MRI, we also performed dynamic perfusion and permeability imaging in order to measure blood flow, blood volume and vascular permeability at a voxel level. PET was performed using and radiolabelled [11C] temozolomide [39,40], thus allowing for the visualization of the spatial distribution of the drug (cf. figure 1). Parametric maps were generated for cerebral blood flow, volume and permeability [18] as well as for the standardized uptake value [41] of radiolabelled temozolomide and quantified within regions of interest identified by the clinician.
2.1. Modelling perfusion and tissue diffusion
2.1.1. Model description
In this work, we model a small block of rodent brain tissue (0.6 × 0.6 × 0.6 mm) that hosts—and is perfused by—a highly tortuous interconnected network of flowing capillary elements (figure 2a). For simulation purposes, the network itself is partitioned into nodes (junctions) and edges (connections) and each element (node/edge) is assigned a range of intrinsic attributes (radius, length, vessel type, volume and conductivity). This model was created using structural images with fluorescein-labelled blood. A three-dimensional mask of the vasculature was obtained from the angiogram and the graph and a mesh of the vasculature was generated [38]. This provided an accurate three-dimensional vascular structure in which to carry out the flow simulations. Summary statistics for the size distribution of the vessel radii are given in table 1.
Figure 2.

(a) Capillary network extracted from optical imaging of vessels surrounding a brain tumour: arterioles are coloured in red, venules in blue and capillary elements in purple. (b) The same network surrounded by an 80 × 80 × 80 cubic mesh grid to model the tissue surrounding the vessels—only a slice of the grid is displayed in the figure.
Table 1.
Summary statistics for the vessel size distributions in the three-dimensional vasculature shown in figure 2.
| minimum vessel radius | 2 µm |
| maximum vessel radius | 43 µm |
| mean vessel radius | 6 µm |
| median vessel radius | 5 µm |
| standard deviation | 3.8 µm |
Several attributes can be extracted directly from optical imaging (two-photon and optical coherence tomography) of the rodent brain and these have been incorporated into our computational (i.e. in silico) vascular bed. We note that, in contrast with our earlier modelling studies, the network used here is completely lattice-free and inlet and outlet pressure boundary conditions can be assigned with great flexibility. While we have assumed a cylindrical geometry for capillary elements at present, it should be noted that this assumption can be easily relaxed if image data suggests otherwise.
The host tissue surrounding the vessels is modelled using a regular cubic mesh comprising 30 × 30 × 30 cubes, of equal edge length hx, hy, hz. Given the scale of the block of brain tissue (0.6 × 0.6 × 0.6 mm), this means that the spatial grid size is of order 20 µm (figure 2b). Beginning with a full mesh we then proceed to remove all blocks that lie completely within vessels and identify all vessel segments that provide capillary surface area to each remaining tissue element—this information is used later when calculating the diffusive flux from a vessel to an adjacent cube (and vice versa).
2.1.2. Flow and diffusion
Computing the nodal pressure field during the full simulation is an essential component of the process and is used to update the tracer concentration within each capillary segment at each time step. We fix an injection rate for the blood flow (although constant pressure drop simulations can also be readily considered), which is effectively used as a boundary condition to determine the nodal pressure distribution and elementary flows within the system. We assume that for each vascular element of shape factor G (the ratio of cross-sectional area to the square of the perimeter), length L and cross-section A, the flow Q is given by a Poiseuille-type law
where g is the element conductance, μ is the fluid viscosity in the element and ΔP the pressure difference acting across the element.
By applying the appropriate pressure gradient across the network, the pressure field inside a network can be obtained by applying the mass conservation law at each node i (assuming that the flowing fluid is incompressible)
where Qi,j is the flow between node i and node j. We use Cholesky factorization to solve this system of linear equations and determine the pressure value at each node. For this work, we assume the flow of a simple Newtonian fluid, although complex blood rheology can be incorporated straightforwardly (see [31] for full details).
In the absence of drug leakage across the vessel membrane, we can apply mass conservation at nodes to update the contrast agent concentrations within each capillary over time. The initial concentration is set to zero for all elements and we then inject a benign contrast agent into the inlet arterioles at dimensionless unit concentration.
If we consider the configuration depicted in figure 3, where Ci and Qi,j represent, respectively, the concentration and the flow in capillary element i; the new tracer concentration after a time step Δt in capillary 1 is given by
where Cold is the old tracer concentration and V1 the volume of capillary 1.
Figure 3.
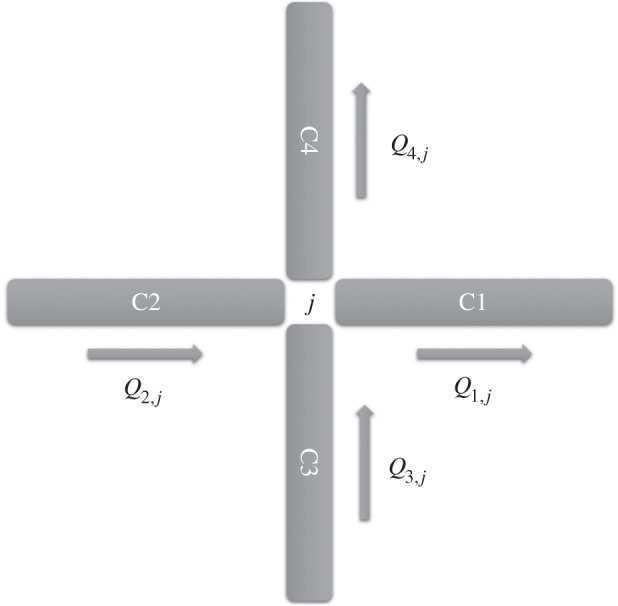
A configuration of elementary flows corresponding to four connected edges (vessels) meeting at a node j (junction). Mass conservation dictates that Q1,j + Q4,j = Q2,j + Q3,j.
However, when tracer leakage in the surrounding tissue is considered, this approach needs to be amended to account for the diffusion of contrast agent into the host tissue. To satisfy mass conservation in this scenario, we need to consider (i) the convective flow of tracer in the vessels (as described above); (ii) the diffusive flux of agent from the vessels to the tissue (and vice versa); (iii) diffusive transport between tissue blocks; and (iv) tracer decay in the tissue.
These four main flow mechanisms are expressed as a coupled system of differential equations as described below
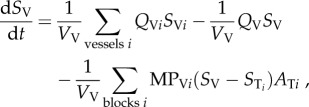 |
2.1 |
| 2.2 |
where SV = vessel concentration; Vv = vessel volume; Qv = flow in vessel V; MPVi = vessel membrane permeability (MP) between vessel V and tissue block i; STi = tissue block i concentration, ATi = contact area between vessel v and tissue block i; ST = tissue block T concentration; VT = tissue block T volume; DT = diffusion coefficient in tissue; σ = tissue intake coefficient.
The term 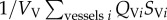 describes the mass flow of drug carried to vessel V by its neighbouring vessels Vi via convection;
describes the mass flow of drug carried to vessel V by its neighbouring vessels Vi via convection; 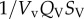 refers to the mass flow of drug leaving vessel V via convection;
refers to the mass flow of drug leaving vessel V via convection; 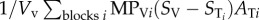 refers to the mass flow of drug leaving vessel V via diffusion into the surrounding tissue; DTΔST is the expression of diffusive flow between tissue blocks;
refers to the mass flow of drug leaving vessel V via diffusion into the surrounding tissue; DTΔST is the expression of diffusive flow between tissue blocks; 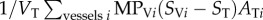 refers to the mass flow leaving the tissue block to its surrounding vessels via diffusion, and σST describes the mass flow of drug consumed by the tissue.
refers to the mass flow leaving the tissue block to its surrounding vessels via diffusion, and σST describes the mass flow of drug consumed by the tissue.
The elementary flows QVi and QV are obtained after solving the pressure field in the vasculature network, and they are proportional to the blood flow rate Q, which is treated as an input. We also note that a homogeneous MP is assumed in the entire network.
Solving this coupled system using finite difference methods yields the contrast agent concentration in each vessel v and tissue block (i, j, k) at every time step Δt as described in the following scheme
 |
 |
To ensure mass conservation of the drug in the vasculature system, the time step has to be chosen carefully to ensure that the dimensionless drug concentration in every vessel and tissue block remains within the interval [0, 1]. Therefore, we consider the case where every vessel and tissue block gets a drug inflow of concentration 1 from its surrounding vessels and tissue blocks whilst assuming a zero drug outflow. This yields the following expression for the time step:
 |
In fact, for the computational results here, 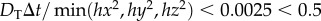 was always satisfied for the range of parameters we used.
was always satisfied for the range of parameters we used.
3. Results
In order to determine the most important factors affecting tracer distribution within a tissue voxel, a range of simulations was undertaken by varying flow rate, transmural transport coefficient (i.e. vessel permeability), and extracellular matrix diffusion coefficient. A summary of the parameter values used in the simulations is given in table 2 (cf. [26,33,35]).
Table 2.
Summary of the parameter values used in the computational simulations presented in figures 4–7. The precise values used in each simulation are given in the legend of the relevant figure.
| flow rate (m3 s−1) | 10−7, 10−8, 10−9 |
| diffusion coefficient (m2 s−1) | 10−5, 10−6, 10−7, 10−8 |
| vessel permeability (m s−1) | 1, 0.1, 0.01, 0.001 |
| tissue intake (s−1) | 0 |
Images of tracer evolution for all parameter combinations explored are shown in figure 4, with each row corresponding to a given set of input parameters (see figure caption for parameter values).
Figure 4.

Five simulations displaying tracer concentrations in vessels and tissue in after continuous drug injection for 10 s. The left-hand column shows tracer in the entire system block, the middle column shows tracer in vessels only, and the right-hand column shows tracer in tissue only. Row 1 is the baseline simulation case and each other row corresponds to a change of a single parameter (highlighted in bold) as follows: row 1: flow rate: 10−8 m3 s−1, vasculature permeability: 10−3 m s−1, tissue diffusion coeff.: 10−6 m2 s−1 (baseline); row 2: flow rate: 10−8 m3 s−1, vasculature permeability: 0.01 m s−1, tissue diffusion coeff.: 10−6 m2 s−1; row 3: flow rate: 10−8 m3 s−1, vasculature permeability: 0.1 m s−1, tissue diffusion coeff.: 10−6 m2 s; row 4: flow rate: 10−7 m3 s−1, vasculature permeability: 0.1 m s−1, tissue diffusion coeff.: 10−6 m2 s−1; row 5: flow rate: 10−7 m3 s−1, vasculature permeability: 0.1 m s−1, tissue diffusion coeff.: 10−5 m2 s−1.
We begin by investigating the combined effects of vessel permeability and ECM diffusion upon the average vessel concentration at a fixed flow rate of 10−8 m3 s−1 (this average is taken over all vessels in the voxel). Figure 5a, corresponding to slow diffusion through the ECM (DT = 10−7 m2 s−1), shows that contrast agent builds up rapidly within the capillary network following infusion. The build-up of agent in the bed is faster when the capillaries are less leaky, as expected: however, we observe that the vessel concentration asymptotes towards a steady-state value that is insensitive to vessel permeability. By contrast, under conditions of faster tissue diffusion (DT = 10−6 m2 s−1), we find that the steady-state vessel concentration increases with decreasing vessel permeability (figure 5b)—the rate of diffusive transport through the tissue block is now relatively high and so contrast agent is efficiently removed from the vasculature throughout its transit.
Figure 5.

(a) The evolution of average vessel concentration in the voxel during continuous drug injection for various vessel membrane permeabilities (slow diffusion coefficient in tissue DT = 10−7 m2 s−1, and flow rate (FR) = 10−8 m3 s−1). (b) The evolution of average vessel concentration in the voxel during continuous drug injection for various vessel membrane permeabilities (fast diffusion coefficient in tissue DT = 10−6 m2 s−1, and FR = 10−8 m3 s−1).
Corresponding graphs of the average tissue concentrations are shown in figures 6a,b. For slow tissue diffusion, the results exhibit the opposite trend to that observed for vessel concentration (i.e. as vessel permeability increases, average tissue concentration increases more rapidly). When tissue diffusion is fast, however, an interesting effect is observed: we find that the highest average level of contrast agent in the tissue corresponds to an intermediate value of vessel permeability. Moreover, it can be seen that all of the simulations yield tissue concentrations that are far smaller than those seen under slow diffusion conditions. It appears that this combination optimizes the vascular supply of contrast agent close to the inlet of the voxel, where it then diffuses rapidly through the tissue and is quickly re-captured via diffusive transport from tissue into low-concentration, downstream vessels; effectively removing it from the system.
Figure 6.

(a) The evolution of average tissue concentration in the voxel during continuous drug injection for various vessel membrane permeabilities (slow diffusion coefficient in tissue DT = 10−7 m2 s−1, and flow rate (FR) = 10−8 m3 s−1). (b) The evolution of average tissue concentration in the voxel during continuous drug injection for various vessel membrane permeabilities (fast diffusion coefficient in tissue DT = 10−6 m2 s−1, and FR = 10−8 m3 s−1).
Finally, we have also studied the effect of flow rate on tracer delivery to the tissue (figure 7). Here, we observe that average tissue concentrations asymptote to higher values at higher flow rates.
Figure 7.

The evolution of average tissue concentration in the voxel during continuous drug injection for a fixed vessel membrane permeability (MP = 0.01 m s−1) and diffusion coefficient in tissue (DT = 10−6 m2 s−1) and three flow rates (FR): 10−7 m3 s−1, 10−8 m3 s−1, 10−9 m3 s−1.
Hence, we can conclude that all three physiological parameters investigated thus far—flow rate, vessel permeability and tissue diffusion coefficient—interact nonlinearly to produce the observed average tracer concentration in the voxel. Preliminary results from the modelling indicate that delivery of the agent to the tumour bed is affected by flow, permeability and diffusion in the interstitial space. We find that reductions in the permeability of the vasculature, as would be typified through restoration of the BBB in response to anti-angiogenic therapy, could potentially reduce the delivery of the drug to the tumour, while increased flow could increase drug delivery.
4. Discussion and conclusion
Mathematical modelling combined with empirical data from in vivo models can provide insights into the role that the vascular architecture, permeability and microenvironment heterogeneity can play in drug distribution within a tumour bed during anti-angiogenic therapy. In humans, PET/MR with radiolabelled drugs (such as temozolomide) can be used to non-invasively validate these models and provide patient specific guidance for therapy. As described above, our modelling suggests that under certain conditions of vascular flow and diffusion through the interstitial space, a decrease in the permeability of the BBB, such as resulting from anti-angiogenic therapies, might actually result in reductions in the drug delivered to the tumours, despite normalization of the vasculature.
This proof-of-concept allows us to explore the use of mathematical models combined with the parameters measured using advanced MR images to predict drug delivery. This modelling will serve as an important link between our optical microscopic measurements of the tumour microenvironment (vascular network, permeability and accessible interstitial space) and the macroscopic MRI, and will ultimately validate our use of MRI to predict drug delivery. In the future, we hope to take our realistic computational models of the vascular and interstitial spaces (incorporating detailed empirical information on vascular perfusion, permeability and interstitial transport) [37–40], and then quantitatively test the ability of several advanced MRI approaches to assess these microenvironmental features from our non-invasive MR datasets [17]. We can then directly validate, in our animal models, the ability of the Model + MRI data to provide specific regional assessment of drug delivery, assessed at both microscopic levels statically using PET. Finally, this approach can be validated in our human patients with the ultimate goal to use patient specific MRI parameters of vascular permeability, flow and diffusion (all parameters easily obtain with non-invasive MRI) to inform the Model + MRI and predict the patient specific response to a therapeutic agent.
Authors' contributions
A.B. and M.W. carried out the computational simulations, participated in data analysis and drafted the manuscript; Y.-F.Y. and C.C. helped with the data acquisition and analysis; patient data were obtained by T.T.B. and E.R.G. as part of a clinical trial; they also helped with the study design and manuscript; S.Mc., M.A.J.C., J.K.-C., T.D., D.B. and B.R. conceived of the study, designed the study, coordinated the study, participated in data analysis and helped draft the manuscript. All authors gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
S.M.c. and M.A.J.C. gratefully acknowledge support of EPSRC grant no. EP/N014642/1 (‘EPSRC Centre for Multiscale Soft Tissue Mechanics—With Application to Heart & Cancer’). M.A.J.C. would like to thank the Isaac Newton Institute for Mathematical Sciences for its hospitality during the programme ‘Coupling geometric PDEs with physics for cell morphology, motility and pattern formation’ supported by EPSRC grant no. EP/K032208/1.J.K.-C., B.R. and Y.-F.Y. gratefully acknowledge NIH/NCI grant no. U01CA154601, E.R.G. acknowledges NIH/NCI grant no. K23CA169021 and C.C. acknowledges NIH/NIBIB grant no. R01EB014894. T.T.B. was supported by NIH/NCI grant R01-R01CA129371-01.
References
- 1.Stupp R, et al. European Organisation for R, Treatment of Cancer Brain T, Radiation Oncology G, National Cancer Institute of Canada Clinical Trials G. 2009. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466. ( 10.1016/S1470-2045(09)70025-7) [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. 2008. Malignant gliomas in adults. N. Engl. J. Med. 359, 492–507. ( 10.1056/NEJMra0708126) [DOI] [PubMed] [Google Scholar]
- 3.Omuro AM. 2008. Exploring multi-targeting strategies for the treatment of gliomas. Curr. Opin. Investig. Drugs 9, 1287–1295. [PubMed] [Google Scholar]
- 4.Reardon DA, Wen PY. 2006. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist 11: 152–164. ( 10.1634/theoncologist.11-2-152) [DOI] [PubMed] [Google Scholar]
- 5.Kole AJ, et al. In press. Concurrent chemoradiotherapy versus radiotherapy alone for ‘biopsy-only’ glioblastoma multiforme. Cancer ( 10.1002/cncr.30063) [DOI] [PubMed] [Google Scholar]
- 6.Parsons DW, et al. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. ( 10.1126/science.1164382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batchelor TT, et al. 2013. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc. Natl Acad. Sci. USA. 110, 19 059–19 064. ( 10.1073/pnas.1318022110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leunig M, Jain R. 1996. Tumor angiogenesis and interstitial hypertension Cancer Res. 56, 4264–4266. [PubMed] [Google Scholar]
- 9.Jain RK. 2001. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat. Med. 7, 987–989. ( 10.1038/nm0901-987) [DOI] [PubMed] [Google Scholar]
- 10.Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. 2011. Delivery of molecular and nanoscale medicine to tumours: transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2, 281–298. ( 10.1146/annurev-chembioeng-061010-114300) [DOI] [PubMed] [Google Scholar]
- 11.Jain RK. 1994. Barriers to drug delivery in solid tumors. Sci. Am. 271, 58–65. ( 10.1038/scientificamerican0794-58) [DOI] [PubMed] [Google Scholar]
- 12.Sorensen AG, et al. 2012. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 72, 402–407. ( 10.1158/0008-5472.CAN-11-2464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain RK. 2013. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J. Clin. Oncol. 31, 2205–2218. ( 10.1200/JCO.2012.46.3653) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van der Veldt AA, et al. 2012. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell 21, 82–91. ( 10.1016/j.ccr.2011.11.023) [DOI] [PubMed] [Google Scholar]
- 15.Arjaans M, et al. 2013. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res. 73, 3347–3355. ( 10.1158/0008-5472.CAN-12-3518) [DOI] [PubMed] [Google Scholar]
- 16.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. 2000. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 60, 2497–2503. [PubMed] [Google Scholar]
- 17.Kut C, Grossman SA, Blakeley J. 2015. How critical is the blood-brain barrier to the development of neurotherapeutics? JAMA Neurol. 72, 381–382. ( 10.1001/jamaneurol.2014.3736) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalpathy-Cramer J, Gerstner ER, Emblem KE, Andronesi OC, Rosen B. 2014. Advanced magnetic resonance imaging of the physical processes in human glioblastoma. Cancer Res. 74, 4622–4637. ( 10.1158/0008-5472.CAN-14-0383) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kothari PD, et al. 2013. Longitudinal restriction spectrum imaging is resistant to pseudoresponse in patients with high-grade gliomas treated with bevacizumab. Am. J. Neuroradiol. 34, 1752–1757. ( 10.3174/ajnr.A3506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellingson BM, Malkin MG, Rand SD, Laviolette PS, Connelly JM, Mueller WM, Schmainda KM. 2011. Volumetric analysis of functional diffusion maps is a predictive imaging biomarker for cytotoxic and anti-angiogenic treatments in malignant gliomas. J. Neurooncol. 102, 95–103. ( 10.1007/s11060-010-0293-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerstner ER, Chen PJ, Wen PY, Jain RK, Batchelor TT, Sorensen G. 2010. Infiltrative patterns of glioblastoma spread detected via diffusion MRI after treatment with cediranib. Neuro. Oncol. 12, 466–472. ( 10.1093/neuonc/nop051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakadz˘ić S, et al. 2011. Cerebral blood oxygenation measurement based on oxygen-dependent quenching of phosphorescence. J. Vis. Exp. 51, e1694 ( 10.3791/1694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devor A, et al. 2012. Frontiers in optical imaging of cerebral blood flow and metabolism. J. Cereb. Blood Flow Metab. 32, 1259–1276. ( 10.1038/jcbfm.2011.195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magnain C, et al. 2014. Blockface histology with optical coherence tomography: a comparison with Nissl staining. Neuroimage 84, 524–533. ( 10.1016/j.neuroimage.2013.08.072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yucel MA, Evans KC, Selb J, Huppert TJ, Boas DA, Gagnon L. 2014. Validation of the hypercapnic calibrated fMRI method using DOT-fMRI fusion imaging. Neuroimage 102, 729–735. ( 10.1016/j.neuroimage.2014.08.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gagnon L, et al. 2015. Quantifying the microvascular origin of BOLD-fMRI from first principles with two-photon microscopy and an oxygen-sensitive nanoprobe. J. Neurosci. 35, 3663–3675. ( 10.1523/JNEUROSCI.3555-14.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaseen MA, Srinivasan VJ, Gorczynska I, Fujimoto JG, Boas DA, Sakadzic S. 2015. Multimodal optical imaging system for in vivo investigation of cerebral oxygen delivery and energy metabolism. Biomed Opt. Exp. 6, 4994–5007. ( 10.1364/BOE.6.004994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emblem KE, et al. 2013. Vessel architectural imaging identifies cancer patient responders to anti-angiogenic therapy. Nat. Med. 19, 1178–1183. ( 10.1038/nm.3289) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaplain MA, Stuart AM. 1991. A mathematical model for the diffusion of tumour angiogenesis factor into the surrounding host tissue. IMA J. Math. Appl. Med. Biol. 8, 191–220. ( 10.1093/imammb/8.3.191) [DOI] [PubMed] [Google Scholar]
- 30.McDougall SR, Anderson AR, Chaplain MA, Sherratt JA. 2002. Mathematical modelling of flow through vascular networks: implications for tumour-induced angiogenesis and chemotherapy strategies. Bull. Math. Biol. 64, 673–702. ( 10.1006/bulm.2002.0293) [DOI] [PubMed] [Google Scholar]
- 31.McDougall SR, Anderson AR, Chaplain MA. 2006. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J. Theor. Biol. 241, 564–589. ( 10.1016/j.jtbi.2005.12.022) [DOI] [PubMed] [Google Scholar]
- 32.Machado MJ, Watson MG, Devlin AH, Chaplain MA, McDougall SR, Mitchell CA. 2011. Dynamics of angiogenesis during wound healing: a coupled in vivo and in silico study. Microcirculation 18, 183–197. ( 10.1111/j.1549-8719.2010.00076.x) [DOI] [PubMed] [Google Scholar]
- 33.McDougall SR, Watson MG, Devlin AH, Mitchell CA, Chaplain MA. 2012. A hybrid discrete-continuum mathematical model of pattern prediction in the developing retinal vasculature. Bull. Math. Biol. 74, 2272–2314. ( 10.1007/s11538-012-9754-9) [DOI] [PubMed] [Google Scholar]
- 34.van de Ven AL, Wu M, Lowengrub J, McDougall SR, Chaplain MA, Cristini V, Ferrari M, Frieboes HB. 2012. Integrated intravital microscopy and mathematical modeling to optimize nanotherapeutics delivery to tumors. AIP Adv. 2, 11208 ( 10.1063/1.3699060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watson MG, McDougall SR, Chaplain MAJ, Devlin AH, Mitchell CA. 2012. Dynamics of angiogenesis during murine retinal development: a coupled in vivo and in silico study. J. R. Soc. Interface 9, 2351–2364. ( 10.1098/rsif.2012.0067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu M, Frieboes HB, McDougall SR, Chaplain MAJ, Cristini V, Lowengrub J. 2013. The effect of interstitial pressure on tumor growth: coupling with the blood and lymphatic vascular systems. J. Theor. Biol. 320, 131–151. ( 10.1016/j.jtbi.2012.11.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu M, Frieboes HB, Chaplain MA, McDougall SR, Cristini V, Lowengrub JS. 2014. The effect of interstitial pressure on therapeutic agent transport: coupling with the tumor blood and lymphatic vascular systems. J. Theor. Biol. 355, 194–207. ( 10.1016/j.jtbi.2014.04.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang Q, Sakadzic S, Ruvinskaya L, Devor A, Dale AM, Boas DA. 2008. Oxygen advection and diffusion in a three-dimensional vascular anatomical network. Opt. Exp. 16, 17530–17541. ( 10.1364/OE.16.017530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosso L, Brock CS, Gallo JM, Saleem A, Price PM, Turkheimer FE, Aboagye EO. 2009. A new model for prediction of drug distribution in tumor and normal tissues: pharmacokinetics of temozolomide in glioma patients. Cancer Res. 69, 120–127. ( 10.1158/0008-5472.CAN-08-2356) [DOI] [PubMed] [Google Scholar]
- 40.Moseley CK, Carlin SM, Neelamegam R, Hooker JM. 2012. An efficient and practical radiosynthesis of [11C]temozolomide. Org. Lett. 14, 5872–5875. ( 10.1021/ol302791x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kinahan PE, Fletcher JW. 2010. PET/CT Standardized uptake values (SUVs) in clinical practice and assessing response to therapy. Semin. Ultrasound CT MR 31, 496–505. ( 10.1053/j.sult.2010.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]