

The title structure represents the tetragonal polymorph (the other known structure being monoclinic) and is isotypic with its [MBr4] analogues (M = Co, Ni, Zn).
Keywords: crystal structure, 1-ethyl-3-methyl imidazolium bromide, ionothermal synthesis, tetrabromidocadmate, supramolecular organization
Abstract
Both unique Cd atoms in the tetragonal polymorph of bis(1-ethyl-3-methylimidazolium) tetrabromidocadmate, (C6H11N2)2[CdBr4], occupy special positions (site symmetry -4). The crystal structure consists of isolated tetrahedral [CdBr4]2− anions which are surrounded by 1-ethyl-3-methylimidazolium cations. The methyl and ethyl side chains of the cations show positional disorder in a 0.590 (11):0.410 (11) ratio. In the crystal, (C6H11N2)+ cations display three weak C—H⋯Br hydrogen-bond interactions through the imidazolium ring H atoms with the Br− ligands of the surrounding complex anions. The alkyl groups of the side chains are not involved in hydrogen bonding.
Chemical context
Laboratories around the world have used ionic liquids to prepare many different types of solids, ranging from nanoparticles of different types, to semiconductors, and inorganic and organic solids (Morris, 2009 ▸). In an attempted synthesis of mineral-related arsenates, the ionic liquid 1-ethyl-3-methylimidazolium bromide (eminBr), C6H11BrN2, was tested as a solvent and template. C6H11BrN2 has a wide liquid range (despite being a solid at room temperature, with a melting point of 356 K), low vapour pressure and has been used extensively for ionothermal synthesis because it is a relatively polar solvent.
The title compound, (C6H11N2)2[CdBr4], was obtained under ionothermal conditions using eminBr as the solvate. The SEM–EDS study of the title compound showed small amounts of a cadmium–manganese arsenate in the form of small needle-like crystals up to maximal 15 µm on the top of the plate-like crystals of the title compound (Fig. 1 ▸). This phase is present in very small amounts and therefore could not be identified using powder or single-crystal X-ray diffraction techniques. The powder pattern indicated the tetragonal polymorph of the title compound as the main phase and the monoclinic polymorph (Gou et al., 2016 ▸) as a minority phase.
Figure 1.

Back-scattered scanning electromicrograph of leaf-like (C6H11N2)2[CdBr4]. The small needle-like crystals on the top are from an unidentified Cd/Mn arsenate.
Structural commentary
Emim, C6H11N2 +, cations together with [CdBr4]2− anions as discrete tetrahedra are the main structural building units (Fig. 2 ▸). The imidazolium ring is, as expected, a planar, slightly distorted pentagon. The deviation of the ring atoms from the least-squares plane is smaller than 0.006 (7) Å. The bond lengths of 1.356 (8) and 1.297 (7) Å for the N1—C1 and C1—N2 bonds, respectively, indicate conjugated double-bond character, having one bond slightly longer than the usual C=N double-bond length, 1.27 Å. The N1—C2 and N2—C3 bond lengths [1.360 (7) and 1.359 (8) Å] are shorter than a typical C—N single bond (1.472 ± 6 Å) and close to the shortened (partial double bond) in heterocyclic systems, 1.352 ± 5 Å, while the bond length of 1.373 (9) Å for C2—C3 is slightly longer than a typical C=C double bond of 1.337 ± 6 Å (Macgillavry & Rieck, 1968 ▸). The alkyl groups of the side chains showed strong anisotropic atomic displacements during refinement, suggesting a statistical positional disorder that was taken into account for the final model (Fig. 2 ▸). The carbon atoms C4, C5, C6 and C7 from the disordered alkyl groups of side chains are also planar and the largest deviation from the least-squares plane through the imidazolium ring atoms is 0.163 (16) Å for C7 and −0.949 (19) Å for C6, while C5 and C4 are just −0.013 (1) and 0.039 (1) Å, respectively, out of plane.
Figure 2.

A view of the molecular entities in the structure of (C6H11N2)2[CdBr4]. Displacement ellipsoids are drawn at the 50% probability level. H atoms are represented as small spheres of arbitrary radius. C—H⋯Br hydrogen-bonding interactions are shown with dashed blue lines. Disordered alkyl groups are distinguished by solid and dotted bonds, together with the C and H atoms being shown in different colours. [Symmetry codes: (a) y −  , −x + , −z + ; (b) −y + , x + , −z + ; (c) −x, −y +
, −x + , −z + ; (b) −y + , x + , −z + ; (c) −x, −y +  , z; (d) −y +
, z; (d) −y +  , x + , −z + ; (e) y − , −x + , −z + ; (f) −x, −y +
, x + , −z + ; (e) y − , −x + , −z + ; (f) −x, −y +  , z.]
, z.]
Both unique Cd atoms occupy special positions (on a fourfold rotoinversion axis parallel to the c axis, site symmetry 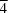 ). Consequently both tetrabromidocadmate anions possess crystallographically imposed symmetry and therefore, each Cd atom bonds to four symmetry-related Br atoms (Fig. 2 ▸). The Cd1—Br1 bond length of 2.5745 (6) Å in the almost regular tetrahedral configuration of the [Cd1Br4]2− anion is slightly shorter than 2.5806 (5) Å for the [Cd2Br4]2− anion. The Br—Cd—Br bond angles are 109.14 (3) and 109.64 (2)° in [Cd1Br4]2− but 107.88 (1) and 112.71 (3)° in the slightly more distorted [Cd2Br4]2– anion. The angular range for both anions is comparable with those reported by Sharma et al. (2006 ▸).
). Consequently both tetrabromidocadmate anions possess crystallographically imposed symmetry and therefore, each Cd atom bonds to four symmetry-related Br atoms (Fig. 2 ▸). The Cd1—Br1 bond length of 2.5745 (6) Å in the almost regular tetrahedral configuration of the [Cd1Br4]2− anion is slightly shorter than 2.5806 (5) Å for the [Cd2Br4]2− anion. The Br—Cd—Br bond angles are 109.14 (3) and 109.64 (2)° in [Cd1Br4]2− but 107.88 (1) and 112.71 (3)° in the slightly more distorted [Cd2Br4]2– anion. The angular range for both anions is comparable with those reported by Sharma et al. (2006 ▸).
Infrared spectroscopy
Fourier-transform infrared (FT–IR) absorption single-crystal infrared spectra were recorded on a Bruker Tensor 27 FT–IR spectrophotometer with a mid-IR glowbar light source and KBr beam splitter, attached to a Hyperion2000 FT–IR microscope with a liquid nitrogen-cooled mid-IR broad band MCT detector. A total of 128 scans were accumulated between 4000 and 550 cm−1 using a circular sample aperture (100 µm diameter) and ATR 15 × objective.
The title compound shows characteristic bands of the imidazolium ring and the alkyl chains (Barbara, 2004 ▸; Nakamoto, 1978 ▸) (Fig. 3 ▸). The bands at 3134 and 3101 cm−1 can be attributed to aromatic C—H stretching (Tait & Osteryoung, 1984 ▸). Their relatively low values confirm the presence of weak hydrogen bonds. A higher wave number would indicate a diminution or absence of hydrogen bonds (Larsen et al., 2000 ▸). The band at 2985 cm−1 can be attributed to aliphatic C—H stretching (Tait & Osteryoung, 1984 ▸); aliphatic C—H bending vibrations [δ(CH2), δ(CH3), δas(CH3)] are located between 1470 and 1380 cm−1 (Katsyuba et al., 2004 ▸) and mostly represented by the band at 1460 cm−1. The band at 1578 cm−1 is assigned to the C=C and C—N stretching vibrations of the imidazolium ring. Bands centred at 1342 and 1162 cm−1, respectively, represent the stretching vibrations between the alkyl chains and N atoms (Katsyuba et al., 2004 ▸). All bands below 850 cm−1 can be attributed to the out-of-plane vibrations of the imidazolium cation (Katsyuba et al., 2004 ▸). The most intense bands are located at 854, 775 and 621 cm−1. Even if there is no water in the structure of (C6H11N2)2[CdBr4], O—H vibrations may still be present because of the hygroscopic character of the ionic liquid.
Figure 3.

FT–IR spectrum of (C6H11N2)2[CdBr4].
Supramolecular features
There are no significant interactions between [Cd2Br4]2– anions, except a short Br1⋯Br1 contact which amounts to 3.764 (2) Å. The crystal packing of the cations and anions in a three-dimensional network is realized through C—H⋯Br interactions (Figs. 2 ▸ and 4 ▸, Table 1 ▸) involving the imidazolium ring H atoms (H1, H2 and H3), but not the H atoms of the alkyl side chains. Larsen et al. (2000 ▸) found that the imidazolium cation is often disordered whereby the disorder can take many different forms. They also have found that positional disorder of the cations in their crystal structures is a direct indicator of packing inefficiency, i.e. packing inefficiency becomes reflected in disorder when cation/anion interactions are reduced essentially to the level of van der Waals or very weak hydrogen-bonding-type forces. The resulting network in the title structure has a channel structure defined by the organization of the imidazolium cations, with the [CdBr4]2– anions residing in the channels (Fig. 5 ▸).
Figure 4.

The packing of the structure of (C6H11N2)2[CdBr4], viewed down the a axis, showing the tetrahedral [CdBr4]2− anions linked to the emim, [C6H11N2]+, cations by hydrogen-bonding interactions. C and N atoms are presented as black and blue spheres, respectively, and H atoms as grey small spheres.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C1—H1⋯Br2i | 0.93 | 2.77 | 3.679 (6) | 167 |
| C2—H2⋯Br1ii | 0.93 | 2.93 | 3.824 (7) | 161 |
| C3—H3⋯Br1 | 0.93 | 2.90 | 3.753 (6) | 154 |
Symmetry codes: (i) 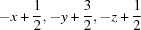 ; (ii)
; (ii) 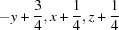 .
.
Figure 5.

The projection of the structure of (C6H11N2)2[CdBr4], viewed down the c axis, normal to the channels formed by the supramolecular organization of the imidazolium cations.
Database survey
Tetragonal (C6H11N2)2[CdBr4] is isotypic with (C6H11N2)2[CoBr4] and (C6H11N2)2[NiBr4] (Hitchcock et al., 1993 ▸), as well as (C6H11N2)2[ZnBr4] (Zhou et al., 2010 ▸; Zhang & Liu, 2012 ▸). However, these three structures do not show any disorder of the imidazolium cations. The crystal structure of the monoclinic (C6H11N2)2[CdBr4] polymorph has also been reported recently (Gou et al., 2016 ▸).
Synthesis and crystallization
A 1 g mixture of CdO, Mn(NO3)2·H2O, As2O5 in the molar ratio 2:2:1 was mixed with 2 g of molten emimBr and placed in a teflon container into a steel autoclave. A heating regime with three steps was chosen: the autoclaves were heated from 293 to 493 K (four h), held at 493 K for 72 h, and finally cooled to room temperature within 99 h. The obtained products were washed with ethanol, filtered and dried in the air at room temperature. The title compound crystallized as leafy-like crystals (yield ca 85%) together with crystals of the monoclinic polymorph (yield ca 10%) and small amounts of a yet unidentified Cd/Mn-arsenate (single-crystal size 10 µm). The crystals of tetragonal (C6H11N2)2[CdBr4]) are no longer than 0.15 mm in length.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. The imidazolium cation was modelled as disordered having approximate twofold rotation symmetry. The two orientations of the disordered cation are related to each other by a 180° rotation around the pseudo-twofold symmetry axis lying in the ring plane, connecting the C1 and bisecting the opposite C2—C3 bonds in the imidazolium ring. This causes a positional disorder of the methyl and ethyl side chains, with a site occupation ratio of 0.590 (11):0.410 (11). All hydrogen atoms attached to C atoms were placed in geometrically calculated positions and refined using a riding model, with C—H = 0.96 Å and U iso(H) = 1.5U eq(C) for methyl H atoms, C—H = 0.97 Å and U iso(H) = 1.2U eq(C) for methylene H atoms, and C—H = 0.93 Å and U iso(H) = 1.2U eq(C) for imidazolium ring H atoms.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | (C6H11N2)2[CdBr4] |
| M r | 654.38 |
| Crystal system, space group | Tetragonal, I41/a |
| Temperature (K) | 100 |
| a, c (Å) | 14.691 (2), 20.075 (4) |
| V (Å3) | 4332.8 (12) |
| Z | 8 |
| Radiation type | Mo Kα |
| μ (mm−1) | 8.39 |
| Crystal size (mm) | 0.15 × 0.02 × 0.01 |
| Data collection | |
| Diffractometer | Stoe StadiVari with pixel array detector |
| Absorption correction | Multi-scan (X-AREA and X-RED32; Stoe, 2013 ▸) |
| T min, T max | 0.366, 0.921 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 34206, 3016, 2046 |
| R int | 0.102 |
| (sin θ/λ)max (Å−1) | 0.694 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.037, 0.074, 0.96 |
| No. of reflections | 3016 |
| No. of parameters | 94 |
| No. of restraints | 17 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.89, −0.86 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989016009919/wm5286sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989016009919/wm5286Isup2.hkl
CCDC reference: 1486568
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors gratefully acknowledge financial support by the Austrian Science Foundation (FWF) (grant No. V203-N19) and the Ministry of Education, Science and Technological Development of the Republic of Serbia (grant No. III45007). The authors are also thankful to Dr Martin Ende for assisting during the low-temperature single-crystal X-ray measurement.
supplementary crystallographic information
Crystal data
| (C6H11N2)2[CdBr4]− | Dx = 2.006 Mg m−3 |
| Mr = 654.38 | Mo Kα radiation, λ = 0.71073 Å |
| Tetragonal, I41/a | Cell parameters from 18051 reflections |
| a = 14.691 (2) Å | θ = 5.6–63.4° |
| c = 20.075 (4) Å | µ = 8.39 mm−1 |
| V = 4332.8 (12) Å3 | T = 100 K |
| Z = 8 | Leaf-like, colourless |
| F(000) = 2480 | 0.15 × 0.02 × 0.01 mm |
Data collection
| Stoe StadiVari with pixel array detector diffractometer | 3016 independent reflections |
| Radiation source: IµS microfocus source | 2046 reflections with I > 2σ(I) |
| Plane graphite monochromator | Rint = 0.102 |
| φ and ω scans | θmax = 29.6°, θmin = 2.8° |
| Absorption correction: multi-scan (X-AREA and X-RED; Stoe, 2013) | h = −17→20 |
| Tmin = 0.366, Tmax = 0.921 | k = −12→20 |
| 34206 measured reflections | l = −27→27 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.037 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.074 | H-atom parameters constrained |
| S = 0.96 | w = 1/[σ2(Fo2) + (0.0344P)2] where P = (Fo2 + 2Fc2)/3 |
| 3016 reflections | (Δ/σ)max = 0.001 |
| 94 parameters | Δρmax = 0.89 e Å−3 |
| 17 restraints | Δρmin = −0.86 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| Cd1 | 0.0000 | 0.2500 | 0.1250 | 0.02409 (13) | |
| Cd2 | 0.0000 | 0.7500 | 0.3750 | 0.02503 (13) | |
| Br1 | 0.00834 (3) | 0.39254 (4) | 0.05065 (3) | 0.04833 (17) | |
| Br2 | 0.12129 (4) | 0.66831 (3) | 0.30377 (3) | 0.03715 (13) | |
| N1 | 0.3058 (3) | 0.5397 (3) | 0.2191 (3) | 0.0513 (12) | |
| N2 | 0.1979 (3) | 0.5755 (3) | 0.1517 (3) | 0.0505 (12) | |
| C1 | 0.2669 (4) | 0.6082 (4) | 0.1840 (3) | 0.0464 (13) | |
| H1 | 0.2864 | 0.6685 | 0.1832 | 0.056* | |
| C2 | 0.2585 (4) | 0.4620 (4) | 0.2067 (3) | 0.0526 (14) | |
| H2 | 0.2708 | 0.4044 | 0.2237 | 0.063* | |
| C3 | 0.1891 (4) | 0.4850 (4) | 0.1641 (3) | 0.0501 (14) | |
| H3 | 0.1447 | 0.4463 | 0.1471 | 0.060* | |
| C4 | 0.38352 (8) | 0.54777 (5) | 0.26324 (6) | 0.073 (2) | |
| H41 | 0.3957 | 0.4900 | 0.2839 | 0.109* | 0.590 (11) |
| H42 | 0.3710 | 0.5923 | 0.2970 | 0.109* | 0.590 (11) |
| H43 | 0.4362 | 0.5664 | 0.2381 | 0.109* | 0.590 (11) |
| H4A | 0.3912 | 0.4890 | 0.2845 | 0.087* | 0.410 (11) |
| H4B | 0.4364 | 0.5574 | 0.2355 | 0.087* | 0.410 (11) |
| C5 | 0.13928 (7) | 0.62737 (8) | 0.11001 (5) | 0.090 (3) | |
| H51 | 0.0914 | 0.5888 | 0.0926 | 0.136* | 0.410 (11) |
| H52 | 0.1721 | 0.6536 | 0.0738 | 0.136* | 0.410 (11) |
| H53 | 0.1110 | 0.6756 | 0.1357 | 0.136* | 0.410 (11) |
| H5A | 0.1300 | 0.5909 | 0.0703 | 0.109* | 0.590 (11) |
| H5B | 0.0821 | 0.6312 | 0.1324 | 0.109* | 0.590 (11) |
| C6 | 0.3888 (12) | 0.6105 (11) | 0.3126 (9) | 0.066 | 0.410 (11) |
| H61 | 0.4458 | 0.6040 | 0.3356 | 0.099* | 0.410 (11) |
| H62 | 0.3396 | 0.6016 | 0.3433 | 0.099* | 0.410 (11) |
| H63 | 0.3849 | 0.6705 | 0.2938 | 0.099* | 0.410 (11) |
| C7 | 0.1525 (10) | 0.7140 (9) | 0.0898 (8) | 0.085 | 0.590 (11) |
| H71 | 0.1101 | 0.7285 | 0.0550 | 0.127* | 0.590 (11) |
| H72 | 0.2135 | 0.7206 | 0.0733 | 0.127* | 0.590 (11) |
| H73 | 0.1434 | 0.7546 | 0.1267 | 0.127* | 0.590 (11) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cd1 | 0.02423 (18) | 0.02423 (18) | 0.0238 (3) | 0.000 | 0.000 | 0.000 |
| Cd2 | 0.02750 (19) | 0.02750 (19) | 0.0201 (3) | 0.000 | 0.000 | 0.000 |
| Br1 | 0.0365 (3) | 0.0530 (3) | 0.0555 (3) | −0.0060 (2) | −0.0071 (2) | 0.0323 (3) |
| Br2 | 0.0464 (3) | 0.0296 (2) | 0.0354 (3) | −0.00354 (19) | 0.0193 (2) | −0.00600 (19) |
| N1 | 0.048 (3) | 0.055 (3) | 0.051 (3) | −0.016 (2) | 0.014 (2) | −0.005 (2) |
| N2 | 0.056 (3) | 0.050 (3) | 0.045 (3) | −0.008 (2) | 0.013 (2) | −0.017 (2) |
| C1 | 0.053 (3) | 0.041 (3) | 0.045 (3) | −0.010 (2) | 0.024 (3) | −0.009 (2) |
| C2 | 0.052 (4) | 0.046 (3) | 0.060 (4) | −0.010 (3) | 0.008 (3) | −0.003 (3) |
| C3 | 0.060 (4) | 0.039 (3) | 0.051 (4) | −0.009 (2) | 0.009 (3) | −0.015 (3) |
| C4 | 0.060 (4) | 0.079 (5) | 0.079 (5) | −0.039 (4) | −0.013 (4) | 0.020 (4) |
| C5 | 0.135 (8) | 0.054 (4) | 0.082 (6) | 0.022 (4) | −0.028 (5) | 0.001 (4) |
| C6 | 0.065 | 0.054 | 0.079 | −0.017 | −0.022 | 0.008 |
| C7 | 0.081 | 0.059 | 0.115 | −0.022 | −0.051 | 0.046 |
Geometric parameters (Å, º)
| Cd1—Br1i | 2.5745 (6) | C4—H42 | 0.9596 (11) |
| Cd1—Br1ii | 2.5745 (6) | C4—H43 | 0.9633 (8) |
| Cd1—Br1 | 2.5745 (6) | C4—H4A | 0.9691 (8) |
| Cd1—Br1iii | 2.5745 (6) | C4—H4B | 0.9666 (8) |
| Cd2—Br2iv | 2.5806 (5) | C5—C7 | 1.350 (11) |
| Cd2—Br2v | 2.5806 (5) | C5—H51 | 0.9681 (8) |
| Cd2—Br2 | 2.5806 (5) | C5—H52 | 0.9543 (10) |
| Cd2—Br2vi | 2.5806 (5) | C5—H53 | 0.9706 (9) |
| N1—C1 | 1.356 (8) | C5—H5A | 0.9700 (8) |
| N1—C2 | 1.360 (7) | C5—H5B | 0.9552 (8) |
| N1—C4 | 1.450 (6) | C6—H42 | 0.488 (16) |
| N2—C1 | 1.297 (7) | C6—H61 | 0.9600 |
| N2—C3 | 1.359 (8) | C6—H62 | 0.9600 |
| N2—C5 | 1.421 (6) | C6—H63 | 0.9600 |
| C1—H1 | 0.9300 | C7—H52 | 0.987 (16) |
| C2—C3 | 1.373 (9) | C7—H53 | 1.241 (15) |
| C2—H2 | 0.9300 | C7—H71 | 0.9600 |
| C3—H3 | 0.9300 | C7—H72 | 0.9600 |
| C4—C6 | 1.355 (18) | C7—H73 | 0.9600 |
| C4—H41 | 0.9610 (8) | ||
| Br1i—Cd1—Br1ii | 109.14 (3) | H4A—C4—H4B | 106.88 (8) |
| Br1i—Cd1—Br1 | 109.638 (17) | C7—C5—N2 | 126.5 (5) |
| Br1ii—Cd1—Br1 | 109.638 (17) | C7—C5—H51 | 123.3 (5) |
| Br1i—Cd1—Br1iii | 109.638 (17) | N2—C5—H51 | 109.8 (2) |
| Br1ii—Cd1—Br1iii | 109.638 (17) | C7—C5—H52 | 46.9 (8) |
| Br1—Cd1—Br1iii | 109.14 (3) | N2—C5—H52 | 111.0 (2) |
| Br2iv—Cd2—Br2v | 112.71 (3) | H51—C5—H52 | 109.27 (10) |
| Br2iv—Cd2—Br2 | 107.878 (14) | C7—C5—H53 | 62.1 (8) |
| Br2v—Cd2—Br2 | 107.878 (14) | N2—C5—H53 | 109.7 (2) |
| Br2iv—Cd2—Br2vi | 107.878 (14) | H51—C5—H53 | 107.93 (9) |
| Br2v—Cd2—Br2vi | 107.878 (14) | H52—C5—H53 | 109.06 (12) |
| Br2—Cd2—Br2vi | 112.71 (3) | C7—C5—H5A | 107.1 (7) |
| C1—N1—C2 | 108.3 (5) | N2—C5—H5A | 105.9 (2) |
| C1—N1—C4 | 126.1 (4) | H51—C5—H5A | 43.85 (3) |
| C2—N1—C4 | 125.7 (5) | H52—C5—H5A | 70.62 (6) |
| C1—N2—C3 | 110.2 (6) | H53—C5—H5A | 141.27 (15) |
| C1—N2—C5 | 124.7 (5) | C7—C5—H5B | 102.3 (8) |
| C3—N2—C5 | 125.1 (5) | N2—C5—H5B | 106.7 (2) |
| N2—C1—N1 | 108.3 (5) | H51—C5—H5B | 64.21 (5) |
| N2—C1—H1 | 125.8 | H52—C5—H5B | 141.30 (18) |
| N1—C1—H1 | 125.8 | H53—C5—H5B | 47.87 (4) |
| N1—C2—C3 | 106.6 (6) | H5A—C5—H5B | 107.23 (8) |
| N1—C2—H2 | 126.7 | C4—C6—H42 | 29.2 (17) |
| C3—C2—H2 | 126.7 | C4—C6—H61 | 109.5 |
| N2—C3—C2 | 106.6 (5) | H42—C6—H61 | 136.1 |
| N2—C3—H3 | 126.7 | C4—C6—H62 | 109.5 |
| C2—C3—H3 | 126.7 | H42—C6—H62 | 86.2 |
| C6—C4—N1 | 123.2 (8) | H61—C6—H62 | 109.5 |
| C6—C4—H41 | 105.9 (7) | C4—C6—H63 | 109.5 |
| N1—C4—H41 | 109.7 (2) | H42—C6—H63 | 102.6 |
| C6—C4—H42 | 14.4 (8) | H61—C6—H63 | 109.5 |
| N1—C4—H42 | 109.7 (2) | H62—C6—H63 | 109.5 |
| H41—C4—H42 | 109.44 (11) | C5—C7—H52 | 44.9 (4) |
| C6—C4—H43 | 98.2 (8) | C5—C7—H53 | 43.7 (4) |
| N1—C4—H43 | 109.6 (2) | H52—C7—H53 | 88.7 (8) |
| H41—C4—H43 | 109.13 (9) | C5—C7—H71 | 109.5 |
| H42—C4—H43 | 109.23 (11) | H52—C7—H71 | 98.7 |
| C6—C4—H4A | 106.1 (7) | H53—C7—H71 | 108.7 |
| N1—C4—H4A | 106.8 (2) | C5—C7—H72 | 109.5 |
| H41—C4—H4A | 3.993 (3) | H52—C7—H72 | 72.9 |
| H42—C4—H4A | 108.60 (11) | H53—C7—H72 | 139.5 |
| H43—C4—H4A | 112.92 (9) | H71—C7—H72 | 109.5 |
| C6—C4—H4B | 106.0 (8) | C5—C7—H73 | 109.5 |
| N1—C4—H4B | 106.9 (2) | H52—C7—H73 | 148.2 |
| H41—C4—H4B | 103.25 (8) | H53—C7—H73 | 68.9 |
| H42—C4—H4B | 117.45 (12) | H71—C7—H73 | 109.5 |
| H43—C4—H4B | 8.460 (7) | H72—C7—H73 | 109.5 |
| C3—N2—C1—N1 | −0.2 (6) | C5—N2—C3—C2 | −179.7 (4) |
| C5—N2—C1—N1 | 179.1 (4) | N1—C2—C3—N2 | 0.8 (6) |
| C2—N1—C1—N2 | 0.7 (6) | C1—N1—C4—C6 | 51.2 (10) |
| C4—N1—C1—N2 | −178.3 (4) | C2—N1—C4—C6 | −127.6 (10) |
| C1—N1—C2—C3 | −0.9 (6) | C1—N2—C5—C7 | 10.0 (12) |
| C4—N1—C2—C3 | 178.0 (4) | C3—N2—C5—C7 | −170.9 (11) |
| C1—N2—C3—C2 | −0.4 (6) |
Symmetry codes: (i) y−1/4, −x+1/4, −z+1/4; (ii) −y+1/4, x+1/4, −z+1/4; (iii) −x, −y+1/2, z; (iv) −y+3/4, x+3/4, −z+3/4; (v) y−3/4, −x+3/4, −z+3/4; (vi) −x, −y+3/2, z.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C1—H1···Br2vii | 0.93 | 2.77 | 3.679 (6) | 167 |
| C2—H2···Br1viii | 0.93 | 2.93 | 3.824 (7) | 161 |
| C3—H3···Br1 | 0.93 | 2.90 | 3.753 (6) | 154 |
Symmetry codes: (vii) −x+1/2, −y+3/2, −z+1/2; (viii) −y+3/4, x+1/4, z+1/4.
References
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst. 32, 115–119.
- Barbara, S. (2004). In Infrared Spectroscopy: Fundamentals and Applications. New York: Wiley.
- Dowty, E. (2000). ATOMS for Windows. Shape Software, Kingsport, Tennessee, USA.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Gou, L., Liu, D., Zhao, K. & Yang, M. Y. (2016). Z. Kristallogr. New Cryst. Struct. 231, 271–272.
- Hitchcock, P. B., Seddon, K. R. & Welton, T. (1993). J. Chem. Soc. Dalton Trans. pp. 2639–2643.
- Katsyuba, S. A., Dyson, P. J., Vandyukova, E. E., Chernova, A. V. & Vidiš, A. (2004). Helv. Chim. Acta, 87, 2556–2565.
- Larsen, A. S., Holbrey, J. D., Tham, F. S. & Reed, C. A. (2000). J. Am. Chem. Soc. 122, 7264–7272.
- Macgillavry, C. H. & Rieck, G. D. (1968). Editors. International Tables for X-ray crystallography, Vol. III, Physical and Chemical Tables, General editor: K. Lonsdale, pp. 273–285. Birmingham, England: IUCr, The Kynoch Press.
- Morris, R. E. (2009). Chem. Commun. pp. 2990–2998. [DOI] [PubMed]
- Nakamoto, K. (1978). In Infrared and Raman Spectra of Inorganic and Coordination Compounds. New York: Wiley.
- Sharma, R. P., Sharma, R., Bala, R., Salas, J. M. & Quiros, M. (2006). J. Mol. Struct. 794, 341–347.
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Stoe (2013). X-AREA and X-RED32. Stoe & Cie, Darmstadt, Germany.
- Tait, S. & Osteryoung, R. A. (1984). Inorg. Chem. 23, 4352–4360.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Zhang, X. C. & Liu, B. (2012). Bull. Chem. Soc. Ethiop. 26, 407–414.
- Zhou, W. W., Zhao, W., Song, M. J., Bao, X. & Wang, F. W. (2010). Z. Kristallogr. New Cryst. Struct. 225, 801–802.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989016009919/wm5286sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989016009919/wm5286Isup2.hkl
CCDC reference: 1486568
Additional supporting information: crystallographic information; 3D view; checkCIF report