

Abstract
Background
BRAF mutations act as an oncogenic driver via the mitogen-activated protein kinase (MAPK) pathway in non-small cell lung cancer (NSCLC). BRAF inhibition has demonstrated antitumor activity in patients with BRAF V600E (Val600Glu)–mutant NSCLC. Dual MAPK pathway inhibition with BRAF and MEK inhibitors in BRAF V600E–mutant NSCLC may improve efficacy over BRAF-inhibitor monotherapy based on observations in BRAF V600–mutant melanoma.
Methods
In this phase 2, multicenter, nonrandomized, open-label study of patients with pretreated metastatic BRAF V600E–mutant NSCLC, antitumor activity and safety of oral dabrafenib (150 mg twice daily) plus oral trametinib (2 mg once daily) were evaluated. Adult patients (≥ 18 years) with documented progression following at least one prior platinum-based chemotherapy and no more than three prior systemic anticancer therapies were enrolled. Patients with prior BRAF or MEK inhibitor treatment were ineligible. Patients with brain metastases were permitted to enroll only if the lesions were asymptomatic, untreated (or stable > 3 weeks after local therapy if treated), and measured < 1 cm. The primary endpoint was investigator-assessed overall response, which was assessed by intention-to-treat in the protocol-defined population (≥ second-line); safety was also assessed in this population. The study is ongoing but no longer recruiting patients. This trial is registered with ClinicalTrials.gov, number NCT01336634.
Findings
Fifty-seven patients previously treated with systemic chemotherapy for metastatic BRAF V600E–mutant NSCLC were enrolled. The investigator-assessed overall response was 63·2% (36 of 57; 95% CI 49·3–75·6). Serious adverse events were reported in 32 (56%) of 57 patients and included pyrexia (16%; 9 of 57), anemia (5%; 3 of 57), confusional state (4%; 2 of 57), decreased appetite (4%; 2 of 57), hemoptysis (4%; 2 of 57), hypercalcemia (4%; 2 of 57), nausea (4%; 2 of 57), and cutaneous squamous cell carcinoma (4%; 2 of 57). Common grade 3/4 AEs included neutropenia (9%; 5 of 57), hyponatremia (7%; 4 of 57), and anemia (5%; 3 of 57).
Interpretation
Dabrafenib plus trametinib represents a new targeted therapy with robust antitumor activity and a manageable safety profile in patients with BRAF V600E–mutant NSCLC.
Funding
GlaxoSmithKline.
Introduction
Non-small cell lung cancer (NSCLC), which constitutes approximately 85% of all lung cancers, remains a leading cause of cancer-related deaths globally.1 Recently, progress has been made in characterizing oncogenic driver mutations that contribute to the molecular pathogenesis of lung cancers, including activating mutations in EGFR and ALK rearrangements. This has led to rapid development of targeted therapeutics and a more personalized approach to NSCLC treatment.2,3
Activating mutations in the BRAF gene, generally mutually exclusive from EGFR mutations or ALK rearrangements, act as an alternative oncogenic driver in NSCLC. The most common of these mutations, BRAF V600E (Val600Glu), is observed in 1% to 2% of lung adenocarcinomas.4–7 Although the prognostic implications of BRAF V600E mutation are unclear, several studies have associated BRAF V600E with poor outcomes and lower response rates to platinum-based chemotherapy in patients with NSCLC compared with patients with NSCLC without BRAF mutations.8,9 Furthermore, in a recent analysis, one-half of 106 BRAF-mutant patients received only best supportive care in a real-world second-line treatment setting.5 Therefore, more effective targeted therapies are needed for these patients with limited therapeutic options.
The current phase 2 study reports on the second (cohort B) of three sequentially enrolled cohorts. In Cohort A, the antitumor activity of a selective BRAF inhibitor, dabrafenib, was evaluated exclusively in patients with BRAF V600E–mutant NSCLC.10 Dabrafenib demonstrated clinical activity with an overall confirmed response of 33% (95% CI 23–45) and median progression-free survival of 5·5 months in patients with previously treated NSCLC.
In a preclinical study, dabrafenib plus trametinib synergistically inhibited cell growth in a BRAF V600E–mutant lung carcinoma cell line (MV522; data on file). Clinically, BRAF plus MEK inhibition has demonstrated an increased overall response, progression-free survival, and overall survival (OS) compared with BRAF-inhibitor monotherapy in patients with BRAF V600–mutant metastatic melanoma.11–13
Cohort B, discussed herein, represents the first examination, to our knowledge, of the clinical activity and safety of the combination BRAF inhibitor dabrafenib plus the MEK inhibitor trametinib in patients with previously treated metastatic BRAF V600E–mutant NSCLC (dabrafenib 150 mg twice daily plus trametinib 2 mg once daily, doses successfully used to treat melanoma11). An additional cohort of this study (cohort C) has enrolled treatment-naive patients with BRAF V600E–mutant NSCLC treated with dabrafenib plus trametinib, and the patients are now being followed up for response and progression-free survival.
Research in context
Evidence before the study
Delineation of the contributions of oncogenic driver mutations to the molecular pathogenesis of non-small lung cancer (NSCLC) has led to direct therapeutic targeting of aberrant signaling pathways and a more personalized approach to treatment. This has led to the approval of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, anaplastic lymphoma kinase (ALK) inhibitors, and ROS1 inhibitors for the treatment of patients with activating mutations in EGFR, ALK rearrangement, and ROS1 rearrangements respectively.
Mutations in the BRAF gene, which encodes for a serine/threonine kinase at the top of the mitogen-activated protein kinase pathway, are generally mutually exclusive from EGFR mutations and ALK rearrangement and acts as an oncogenic driver in NSCLC. The most common BRAF mutation, BRAF V600E (Val600Glu), has been associated with more aggressive tumors which provides a strong rationale for targeting of this pathway in patients with BRAF V600E–mutant NSCLC. Indeed, the use of BRAF inhibitors has demonstrated clinical activity, including a 33% overall response and 5·5-month median progression-free survival, in patients treated with dabrafenib monotherapy in cohort A of the current phase 2 trial and 42% overall response and 7·3-month median progression-free survival in patients treated with vemurafenib as part of a prospective basket study. The combination of BRAF inhibitors with MEK inhibition has demonstrated superior efficacy compared with BRAF inhibitor monotherapy in patients with BRAF-mutant metastatic melanoma, potentially contributing to sustained pathway inhibition and delay or prevention of resistance. Further, addition of the MEK inhibitor trametinib to dabrafenib led to synergistic antitumor activity in a BRAF–mutant human lung cancer cell line, suggesting that combination BRAF and MEK inhibition could potentially provide increased benefit compared with BRAF inhibitor monotherapy in patients with BRAF V600E–mutant NSCLC.
We searched PubMed for studies of combined BRAF and MEK inhibition for the treatment of patients with BRAF V600E–mutant NSCLC, without date limitations. We used the search terms “dabrafenib AND trametinib” and “vemurafenib AND cobimetinib” both with “non-small cell lung cancer” OR “NSCLC.” No clinical studies were identified utilizing combined BRAF and MEK inhibition in patients with BRAF V600E-mutant NSCLC.
Added value of this study
We found that combination dabrafenib plus trametinib had substantial antitumor activity (proportion with overall response 63%) in patients with BRAF V600E–mutant NSCLC. Further, responses were durable, with a median progression-free survival of 9.7 months, and the safety profile was tolerable.
Implications of all the available evidence
To our knowledge, this is the first trial to assess combination BRAF and MEK inhibition in patients with BRAF V600E–mutant NSCLC. Notably, the overall response and median progression-free survival observed with combination dabrafenib plus trametinib were higher when compared indirectly with dabrafenib monotherapy, utilized in cohort A of this study. Although cross-trial comparisons should be undertaken with caution, the clinical activity observed in the current study appears similar to that observed for other targeted therapies, including EGFR-tyrosine kinase inhibitors and ALK inhibitors in selected patient populations. Moreover, the rarity of this patient population renders the potential conduct of a randomized trial is extremely challenging. Therefore, the results presented herein have a strong potential to change the management of patients with BRAF V600E–mutant NSCLC, a population with an unmet medical need.
Methods
Study design and participants
This study was part of a continuing phase 2, multicenter, non-randomized, open-label study. Adult patients (≥18 years) with stage IV BRAF V600E–mutant NSCLC, documented tumor progression following at least one platinum-based chemotherapy regimen (based on medical history), and no more than three prior systemic treatments for metastatic NSCLC were enrolled. BRAF V600E mutational status was determined based on local testing in Clinical Laboratory Improvement Amendments–approved (or its equivalent outside the United States) laboratories. Patients had to have measurable disease according to Response Evaluation Criteria In Solid Tumors (RECIST) v1·1, an Eastern Cooperative Oncology Group (ECOG) performance status ≤2 and an estimated life expectancy ≥ 3 months. Patients with prior BRAF- or MEK-inhibitor therapy were ineligible. Patients with brain metastases were ineligible unless they were asymptomatic, were untreated, and had measured <1 cm, or, if treated, were clinically and radiographically stable three weeks after local therapy. For full inclusion/exclusion criteria see the Supplementary Appendix p. 4.
This study was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was approved by the institutional review board at each institution. All patients provided written informed consent.
Procedures
Patients were treated with oral dabrafenib (150 mg twice daily) plus oral trametinib (2 mg once daily) in continuous 21-day cycles until disease progression, unacceptable adverse events (AEs), consent withdrawal, or death. Patients with progressive disease were permitted to continue treatment if they had a confirmed response according to RECIST v1·1 or had stable disease (SD) lasting for ≥12 weeks during study treatment, and the investigator believed they were clinically benefiting from therapy. Dose modifications or interruptions were used to manage intolerable grade ≥2 AEs. Dose modification guidelines are included in the Supplementary Appendix p. 4.
Radiological disease assessments by computed tomography using RECIST v1·1 were performed at baseline, at week 6, every 6 weeks until week 36, and then every 12 weeks, and the responses were confirmed by repeat assessment 4 to 7 weeks after initial response. RECIST scans were reviewed by an independent review committee (IRC). All patients who discontinued study medication were followed up for subsequent treatment(s) and survival every 12 weeks, until death or study completion. Patients were evaluated for safety at least once every 3 weeks. AEs, laboratory values (hematology and clinical chemistry), and vital signs were graded according to the Common Terminology Criteria for Adverse Events v4·0. Cutaneous squamous carcinoma was required to be reported as at least grade 3. The protocol also required certain events (grade 2 or worse pyrexia with symptoms, LVEF decrease, and others reported as protocol specified SAEs regardless if they meet the standard definition of SAE). Complete details on study assessment are included in the Supplementary Appendix p. 4. The cut-off date for safety and efficacy data was 7 October 2015.
Outcomes
The primary endpoint was investigator-assessed overall response, defined as the percentage of patients with a confirmed complete response (CR) or partial response (PR) according to RECIST v1·1. Secondary endpoints (defined in the Supplementary Appendix p. 4) included progression-free survival based on investigator-assessed disease response, duration of response (DOR) based on investigator-assessed confirmed response, OS, safety and tolerability, and pharmacokinetic assessment.
Statistical analysis
A 2-stage Green-Dahlberg14 design was used to monitor patients for clinical response during the study to enable early stopping for futility if sufficient clinical activity was not demonstrated. An interim analysis was planned after 20 patients had at least two post-baseline scans or withdrew from the study before response was assessed. The null hypothesis was that the overall response was not clinically meaningful (≤30%), and the alternative hypothesis was that the overall response was ≥55% for dabrafenib plus trametinib in second- to fourth-line patients with BRAF V600E–mutant NSCLC. The trial could be terminated for futility after enrollment of 20 patients if a confirmed response was not observed in ≥6 of 20 patients after stage I and ≥18 of 40 patients after both stages. Enrollment of additional patients was permitted per protocol in order to ensure an adequate number of evaluable patients with central confirmation of response assessments and BRAF mutation status. The statistical analyses were based on the planned enrollment of 40 patients and corresponded to a type 1 error of 0·032 and power of 92·2%. These statistical assumptions were not changed by the enrollment of additional patients. Efficacy and safety were evaluated by intention-to-treat in the protocol-defined population (≥ second-line). Patients defined as not assessable either had no postbaseline CT scan or discontinued before 12 weeks without documented progression. For overall response we used the Clopper-Pearson method to calculate 95% CIs. The DOR, progression-free survival, and OS were estimated by medians calculated using the Kaplan-Meier method with corresponding 2-sided CIs calculated using the Brookmeyer-Crowley method.15 A sensitivity analysis for these endpoints was done using an independent review committee and the same methods. An additional sensitivity analysis was done for progression-free survival with the inclusion of clinical progression as an event and the same methods. SAS version 9·4 was used for statistical analyses. This trial is registered at ClinicalTrials.gov, number NCT01336634.
Role of the funding source
This study was sponsored by GlaxoSmithKline; dabrafenib and trametinib are assets of Novartis AG as of 2 March 2015. The study was designed by the academic authors in conjunction with representatives of the sponsor. Data were collected by the sponsor and analyzed in collaboration with the authors. AD’A, PZ, BM, and AU had full access to the raw data. The sponsor was involved in writing of the report. The first and last authors wrote the initial draft; all authors contributed to subsequent drafts and made the decision to submit for publication. The corresponding author had full access to all of the data and the final responsibility to submit for publication. The authors affirm accuracy of the data and fidelity of the study to the protocol. Editorial support was provided by ArticulateScience and funded by Novartis Pharmaceuticals.
Results
Between December 20, 2013 to January 14, 2015, 59 patients were enrolled from 30 centers in nine countries across North America, Europe, and Asia. Because this study enrolled patients who had BRAF V600E mutation based on testing in local laboratories (mentioned in the methods), the exact number of patients with NSCLC screened at the participating institutions for BRAF V600E mutation was not assessed.
Fifty-seven patients previously treated for metastatic disease (one prior regimen [n=38]; two to three prior regimens [n=19]) received dabrafenib plus trametinib and were included in efficacy and safety analyses. Two treatment-naive patients were enrolled due to protocol deviation and are reported separately.
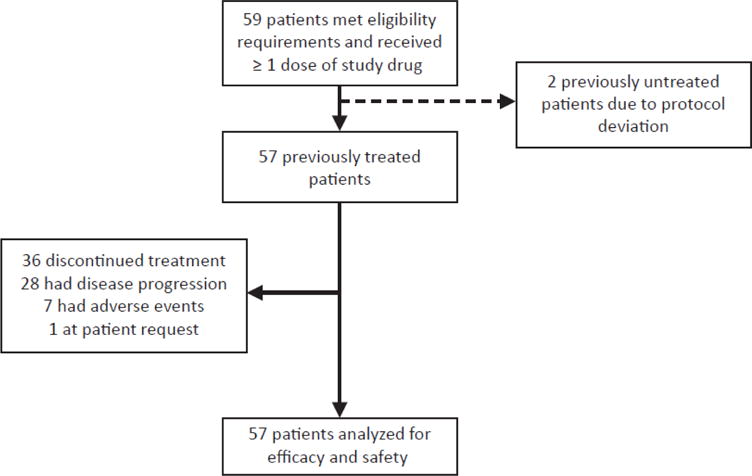
Baseline characteristics for the 57 patients receiving dabrafenib plus trametinib as second-line or later treatment are presented in table 1. A single patient (2%) of 57 had a non-measurable brain metastasis at enrollment and the lesion was asymptomatic. At the cut-off date of 7 October 2015, 21 (37%) of 57 patients remained on treatment. Twenty-eight (49%) of 57 patients discontinued due to disease progression, 7 (12%) of 57 discontinued due to AEs, and 1 (2%) of 57 discontinued at her request (figure 1). Chemotherapy was the most commonly used post-progression therapy (26%; 15 of 57).
Table 1.
Baseline demographics and disease history in ≥ second-line patients
| N=57 | |
|---|---|
|
| |
| Age, median (range), years | 64 (41–88) |
|
| |
| Sex, n (%) | |
| Male | 29 (51) |
| Female | 28 (49) |
|
| |
| Race, n (%) | |
| White | 49 (86) |
| Black | 2 (4) |
| Asian | 4 (7) |
| Mixed | 1 (2) |
| Missing | 1 (2) |
|
| |
| ECOG performance status, n (%) | |
| 0 | 17 (30) |
| 1 | 35 (61) |
| 2 | 5 (9) |
|
| |
| Histology at initial diagnosis, n (%) | |
| Adenocarcinoma* | 56 (98) |
| Large cell | 1 (2) |
|
| |
| History of tobacco use, n (%) | |
| Never smoker | 16 (28) |
| Current smoker | 6 (11) |
| Former smoker | 35 (61) |
|
| |
| Smoking history, n (%) | n = 41 |
| ≤30 pack-years | 22 (54) |
| >30 pack-years | 19 (46) |
|
| |
| Number of prior systemic regimens for metastatic disease, n (%) | |
| 1 | 38 (67) |
| ≥2 | 19 (33) |
Includes 1 patient with adenosquamous carcinoma—predominately adenocarcinoma histology—and 2 patients with lepidic predominant or invasive mucinous adenocarcinoma (formerly known as bronchioalveolar carcinoma). All histology was determined by local pathological report.
Figure 1. CONSORT diagram.
With a median follow-up of 11·6 months (IQR 8·8–15·2), the investigator-assessed confirmed overall response was 63·2% (36 of 57; 95% CI 49·3–75·6), including 2 (3·5%) of 57 patients with CR and 34 (59·6%) of 57 with PR. The investigator-assessed disease control rate (DCR; CR + PR + SD) was 78·9% (45 of 57; 95% CI 66·1–88·6; table 2; figure 2). IRC-assessed overall response and DCR were similar: 63·2% (36 of 57 with PR; 95% CI 49·3–75·6) and 75·4% (43 of 57; 95% CI 62·2–85·9), respectively (table 2). Two patients with confirmed CR by investigator assessment did not have confirmed CR by independent review. The patient with non-measurable brain metastasis at baseline had a non-CR/non-PD response in the brain lesion. No patients had documented new brain metastases as part of their progression.
Table 2.
Antitumor activity in ≥ second-line patients
| Investigator Assessment (N=57) |
Independent Assessment (N=57) |
|
|---|---|---|
|
| ||
| Best response, n (%) | ||
| Complete response (CR) | 2 (3·5) | 0 |
| Partial response (PR) | 34 (59·6) | 36 (63·2) |
| Stable disease (SD) | 9 (15·8) | 4 (7·0) |
| Progressive disease (PD) | 7 (12·3) | 8 (14·0) |
| Non-CR/non-PD | 0 | 3 (5·3) |
| Not evaluable | 5 (8·8) | 6 (10·5) |
|
| ||
| Overall response (CR + PR), n (%) [95% CI] | 36 (63·2) [49·3–75·6] | 36 (63·2) [49·3–75·6] |
|
| ||
| Disease control rate (CR + PR + SD), n (%) [95% CI] | 45 (78·9) [66·1–88·6] | 43 (75·4) [62·2–85·9] |
|
| ||
| Progression-free survival, median (95% CI), months | 9·7 (6·9–19·6) | 8·6 (5·2–19·1) |
|
| ||
| Duration of response, median (95% CI), months | 9·0 (6·9–18·3) | 9·0 (5·8–17·6) |
Figure 2. Tumor responses to dabrafenib + trametinib in BRAF V600E–mutant non-small cell lung cancer.
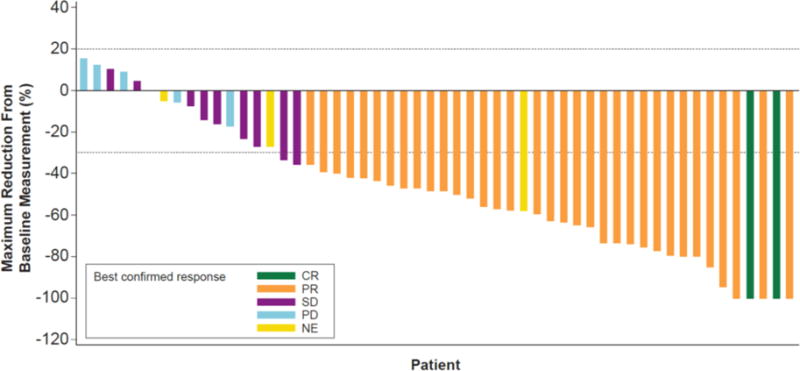
Maximum reduction from baseline sum of lesion diameters by best investigator-assessed confirmed response in ≥ second-line patients (n=57). CR=complete response. NE=not evaluable. PD=progressive disease. SD=stable disease.
The median investigator-assessed progression-free survival was 9·7 months (95% CI 6·9–19·6); 32 of 57 patients (56%) had progressed or died at the time of the analysis (table 2; figure 3), and the 6-month progression-free survival rate was 65% (37 of 57; 95% CI 51–76). The investigator-assessed median DOR was 9·0 months (95% CI 6·9–18·3; table 2; figure 4; appendix p. 6). Notably, at data cut-off, 50% (18 of 36) of confirmed responses were ongoing. Based on IRC assessment, median progression-free survival was 8·6 months (95% CI 5·2–19·1; table 2; appendix p. 5) and median DOR was 9·0 months (95% CI 5·8–17·6; table 2; appendix p. 6). Progression-free survival was also similar when clinical progression was included as an event with a median of 9·7 months (95% CI, 5·7–13·6). Median time to first response was 6 weeks (IQR 6–10 weeks). At data cut-off, 23 (40%) of 57 patients had died. The median OS was immature. Forty-seven of 57 patients were alive at 6 months (6-month OS rate, 82%).
Figure 3. Kaplan-Meier curve of investigator-assessed progression-free survival in ≥ second-line patients.
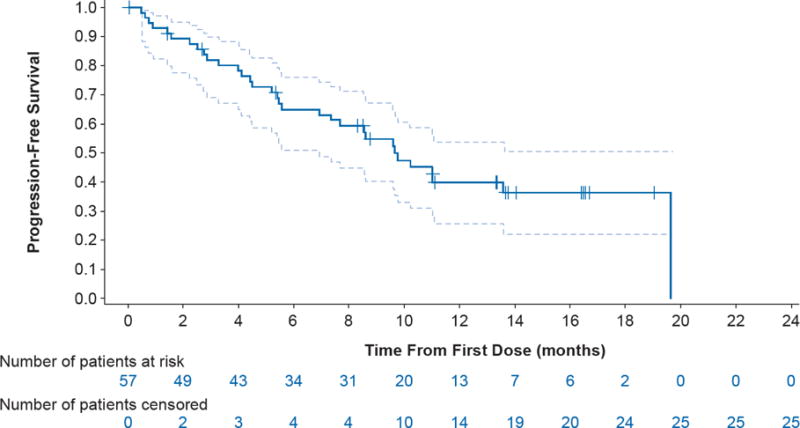
Dashed lines represent 95% CI. Number of patients censored represent cumulative totals.
Figure 4. Duration of response (n=36) based on investigator assessment in ≥ second-line patients.
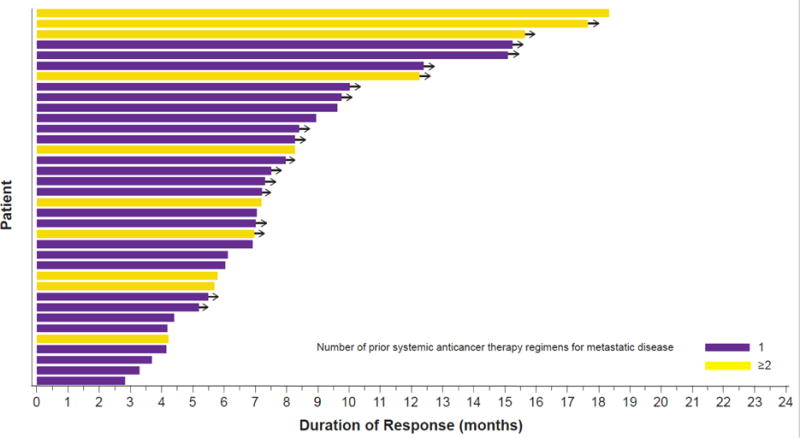
Duration of response by number of prior systemic anticancer therapies for metastatic disease. Yellow bars indicate patients with one prior regimen. Purple bars indicate patients with at least two prior regimens. Arrows denote ongoing response at the time of data cut-off. Note that half (n=18) of the patients had an ongoing response at the time of data cut-off.
Two patients without prior systemic treatment for metastatic disease were enrolled due to protocol deviation. One patient achieved a CR and remained progression-free at data cut-off (received study drug for >16 months). The other patient achieved a PR and progressed after 9·7 months of treatment.
The median duration of treatment for both dabrafenib and trametinib was 10·6 months (IQR 4·2–12·2 months); 30% (17 of 57) of patients received >12 months of treatment (appendix p. 7). AEs led to permanent discontinuation, dose interruption/delay, and dose reduction in 7 (12%) of 57, 35 (61%) of 57 and 20 (35%) of 57 patients, respectively. Thirty-three (58%) and 43 (75%) of 57 patients received ≥80% of the planned dose of dabrafenib and trametinib, respectively. Nearly all patients experienced at least one AE (56 of 57; 98%), and nearly half (28 of 57; 49%) experienced at least one grade 3/4 event. Common AEs of any grade (≥30%) included pyrexia (46%; 26 of 57), nausea (40%; 23 of 57), vomiting (35%; 20 of 57), diarrhea (33%; 19 of 57), asthenia (32%; 18 of 57), and decreased appetite (30%; 17 of 57) (table 3). Common grade 3/4 AEs (≥5%) were neutropenia (9%; 5 of 57), hyponatremia (7%; 4 of 57), and anemia (5%; 3 of 57). The most common serious AEs were pyrexia (16%; 9 of 57), anemia (5%; 3 of 57), confusional state (4%; 2 of 57), decreased appetite (4%; 2 of 57), hemoptysis (4%; 2 of 57), hypercalcemia (4%; 2 of 57), nausea (4%; 2 of 57), and squamous cell carcinoma (SCC) of skin (4%; 2 of 57). Two (4%) of 57 patients died from fatal serious AEs considered unrelated to study treatment by the investigators. One patient with a history of mitral valve replacement and receiving anti-coagulative therapy experienced an episode of ventricular fibrillation, was hospitalized, and developed retroperitoneal hemorrhage, and 1 patient with a history of cranial artery aneurysm experienced a subarachnoid hemorrhage.
Table 3.
Common (>10% all grades or any ≥ grade 3) adverse events in ≥ second-line patients (n=57)
| Common Adverse Events, n (%) | Grade 1–2 | Grade 3 | Grade 4 | Grade 5 |
|---|---|---|---|---|
| Pyrexia | 25 (44) | 1 (2) | 0 | 0 |
| Nausea | 23 (40) | 0 | 0 | 0 |
| Vomiting | 20 (35) | 0 | 0 | 0 |
| Diarrhea | 18 (32) | 1 (2) | 0 | 0 |
| Decreased appetite | 17 (30) | 0 | 0 | 0 |
| Asthenia | 16 (28) | 2 (4) | 0 | 0 |
| Dry skin | 14 (25) | 1 (2) | 0 | 0 |
| Peripheral edema | 13 (23) | 0 | 0 | 0 |
| Chills | 12 (21) | 1 (2) | 0 | 0 |
| Cough | 12 (21) | 0 | 0 | 0 |
| Rash | 11 (19) | 1 (2) | 0 | 0 |
| Arthralgia | 11 (19) | 0 | 0 | 0 |
| Constipation | 10 (18) | 0 | 0 | 0 |
| Fatigue | 9 (16) | 1 (2) | 0 | 0 |
| Blood alkaline phosphatase increased | 9 (16) | 0 | 0 | 0 |
| Dyspnea | 8 (14) | 2 (4) | 0 | 0 |
| Pruritis | 8 (14) | 1 (2) | 0 | 0 |
| Dizziness | 8 (14) | 0 | 0 | 0 |
| Anemia | 7 (12) | 2 (4) | 1 (2) | 0 |
| Weight decreased | 7 (12) | 1 (2) | 0 | 0 |
| Upper abdominal pain | 7 (12) | 0 | 0 | 0 |
| Hypotension | 7 (12) | 0 | 0 | 0 |
| Neutropenia | 6 (11) | 5 (9) | 0 | 0 |
| Chest pain | 6 (11) | 0 | 0 | 0 |
| Dysgeusia | 6 (11) | 0 | 0 | 0 |
| Headache | 6 (11) | 0 | 0 | 0 |
| Muscle spasms | 6 (11) | 0 | 0 | 0 |
| Myalgia | 6 (11) | 0 | 0 | 0 |
| Productive cough | 6 (11) | 0 | 0 | 0 |
| Vertigo | 6 (11) | 0 | 0 | 0 |
| Hyperkeratosis | 5 (9) | 1 (2) | 0 | 0 |
| Weight increased | 5 (9) | 1 (2) | 0 | 0 |
| Back pain | 4 (7) | 0 | 1 (2) | 0 |
| Hemoptysis | 4 (7) | 1 (2) | 0 | 0 |
| Aspartate aminotransferase increased | 3 (5) | 1 (2) | 0 | 0 |
| Blood creatinine increased | 3 (5) | 1 (2) | 0 | 0 |
| Hypophosphatemia | 3 (5) | 1 (2) | 0 | 0 |
| Thrombocytopenia | 3 (5) | 1 (2) | 0 | 0 |
| Hyponatremia | 2 (4) | 3 (5) | 1 (2) | 0 |
| Leukopenia | 2 (4) | 2 (4) | 0 | 0 |
| Alanine aminotransferase increased | 2 (4) | 1 (2) | 0 | 0 |
| Dehydration | 1 (2) | 2 (4) | 0 | 0 |
| Hypertension | 1 (2) | 2 (4) | 0 | 0 |
| Amylase increased | 1 (2) | 1 (2) | 0 | 0 |
| Basal cell carcinoma | 1 (2) | 1 (2) | 0 | 0 |
| Erythema nodosum | 1 (2) | 1 (2) | 0 | 0 |
| Hematuria | 1 (2) | 1 (2) | 0 | 0 |
| Peripheral neuropathy | 1 (2) | 1 (2) | 0 | 0 |
| Pain | 1 (2) | 1 (2) | 0 | 0 |
| Pulmonary embolism | 1 (2) | 1 (2) | 0 | 0 |
| Tubulointerstitial nephritis | 1 (2) | 1 (2) | 0 | 0 |
| Visual impairment | 1 (2) | 1 (2) | 0 | 0 |
| Gamma-glutamyltransferase increased | 0 | 1 (2) | 1 (2) | 0 |
| Hypercalcemia | 0 | 2 (4) | 0 | 0 |
| Respiratory distress | 0 | 1 (2) | 0 | 1 (2) |
| Squamous cell carcinoma of skin | 0 | 2 (4) | 0 | 0 |
| C-reactive protein increased | 0 | 1 (2) | 0 | 0 |
| Cholecystitis acute | 0 | 1 (2) | 0 | 0 |
| Coronary artery stenosis | 0 | 1 (2) | 0 | 0 |
| Febrile neutropenia | 0 | 1 (2) | 0 | 0 |
| Hepatocellular carcinoma | 0 | 1 (2) | 0 | 0 |
| Hip fracture | 0 | 1 (2) | 0 | 0 |
| Incisional hernia | 0 | 1 (2) | 0 | 0 |
| Intestinal obstruction | 0 | 1 (2) | 0 | 0 |
| Legionella infection | 0 | 0 | 1 (2) | 0 |
| Lung neoplasm malignanta | 0 | 1 (2) | 0 | 0 |
| Neoplasm progressionb | 0 | 0 | 0 | 1 (2) |
| Pancytopenia | 0 | 1 (2) | 0 | 0 |
| Pleural effusion | 0 | 1 (2) | 0 | 0 |
| Pyelonephritis | 0 | 1 (2) | 0 | 0 |
| Quadriplegia | 0 | 1 (2) | 0 | 0 |
| Renal failure | 0 | 1 (2) | 0 | 0 |
| Retroperitoneal hemorrhage | 0 | 0 | 0 | 1 (2) |
| Subarachnoid hemorrhage | 0 | 0 | 0 | 1 (2) |
| Ventricular fibrillation | 0 | 0 | 1 (2) | 0 |
One patient had a lung metastasis that did not respond to therapy, was biopsied, and was determined to have a KRAS mutation. This was reported as an AE by the investigator.
One patient was determined by the investigator to have progression that was more severe than typical progression. According to study protocol this can be documented as an AE.
Patients with multiple events in the same category are counted only once in that category. Patients with events in more than 1 category are counted once in each of those categories.
In a post-hoc analysis of response by prior lines of systemic therapy, investigator-assessed response was observed in 26 (68·4%) of 38 patients (95% CI, 51·3–82·5) with 1 prior line vs 10 (52·6%) of 19 patients (95% CI, 28·9–75·6) with 2 to 3 prior lines. Post-hoc analysis of investigator-assessed response by smoking history demonstrated an overall response of 62·5% (10 of 16; 95% CI, 35·4–84·8) in patients with no prior smoking history, 68.6% (24 of 35; 95% CI, 50·7–83·1) in former smokers, and 33% (2 of 6; 95% CI, 4·3–77·7) in current smokers. Of the five patients who had an ECOG performance status of 2 at baseline, 4 (80%) of 5 had a best response of PR and one had a best response of SD. One of the five patients remains on treatment with a PFS of ≈ 25 months; the remaining four patients discontinued due to progressive disease with PFS ranging from 3·5–13·0 months. One of the patients had a fatal SAE of respiratory distress that was associated with the disease under study and considered unrelated to study treatment.
Discussion
The results of this trial, the first evaluation, to our knowledge, of combination BRAF and MEK inhibition in NSCLC, demonstrate the substantial clinical activity of dabrafenib plus trametinib therapy in patients with previously treated BRAF V600E–mutant metastatic NSCLC. The protocol-defined primary objective was met with a confirmed overall response of 63·2%. Responses were durable, with half of confirmed responses ongoing at data cut-off.
The results are particularly noteworthy because of the limited data and clear unmet need of effective targeted therapy for patients with BRAF-mutant NSCLC. Among patients with NSCLC with BRAF mutations, half of the mutations are BRAF V600E, which activate BRAF in its monomeric state and are sensitive to BRAF mutant specific inhibitors. Other BRAF mutations are activating due to either constitutive or RAS-dependent dimer formation or are not activating of BRAF and their relevance to the disease is undefined. Thus BRAF inhibitors are not effective in these patients.8,16
An analysis of patients with BRAF V600–mutant NSCLC who received standard-of-care chemotherapy as second-line treatment in a real-world setting demonstrated poor outcomes: 9% overall response (n=59) and 3·1-month median progression-free survival (n=71).5 BRAF-inhibitor monotherapy has clinical activity in BRAF V600E–mutant NSCLC with the 33% confirmed overall response and 5·5-month progression-free survival observed with dabrafenib monotherapy,10 the 42% overall response (however unconfirmed by repeat imaging) and 7·3-month progression-free survival observed with vemurafenib monotherapy,17 or the 5-month progression-free survival reported in a retrospective analysis of patients treated with dabrafenib, vemurafenib, or sorafenib.18 However, the overall response and progression-free survival measures need further improvement. The increased efficacy of the combination in NSCLC vs BRAF inhibitor alone (dabrafenib) is similar to observations in BRAF V600–mutant melanoma, for which the greater overall response (69% vs 53%; p=0·0014) and longer progression-free survival (11·0 vs 8·8 months; p=0·0004) significantly favored the dabrafenib plus trametinib arm (vs dabrafenib plus placebo).13 Acquired resistance does occur in patients treated with melanoma and appears to primarily be linked to reactivation of the mitogen activated protein kinase pathway (upregulation of receptor tyrosine kinases, NRAS mutations, activating mutations in MEK1/2, and alterations at the level of BRAF) and adaptations in the PI3K-PTEN-AKT pathway.19 It is currently unknown whether these mechanisms of acquired resistance occur in patients with BRAF V600E–mutant NSCLC treated with BRAF and/or MEK inhibitors and further investigation is warranted and ongoing. In the current study, optional biopsies could be collected at week 6 and at the end of study per protocol. To date, only two post-progression biopsies have been acquired across all cohorts of this trial and further sample acquisition and follow-up is ongoing.
Targeted therapeutics have proven to be a successful strategy in NSCLC harboring oncogenic driver mutations. For example, EGFR tyrosine kinase inhibitors induce high overall response (≈70%) and durable responses (9·7- to 11·0-month median PFS) in treatment-naive patients with activating mutations in EGFR.2,20,21 Similarly, in previously treated patients with ALK rearrangements, ALK inhibitors demonstrate marked efficacy with 65% overall response and median progression-free survival of 7·7 months.3 Further, inhibition of ROS1 with crizotinib has demonstrated clinical activity in patients with ROS-1 rearranged NSCLC with an overall response of 72% and a PFS of 19.2 months.22 Additional personalized therapeutic strategies targeting oncogenic drivers could also be on the horizon including the use of crizotinib in patients with MET exon 14 skipping mutation.23–25 As demonstrated here, dabrafenib plus trametinib appears to have similar clinical activity in a selected patient population. Although the prognostic implications of BRAF V600E mutations in NSCLC remain unclear,5,8,9,26 data from the current study indicate a 6-month OS rate of 82% with a more mature OS planned in a future analysis. Long-term OS data will help determine whether targeted agents can change the natural history of NSCLC, similar to what has been observed in melanoma.27 As it has for other oncogenic drivers, this could change NSCLC treatment by placing increased emphasis on determination of BRAF mutation status at diagnosis to help inform personalized therapeutic decisions. Additionally given the rarity of the mutation rate5 it is infeasible to conduct a randomized clinical trial. Given the mutation rate of 1% for BRAF V600E, approximately 6000 patients may have been screened at the local sites to identify the 59 patients enrolled in this cohort. The potential for advances in liquid biopsy methods to detect oncogenic driver mutations has been tested in NSCLC for more common mutations and in melanoma for BRAF V600E mutations and could provide for enhanced screening capabilities once the technique has been optimized and validated.28 Further, the future inclusion of rare mutations such as BRAF V600E in umbrella trials in patients with NSCLC could help to enhance enrollment and allow for larger trials. Upcoming discussions regarding revisions to the European and United States guidelines for molecular testing in NSCLC could potentially recommend assessment of EGFR, ALK, ROS-1, and BRAF V600E for all patients with lung adenocarcinoma.
Studies suggest that the ALK inhibitor crizotinib may demonstrate better efficacy in treatment-naive vs previously treated patients with ALK rearrangements yielding a modestly higher overall response (74% vs 65%) and a longer median progression-free survival (10·9 vs 7·7 months).3,29 Cohort C of this trial, had completed enrollment at the time of preparation of the current manuscript and patients are being followed for response and progression-free survival. This data will help confirm whether clinical activity of the combination is increased in earlier lines of therapy in BRAF V600E–mutant NSCLC.
Immune checkpoint inhibitors, nivolumab and pembrolizumab, represent a new second-line treatment option for patients with NSCLC: nivolumab without biomarker selection and pembrolizumab in PDL-1–positive patients. Patients with previously treated metastatic primarily non-squamous NSCLC who were treated with nivolumab or pembrolizumab had a median OS of 10·4 to 12·7 months.30–32 However, a response was observed in only a limited subset of patients (≈20%), and no data currently exist regarding the efficacy of these checkpoint inhibitors in patients with BRAF V600–mutant NSCLC. Given the high overall response observed with dabrafenib plus trametinib in patients with previously treated BRAF V600E–mutant NSCLC and the success of other targeted therapies in early lines of treatment, future research will determine the position of dabrafenib plus trametinib as an early treatment option compared to platinum-based chemotherapy or immunotherapy options.
The safety profile demonstrated in the current study was manageable (12% of patients discontinued due to AEs) and similar to the safety profile for dabrafenib plus trametinib in patients with unresectable or metastatic melanoma (all-grade AEs, 95%; grade 3/4 AEs, 35%).13,33 Dabrafenib plus trametinib provides a clear clinical benefit in patients with BRAF V600E–mutant NSCLC, but grade 3/4 AEs were observed in nearly half of patients in this study. Notably, experience with the combination in patients with BRAF V600–mutant melanoma has demonstrated that most grade 3/4 AEs can be managed through dose modification and provides a framework for physicians to manage patients and mitigate risk of unacceptable toxicity. Similar to experience in melanoma, pyrexia was observed in 36% of patients with dabrafenib monotherapy and in 46% of patients with dabrafenib plus trametinib.10 Analysis of pyrexia in melanoma showed a correlation between dabrafenib and hydroxydabrafenib concentrations and pyrexia, although the etiology of the observed increase in incidence with dabrafenib plus trametinib remains unclear.34 Conversely, cutaneous SCC was observed in 10 of 84 (12%) patients with dabrafenib monotherapy and in only 2 of 57 (4%) patients treated with dabrafenib plus trametinib. In previous melanoma studies, combination of MEK and BRAF inhibitors substantially reduced the risk of cutaneous SCC compared with BRAF-inhibitor monotherapy (1%–3% vs 9%–18%).12,13,33 This supports the hypothesis that the addition of MEK-inhibitor to BRAF-inhibitor therapy can block paradoxical activation of MAPK signaling in BRAF wild-type cells and reduce the incidence of cutaneous SCC.11,35 Hemoptysis was reported as an SAE in 2 (4%) of 57 patients in this study, which is consistent with other studies in patients with previously treated lung cancer; 5% to 7% hemoptysis or pulmonary hemorrhage in patients treated with docetaxel alone.36 Hemorrhage has also been observed in patients with metastatic melanoma treated with combination dabrafenib plus trametinib, but the rate of grade 3/4 events was modest and similar between patients treated with the combination and patients receiving dabrafenib monotherapy (dabrafenib plus trametinib [2·0%] vs dabrafenib monotherapy [1.9%]).37 Four (80%) of 5 patients with ECOG performance status of 2 at baseline had a confirmed response and none discontinued due to AEs. Although the sample is small, this suggests that combination dabrafenib plus trametinib could be safely administered to patients with baseline ECOG performance status 2.
Overall, dabrafenib plus trametinib is a promising new therapy for patients with BRAF V600E–mutant NSCLC, with high overall response, prolonged DOR, and manageable toxicity. This is the first report, to our knowledge, demonstrating a highly effective targeted therapy combination strategy in this patient population with few treatment options that can achieve >50% overall response and median progression-free survival of greater than 9 months. The emergence of optimized sequencing strategies and targeted agents including dabrafenib plus trametinib will continue to broaden personalized therapy in NSCLC and improve patient outcomes.
Supplementary Material
Acknowledgments
Supported by GlaxoSmithKline. Dabrafenib and trametinib are assets of Novartis AG as of 2 March 2015.
We thank the patients and their families for participating in this study and Michael Demars, PhD (ArticulateScience, LLC, Hamilton, NJ), for assistance in the preparation of the manuscript.
Footnotes
Contributions
Literature search: PZ
Conception and design: DP, BB, JM, JRR, AD’A, BM, BEJ
Collection and assembly of data: DP, BB, P-JS, EQ, FB, TMK, JM, SN, AU, PZ, BM, BEJ
Data analysis and interpretation: DP, BB, HJMG, P-JS, CSB, FB, TMK, JM, SN, JRR, AU, AD’A, PZ, BM, BEJ
Figure and table development: JRR, AD’A
Manuscript writing: DP, BB, HJMG, P-JS, EQ, CSB, FB, JM, SN, JRR, AU, AD’A, PZ, BM, BEJ
Final approval of manuscript: DP, BB, HJMG, P-JS, EQ, CSB, FB, TMK, JM, SN, JRR, AU, AD’A, PZ, BM, BEJ
Provision of study material or patients: P-JS, CSB
Declaration of interests
DP acts as an advisor for AstraZeneca, Boehringer, Bristol-Myers Squibb, Clovis, Lilly, Merck Sharp & Dohme, Novartis, Pfizer, Roche, and Sanofi and has received research funding from Novartis unrelated to the current trial. BB’s institution received a grant from Novartis for this study. HJMG’s institution has received payments from Eli Lilly, Merck Sharp & Dohme, and Roche. P-JS has been involved in clinical trials for GlaxoSmithKline and Novartis. EQ has received personal fees from AbbVie, Bristol-Myers Squibb, Clovis, Lilly, Pfizer, and Roche, has received grants from Amgen, Boehringer, Bristol-Myers Squibb, Lilly, Merck Sharp & Dohme, Mundipharma, and Roche, and has received non-financial support from Boehringer. CSB’s institution received a grant for this study from Novartis and CSB has received personal fees from Novartis. FB has received personal fees from Novartis. SN has received personal fees from Boehringer, Bristol-Myers Squibb, Eli Lilly, Merck Sharp & Dohme, and Roche. AU, PZ, AD’A, and BM are currently employees of Novartis. AD’A and BM were employees of GlaxoSmithKline during a portion of the study. AD’A and BM own stock in GlaxoSmithKline and Novartis and BM owns stock in Incyte and AstraZeneca. BEJ has received personal fees from Amgen, AstraZeneca, Boehringer, Chugai Pharmaceuticals, Clovis, Genentech, KEW Group, Merck, and Novartis, and shares of post-market revenue for an EGFR genotyping patent. The other authors declare no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT) Lancet. 2016;387:1415–26. doi: 10.1016/S0140-6736(16)00004-0. [DOI] [PubMed] [Google Scholar]
- 6.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046–51. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardarella S, Ogino A, Nishino M, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res. 2013;19:4532–40. doi: 10.1158/1078-0432.CCR-13-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchetti A, Felicioni L, Malatesta S, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29:3574–9. doi: 10.1200/JCO.2011.35.9638. [DOI] [PubMed] [Google Scholar]
- 10.Planchard D, Min Kim T, Mazieres J, Quiox E, Riely G, Barlesi F. Dabrafenib in BRAF V600E–mutant advanced non-small cell lung cancer: an open-label, single arm, multicenter, phase 2 trial. Lancet Oncol. 2016 doi: 10.1016/S1470-2045(16)00077-2. published online Apr 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 13.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 14.Green SJ, Dahlberg S. Planned versus attained design in phase II clinical trials. Stat Med. 1992;11:853–62. doi: 10.1002/sim.4780110703. [DOI] [PubMed] [Google Scholar]
- 15.Brookmeyer R, Crowley J. A confidence interval for the mean survival time. Biometrics. 1982;38:29–41. [Google Scholar]
- 16.Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–83. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol. 2015;10:1451–7. doi: 10.1097/JTO.0000000000000625. [DOI] [PubMed] [Google Scholar]
- 19.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–22. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 20.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 22.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–71. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol. 2016;34:721–30. doi: 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 24.Jorge SE, Schulman S, Freed JA, et al. Responses to the multitargeted MET/ALK/ROS1 inhibitor crizotinib and co-occurring mutations in lung adenocarcinomas with MET amplification or MET exon 14 skipping mutation. Lung Cancer. 2015;90:369–74. doi: 10.1016/j.lungcan.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–9. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen-Ngoc T, Bouchaab H, Adjei AA, Peters S. BRAF alterations as therapeutic targets in non-amall-cell lung cancer. J Thorac Oncol. 2015;10:1396–403. doi: 10.1097/JTO.0000000000000644. [DOI] [PubMed] [Google Scholar]
- 27.Jakob JA, Bassett RL, Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–23. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698–705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–77. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 30.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2015;387:1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 31.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 32.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 34.Menzies AM, Ashworth MT, Swann S, et al. Characteristics of pyrexia in BRAFV600E/K metastatic melanoma patients treated with combined dabrafenib and trametinib in a phase I/II clinical trial. Ann Oncol. 2015;26:415–21. doi: 10.1093/annonc/mdu529. [DOI] [PubMed] [Google Scholar]
- 35.Lito P, Pratilas CA, Joseph EW, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–82. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garon EB, Ciuleanu TE, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384:665–73. doi: 10.1016/S0140-6736(14)60845-X. [DOI] [PubMed] [Google Scholar]
- 37.Mekinist (trametinib) [package insert] Novartis Pharmaceuticals Corporation East Hanover; New Jersey 07936: 2015. Accessed 28 April 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.