

Abstract
Dilated cardiomyopathy (DCM), the third most common cause of heart failure, is often associated with arrhythmias and sudden cardiac death if not controlled. The majority of DCM is of unknown etiology. Protein sialylation is altered in human DCM, with responsible mechanisms not yet described. Here we sought to investigate the impact of clinically relevant changes in sialylation on cardiac function using a novel model for altered glycoprotein sialylation that leads to DCM and to chronic stress-induced heart failure (HF), deletion of the sialyltransferase, ST3Gal4. We previously reported that 12- to 20-week-old ST3Gal4−/− mice showed aberrant cardiac voltage-gated ion channel sialylation and gating that contribute to a pro-arrhythmogenic phenotype. Here, echocardiography supported by histology revealed modest dilated and thinner-walled left ventricles without increased fibrosis in ST3Gal4−/− mice starting at 1 year of age. Cardiac calcineurin expression in younger (16–20 weeks old) ST3Gal4−/− hearts was significantly reduced compared to WT. Transverse aortic constriction (TAC) was used as a chronic stressor on the younger mice to determine whether the ability to compensate against a pathologic insult is compromised in the ST3Gal4−/− heart, as suggested by previous reports describing the functional implications of reduced cardiac calcineurin levels. TAC’d ST3Gal4−/− mice presented with significantly reduced systolic function and ventricular dilation that deteriorated into congestive HF within 6 weeks post-surgery, while constricted WT hearts remained well-adapted throughout (ejection fraction, ST3Gal4−/− = 34 ± 5.2 %; WT = 53.8 ± 7.4 %; p < 0.05). Thus, a novel, sialo-dependent model for DCM/HF is described in which clinically relevant reduced sialylation results in increased arrhythmogenicity and reduced cardiac calcineurin levels that precede cardiomyopathy and TAC-induced HF, suggesting a causal link among aberrant sialylation, chronic arrhythmia, reduced calcineurin levels, DCM in the absence of a pathologic stimulus, and stress-induced HF.
Keywords: Dilated cardiomyopathy, Sialic acid, Echocardiography, Congenital disorders of glycosylation, Arrhythmic heart failure, Animal models, Calcineurin
Introduction
Heart failure (HF) affects more than 23 million people worldwide [12], with dilated cardiomyopathy (DCM) the third most common cause of HF. The European Society of Cardiology defines DCM as, “left ventricular dilation and systolic dysfunction in the absence of abnormal loading conditions or coronary artery disease sufficient to cause global systolic impairment; the right ventricle can also be involved” [21].
Potential causes for DCM include gene mutation, infection (viral myocarditis, Chagas disease), alcoholism, cardiotoxic agents, endocrine dysfunction and nutritional deficiencies. Approximately 25 % of DCM is reported as familial due to different gene mutations. However, approximately 70 % of DCM is considered idiopathic, of unknown etiology and mechanism [22].
Glycosylation is a common co/posttranslational modification in mammalian cells [49] that is essential for protein function such as promoting proper folding, targeting proteins to the correct cellular compartments, contributing to ligand recognition processes, as well as modulating receptor and ion channel functions [17, 49]. Sialic acid, typically the terminal carbohydrate residue attached to N-and O-glycans, is a unique monosaccharide that carries a negative charge at physiologic pH and participates in a variety of cellular functions [17]. Previously, we and others showed that sialic acid, likely through an electrostatic effect, modulates voltage-sensitive ion channel gating through direct, isoform-specific mechanisms [3, 4, 17, 34, 35, 45, 53, 56, 60].
Several DCM risk factors, including smoking, alcohol abuse, obesity, and diabetes, cause changes in protein glycosylation, and data show glycosylation is altered in DCM [2, 33, 36, 64, 69]. Indeed, patients with congenital disorders of glycosylation (CDG) often present with idiopathic cardiac deficits including cardiomyopathy with related arrhythmias [1, 14, 23, 27, 38]. In nearly all of these instances, protein sialylation is altered. Relevant examples in human DCM include measurement of reduced mRNA levels of the CMP sialic acid transporter and a putative sialyltransferase. However, mechanisms by which clinically relevant altered sialylation contribute to DCM have not been described, and there is currently no model available to investigate these mechanisms. Thus, here, gene deletion of the β-galactoside α2,3-sialyltransferase, ST3Gal4, was used as a model for altered cardiac co/posttranslational protein sialylation that leads to a modest DCM in older animals and to chronic stress-induced HF in younger animals. The ST3Gal4 gene product transfers sialic acids preferentially to N-glycoproteins [57], and ST3Gal4 is uniformly expressed in tissues and organs throughout all developmental stages, including development of the mouse heart [20, 37]. Deletion of ST3Gal4 should produce a modest reduction in protein sialylation consistent with that observed in human DCM, thus rendering the ST3Gal4−/− mouse as a model for clinically relevant reductions in protein sialylation. The ST3Gal4−/− model used here was previously characterized by us and others, with data showing that gene deletion of ST3Gal4 (ST3Gal4−/−) leads to reduced von Willebrand factor and platelet counts, prolonged bleeding time, reduced L-selectin binding and L-selectin-dependent leukocyte rolling with increased terminal galactose residue exposure, as well as reductions in cardiomyocyte voltage-gated ion channel sialylation that directly alter channel gating [18–20, 55].
Here using the ST3Gal4−/− mouse model, we provide a potential mechanistic link among aberrant sialylation, altered electrical signaling, calcineurin levels, and cardiomyopathy. Previously, in younger ST3Gal4−/− mice, we showed that reductions in voltage-gated Na+ channel (NaV) and K+ channel (KV) sialylation resulted in direct alterations in NaV and KV gating, reduced cellular/ventricular refractory periods, action potential (AP)/ECG QT segment prolongations, and arrhythmogenic activities [18, 19]. Here, we also found in similarly aged ST3Gal4−/− hearts, reduced expression of the Ca2+-dependent pro-hypertrophic protein, calcineurin. We go on to show in these same ST3Gal4−/− mice, that the partial reduction of α2,3-sialylation is sufficient to induce DCM in older mice and chronic stress-induced HF in younger mice. This initial characterization of the ST3Gal4−/− strain supports its viability as a model for arrhythmic DCM/HF, showing that clinically relevant reduced sialylation causes aberrant cardiac electrical signaling and reduced cardiac calcineurin expression that precede ventricular dilation and stress-induced HF, suggesting a causal link among aberrant sialylation, chronic arrhythmia, reduced calcineurin expression, DCM and stress-induced HF.
Methods
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985) and was approved by the University of South Florida Animal Care and Use Committee.
Generation of ST3Gal4 null mice
ST3Gal4−/− mice were generated by breeding within the ST3Gal4−/+ strain as previously described [18–20]. The ST3Gal4−/+ mouse strain was generously provided by Dr. Jamey Marth. Genotyping was performed using standard methods as previously described [19]. Male ST3Gal4−/−and WT littermates that have the same genetic background, were used throughout this study.
Transthoracic echocardiography
Transthoracic echocardiography was performed on Vevo770 system (VisualSonics) using 30 MHz–25 MHz transducers. Cardiac function was measured using previously published procedures [51, 62]. Briefly, mice were lightly anesthetized with 1–1.5 % isoflurane and maintained 37 °C body temperature. 2-D mode in parasternal long axis and the parasternal short axis at the mid-papillary muscle level were imaged. From this parasternal short axis view, the 2-D guided M-mode across the anterior and posterior wall were recorded. Left ventricular (LV) anterior wall thickness at diastole and systole (LVAW;d and LVAW;s), LV internal dimension at diastole and systole (LVID;d and LVID;s), LV posterior wall thickness at diastole and systole (LVPW;d and LVPW;s), LVMass, and other parameters were measured blindly from three consecutive cardiac cycles using the leading edge method according to the guidelines of the American Society of Echocardiography. Ejection fraction (EF %), fractional shortening (FS %), LV Mass Index, cardiac output (CO), stroke volume (SV), end-diastolic volume (EDV), end-systolic volume (ESV) were calculated. The following formulas were used to calculate EF % and FS %: ; [62]. Successful transverse aortic constriction (TAC) was confirmed in 2-D mode of aortic view first. Trans-constriction peak velocity (V, mm/s) was then assessed using pulsed-wave Doppler with the angle adjusted in line with the direction of the blood flow (generally 0°–20°) (Fig. 4b–c). The aortic peak pressure gradient of aortic constriction (AoPg, mmHg) was calculated using the modified Bernoulli equation: AoPg = 4 V2 [62].
Fig. 4.
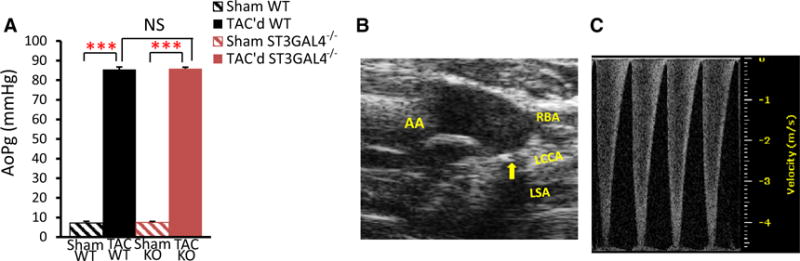
TAC imposed equivalent pressure-load on WT and ST3Gal4−/− mice. a Equivalent pressure-overload was imposed on 16- to 20-week-old ST3Gal4−/− (85.9 ± 1 mmHg; n = 12) and WT littermates (85.5 ± 1 mmHg; n = 7), p > 0.05. b B-mode of the aortic view was imaged 7 days after TAC. Arrow TAC; AA ascending aorta, Rt. RBA right brachiocephalic artery, Lf. LCCA left common carotid artery, LSA. c Pulsed-wave Doppler was used to assess blood flow velocity across the TAC for calculation of AoPg 1 week after surgery
Anatomical and histological analyses
Mice were heparinized and terminally anesthetized in 5 % isoflurane. The whole heart, left ventricle, right ventricle, left atria, right atria, lungs, liver and tibia were removed, rinsed, blotted dry and weighed for anatomical studies. For histological analysis, hearts were arrested in KCl solution first and fixed with 4 % buffered paraformaldehyde, followed by dehydration and paraffin embedding. Tissue sections with 5 μm thickness were prepared and stained with haematoxylin and eosin (H&E) (Thermo Scientific, MA) for morphological analysis or Masson’s trichrome (Sigma-Aldrich, MO) for collagen deposition.
Protein extraction and western blotting
Murine LV tissues were homogenized in radioimmuno-precipitation (RIPA) lysis buffer. Lysate protein concentrations were determined using the bicinchoninic acid (BCA) method (Thermo scientific, MA) and detected using a spectrophotometer. Protein extracts were reduced and denatured in Laemmli sample buffer, followed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore, MA). Transferred blots were blocked with 5 % non-fat dry milk for non-specific bindings, probed with unconjugated primary antibodies against calcineurin A subunit (CnA, BD Pharmingen, CA) [58] and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Santa Cruz, CA), followed by the incubation with HRP conjugated secondary antibodies. Blots were then visualized using enhanced chemiluminescence (ECL, Thermo Scientific, MA) under ChemiDoc XRS+ (Bio-Rad, CA). Signal density measurements were performed using Image Lab software version 5.2.1 (Bio-Rad, CA). GAPDH was used as the protein loading control.
Minimally invasive transverse aortic constriction (TAC)
16- to 20-week-old male WT and ST3Gal4−/− mice were randomly assigned into sham and TAC’d groups. Baseline echocardiography was recorded 2 days before the surgery. Minimally invasive transverse aortic constriction (TAC) was performed as described previously to ensure minimal to no blood loss during surgery, and was successful in preventing any mortality among all cohorts throughout the 6-week experiment [32]. Briefly, mice were anesthetized with isoflurane (2 %)/O2, intubated with 20G intravenous catheter, mechanically ventilated and maintained 37 °C body temperature. A partial left thoracotomy was created followed by thymus retraction. Ligation against a 26-G blunt needle at the level of the aortic arch between the origin of the right innominate artery and left common carotid artery was performed, followed by prompt needle removal. Sham-operated groups received surgery consisting of aortic exposure, but without aortic ligation. AoPg was recorded 1 week after TAC. Data include only TAC’d hearts with AoPg = 85 ± 5 mmHg to ensure consistent pressure overload between groups. Mice were observed for 6 weeks after TAC surgery, followed by euthanasia and tissue collection used for anatomical and histological measurements.
Lectin staining
To determine the relative levels of sialylation, the Erythrina cristagalli lectin (ECL, Vector Lab, CA) was used to probe murine left ventricular sections. ECL binds preferentially to terminal unsubstituted galactose β1,4 N-acetylglucosamine (Galβ1,4GlcNAc) [68]. Biotinylated ECL with an avidin–biotin–fluorophore system was applied on paraformaldehyde-fixed paraffin-embedded WT and ST3Gal4−/− cardiac tissues, as described previously [7, 8, 28, 47, 68]. Specifically, tissue sections were blocked for endogenous biotin (Streptavidin/Biotin blocking kit, Vector Lab, CA) and non-specific binding (Vector Lab, CA) before incubation with 1:200 biotinylated ECL for 2 h at room temperature. Tissue sections were then incubated 2 h at room temperature with 1:1000 Dylight™ 488 conjugated NeutrAvidin (Thermo Scientific, MA) followed by 4,6-diamidino-2-phenylindole (DAPI, Vector Lab, CA) counterstain. No positive staining was observed for the negative controls in which biotinylated lectin, Dylight 488 conjugated neutravidin or both were omitted. Multiple sections from each of two to three hearts per condition were studied, with the data showing staining for a given condition among slices consistent with that shown in Fig. 5.
Fig. 5.

ECL Lectin staining of cardiac tissue. ECL lectin staining (green) of representative slices from WT and ST3Gal4−/− sham and TAC operated hearts, 6 weeks post-surgery, with DAPI (blue) nuclear counterstain overlay. Magnification, ×40. Scale bars—20 μm. a WT Sham, b ST3Gal4−/− sham, c WT TAC’d, d ST3Gal4−/− TAC’d. Both sets of WT samples had minimal ECL staining, while ST3Gal4−/− hearts had similarly increased ECL binding (compared to WT sham and TAC’d heart slices). Significant ECL staining was observed on cardiomyocyte sarcolemma and capillary endothelium, with little to no indication of intracellular staining, consistent with previous reports [6, 47]
Statistical analysis
Data were presented as mean ± SEM. Parametric data were compared using two-tailed Student’s t test, one-way ANOVA with TukeyHSD (homogeneity of variances), Games-Howell (heterogeneity of variances) post hoc tests, or two-way ANOVA using Bonferroni correction. Nonparametric data were compared using Mann–Whitney U test or Kruskal–Wallis one-way analysis of variance with Dunn’s post hoc test using Bonferroni correction. Only p values <0.05 were considered significant. All statistical analyses were performed using SPSS software (IBM, NY).
Results
ST3Gal4−/− mice demonstrate progressively dilated and thinner-walled left ventricles
In agreement with previous studies, young ST3Gal4−/−mice appear normal, fully fertile, active, and are produced in Mendelian ratios among heterozygous mating [20, 55]. By 18 months of age, there was no significant difference in body weight between male ST3Gal4−/− and WT littermates, weighing 49.3 ± 1.7 g (n = 9) and 47.3 ± 1.4 g (n = 8), respectively.
In order to determine how sialylation modulates cardiac function in vivo, echocardiography was performed on the same set of animals at ages 6, 9, 12, and 18 months (Fig. 1; Table 1). ST3Gal4−/− mice (n = 9) demonstrated progressively enlarged EDV (Fig. 1a), dilated LVID;d (Fig. 1b), and thinner LVPW;d (Fig. 1c) compared to WT littermates (n = 8), with each parameter reaching significant differences between WT and ST3Gal4−/− mice by 18 months of age (p < 0.05). While small reductions in ejection fraction were observed in ST3Gal4−/− mice starting at 9 months of age, ST3Gal4−/− systolic function was mostly preserved over the 18-month experiment. No significant differences were measured in LVMass between ST3Gal4−/− and WT littermates (Fig. 1d). No septal defect or great vessel abnormality was observed in either ST3Gal4−/− or WT mice. The gradual cardiac dilation with mostly preserved systolic function suggested that an early-stage dilated cardiomyopathy (DCM) presented in older ST3Gal4−/− mice.
Fig. 1.
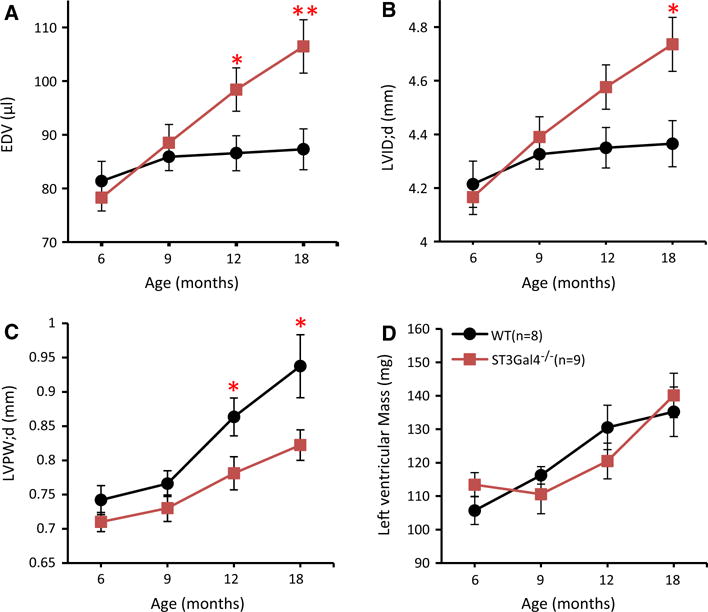
Progressive dilation and thinning of the LV wall were observed in ST3Gal4−/− hearts. Echocardiography was performed on 6, 9, 12, and 18-month-old male mice. ST3Gal4−/− hearts showed progressively enlarged EDV (a) and LVID;d (b) with thinner LVPW;d (c), reaching significance between 12 and 18 months of age compared to age-matched WT littermates. At 18 months: EDV—WT = 87.3 ± 3.8 μl, ST3Gal4−/− = 106.5 ± 5.0 μl; LVID;d—WT = 4.37 ± 0.09 mm, ST3Gal4−/− = 4.74 ± 0.1 mm; LVPW;d—WT = 0.94 ± 0.05 mm, ST3Gal4−/− = 0.82 ± 0.02 mm. d LV mass of ST3Gal4−/− and WT littermates were not significantly different (p > 0.05). *p < 0.05, **p < 0.01. WT (n = 8), black circles/lines; ST3Gal4−/− (n = 9), red squares/lines
Table 1.
Echocardiographic analysis of 18-month-old mice
| WT | ST3Gal4−/− | |
|---|---|---|
| n | 8 | 9 |
| Body weight (g) | 47 ± 1 | 49 ± 2 |
| HR, beats/min | 535 ± 4 | 525 ± 5 |
| Diastole | ||
| LVID (mm) | 4.37 ± 0.09 | 4.74 ± 0.10* |
| LVAW (mm) | 0.95 ± 0.06 | 0.92 ± 0.03 |
| LVPW (mm) | 0.94 ± 0.05 | 0.82 ± 0.02* |
| Systole | ||
| LVID (mm) | 2.89 ± 0.12 | 3.28 ± 0.09* |
| LVAW (mm) | 1.39 ± 0.08 | 1.34 ± 0.06 |
| LVPW (mm) | 1.31 ± 0.07 | 1.18 ± 0.02 |
| LVM (mg) | 135.2 ± 7.4 | 140.1 ± 6.6 |
| LVMi (mg/g) | 2.9 ± 0.1 | 2.9 ± 0.1 |
| LVEF (%) | 64.4 ± 2.8 | 61.3 ± 1.1 |
| %FS | 35.3 ± 2.2 | 32.9 ± 0.7 |
| CO (ml/min) | 30.0 ± 1.8 | 34.2 ± 1.6 |
| SV (μl) | 56.2 ± 3.2 | 65.1 ± 2.9 |
| EDV (μl) | 87.3 ± 3.8 | 106.5 ± 5.0** |
| ESV (μl) | 31.1 ± 2.8 | 41.4 ± 2.6* |
HR heart rate, LVID left ventricular internal dimension, LVAW left ventricular anterior wall, LVPW left ventricular posterior wall, LVM left ventricular mass, LVMi left ventricular mass index, LVEF left ventricular ejection fraction, FS left ventricular fractional shortening, CO cardiac output, SV stroke volume, EDV left ventricular end-diastolic volume, ESV left ventricular end-systolic volume Values are mean ± SEM;
p < 0.05,
p < 0.01
Consistent with echocardiography, H&E staining of 18-month-old ST3Gal4−/− hearts demonstrated a much thinner LV free wall with all four cardiac chambers dilated, most obvious in left and right ventricles (Fig. 2). No apparent defect in the septum, valves or great vessels was observed. No narrowing or obstruction of the coronary artery lumen was seen in the atrium, ventricle or myocardium, nor was there any apparent cell infiltration in WT or ST3Gal4−/− hearts (Fig. 2c). Masson’s trichrome staining in ST3Gal4−/− hearts did not detect any increase in fibrosis (Fig. 2e–f). No statistical difference in heart weight, left ventricular weight, right ventricular weight and left atrial weight between ST3Gal4−/− (n = 5) and WT littermates (n = 4) was found. Together, the echocardiographic data combined with the consistent histological and anatomical data suggest that older ST3Gal4−/− mice manifest a modest dilated cardiomyopathy.
Fig. 2.
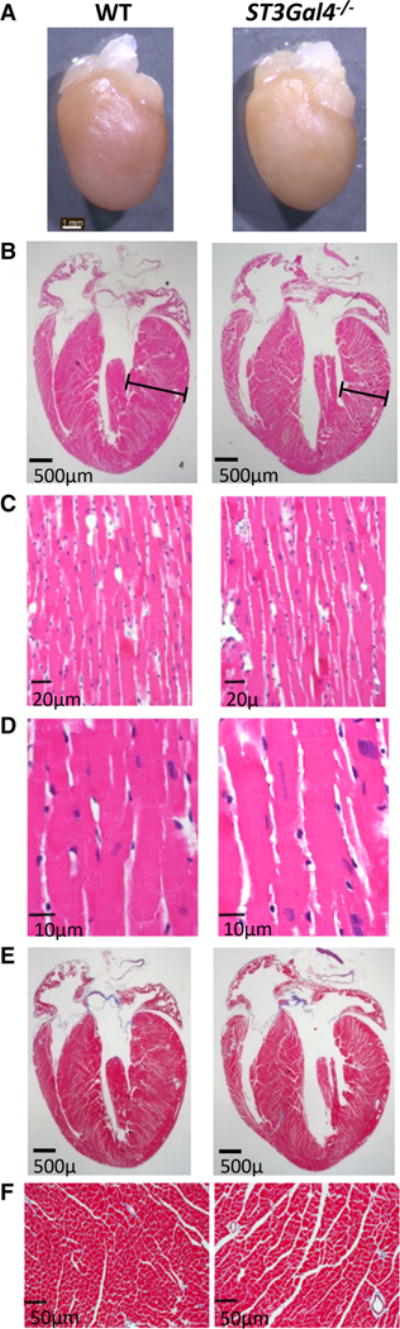
Histological and anatomical analyses support the echocardiographic data. a 18-month-old WT and ST3Gal4−/− hearts. Scale bar 1 mm. In agreement with echocardiography, b H&E staining (×1.25) revealed a thinner left ventricular free wall (bar) with all four chambers dilated, most obvious in ST3Gal4−/− left and right ventricles. Scale bar 0.5 mm. c, d H&E staining at higher magnification (×40 and ×100, respectively). No apparent cell infiltration was found. Scale bar 20 μm in (c) and scale bar 10 μm in d. e, f No significant difference in fibrosis between ST3Gal4−/− and WT hearts was observed (using Masson’s trichrome staining). Scale bar 0.5 mm in e and scale bar 50 μm in f. These experiments were repeated three times, with very similar results
ST3Gal4−/− cardiac calcineurin levels were reduced in younger animals
As shown in Fig. 3, a significant reduction in Ca2+-activated calcineurin subunit A expression was measured in 16- to 20-week-old ST3Gal4−/− hearts compared to WT hearts, suggesting that ST3Gal4 expression impacts Ca2+-dependent transcriptional signaling prior to any indication of impaired contractile function or altered cardiac morphology. Together, the data suggest that ST3Gal4 gene deletion contributes to the early onset of arrhythmic activity [18, 19] and to reduced calcineurin levels.
Fig. 3.
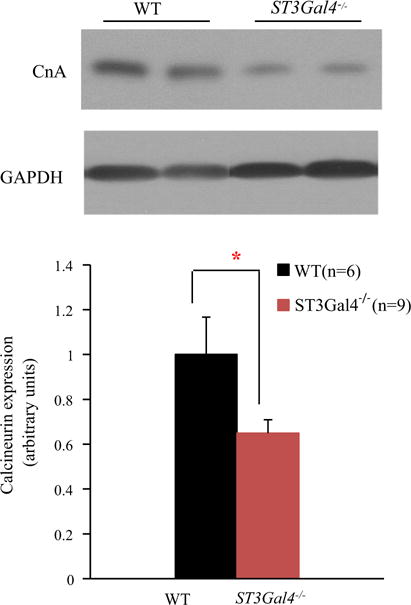
Reduced calcineurin levels were observed in younger ST3Gal4−/− myocytes prior to DCM changes. Top Left ventricular tissue extracts were used to examine calcineurin levels from 16- to 20-week-old WT and ST3Gal4−/−. GAPDH was used as the loading control, with calcineurin subunit A (CnA) expression normalized to WT. Bottom CnA expression in ST3Gal4−/− left ventricles (n = 9) was significantly lower than in WT ventricles (n = 6) (normalized CnA; WT = 1 ± 0.17, ST3Gal4−/− = 0.65 ± 0.06, p < 0.05). *p < 0.05
Pressure overload induces rapid cardiac decompensation in younger ST3Gal4−/− mice
Altered Ca2+-dependent transcription signaling, and specifically a reduction in calcineurin levels, suggest that the ST3Gal4−/− heart may not be as able to compensate against known pathologic stressors, as described previously [39, 66, 70]. Interestingly, we showed above that ST3Gal4 gene deletion contributes to the late-onset of a mild DCM, suggesting that the partial reduction in cardiac sialylation contributes to chronic cardiac dysfunction that slowly leads to altered cardiac structure and contractility. However, the mice were never exposed to chronic environmental or pathological cardiovascular insults such as smoking, alcohol abuse, hypertension, intense exercise, obesity, and/or chronic generalized stress, all of which are risk factors for DCM and HF to which humans are often exposed [22, 29]. Contractile reserve is an important predictor for disease prognosis, especially in asymptomatic patients, and has been defined as, “…a difference between LV function at rest and under load” [24, 50]. Thus, in order to determine whether the reduced calcineurin levels observed in younger ST3Gal4−/− animals results in impaired myocardial contractile reserve, we used a chronic pressure-overload model for stress-induced heart failure, transverse aortic constriction (TAC) [61].
16- to 20-week-old male ST3Gal4−/− and WT mice were randomly and blindly assigned to sham-operated or TAC groups and observed for 6 weeks. At this age there was no indication of altered cardiac structure or reduced systolic function in any mouse (see Fig. 1). Baseline echocardiographic parameters measured before TAC showed no significant differences in cardiac function between WT (n = 7) and ST3Gal4−/− (n = 12) mice (Table 2). Aortic pressure gradients (AoPg) were measured at 1 week post-TAC to determine the degree of pressure-load imposed by aortic constriction and confirmed that the two groups received similar treatment; the measured AoPg for TAC’d ST3Gal4−/− and TAC’d WT being nearly identical (Fig. 4).
Table 2.
Echocardiographic analysis of TAC- and sham-operated mice 6 weeks after surgery
| WT
|
ST3Gal4−/−
|
|||
|---|---|---|---|---|
| Sham | TAC | Sham | TAC | |
| n | 8 | 7 | 10 | 12 |
| Baseline body weight (g) | 31.8 ± 0.9 | 29.1 ± 1.1 | 30.8 ± 0.7 | 30.5 ± 0.9 |
| HR, beats/min | 541 ± 2 | 535 ± 7 | 534 ± 3 | 532 ± 6 |
| Diastole | ||||
| LVID (mm) | 4.37 ± 0.13 | 4.17 ± 0.18 | 4.24 ± 0.05 | 4.80 ± 0.16##,* |
| LVAW (mm) | 0.95 ± 0.02 | 1.12 ± 0.04## | 1.00 ± 0.04 | 0.98 ± 0.02** |
| LVPW (mm) | 0.72 ± 0.02 | 1.15 ± 0.10## | 0.73 ± 0.02 | 0.88 ± 0.03###,** |
| Systole | ||||
| LVID (mm) | 2.77 ± 0.14 | 3.13 ± 0.31 | 3.02 ± 0.04 | 4.10 ± 0.25##,* |
| LVAW (mm) | 1.44 ± 0.03 | 1.56 ± 0.10 | 1.35 ± 0.04 | 1.29 ± 0.05* |
| LVPW (mm) | 1.14 ± 0.04 | 1.48 ± 0.14 | 1.06 ± 0.02 | 1.07 ± 0.04* |
| LVM (mg) | 110.5 ± 7.2 | 162.4 ± 10.1### | 110.2 ± 3.0 | 161.3 ± 11.2### |
| LVMi (mg/g) | 3.5 ± 0.2 | 5.6 ± 0.4### | 3.6 ± 0.1 | 5.3 ± 0.3### |
| EF (%) | 68.9 ± 2.1 | 53.8 ± 7.3 | 59.2 ± 1.5 | 34.0 ± 5.2###,* |
| FS (%) | 38.6 ± 1.6 | 28.6 ± 4.5 | 31.2 ± 1.0 | 16.9 ± 2.9###,* |
| CO (ml/min) | 32.0 ± 1.8 | 21.1 ± 1.7### | 25.6 ± 0.9 | 17.7 ± 1.5### |
| SV (μl) | 59.0 ± 3.3 | 39.4 ± 2.9### | 48.0 ± 1.8 | 33.1 ± 2.7### |
| EDV (μl) | 86.7 ± 6.8 | 79.5 ± 8.3 | 81.0 ± 2.3 | 109.6 ± 8.2##,* |
| ESV (μl) | 27.7 ± 4.1 | 40.1 ± 10.3 | 33.1 ± 1.5 | 76.5 ± 10.3##,* |
n number of mice in each group, WT wild type, HR heart rate, LVID left ventricular internal dimension, LVAWleft ventricular anterior wall, LVPW left ventricular posterior wall, LVM left ventricular mass, LVMi left ventricular mass index: LVMi , EF left ventricular ejection fraction, FS left ventricular fractional shortening, CO cardiac output, SV stroke volume, EDV left ventricular end-diastolic volume, ESV left ventricular end-systolic volume Values are presented as mean ± SEM.
p < 0.05 versus TAC’d WT;
p < 0.01 versus TAC’d WT;
p < 0.05 versus sham;
p < 0.01 versus sham; and
p < 0.001 versus sham
ECL lectin staining was used to examine the cardiomyocyte sialylation level in ST3Gal4−/− heart slices before and after TAC (Fig. 5). ECL staining was much greater in ST3Gal4−/− hearts compared to WT hearts, indicating that there are fewer sialic acid attached to ST3Gal4−/− cardiac glycans. That is, ECL binding to the ST3Gal4−/− tissue should increase because a greater number of galactose residues will be exposed as sialylation is reduced. The data also show that there was no apparent change in myocyte sialylation levels in WT or ST3Gal4−/− hearts following TAC surgeries, suggesting that there is no inherent remodeling of sialylation in response to pressure-overload.
However, TAC’d ST3Gal4−/− cardiac function declined rapidly, reaching significant differences (as compared to TAC’d WT) within 1 week post-surgery that continued to deteriorate throughout the observation period, resulting in the development of left congestive heart failure by the sixth week post-surgery (Fig. 6; Table 2). Systolic function remained within the normal range for TAC’d WT and remained normal and relatively constant in sham WT and sham ST3Gal4−/− mice during the 6-week experiment. No mortality was observed throughout the 6-week TAC experiments.
Fig. 6.
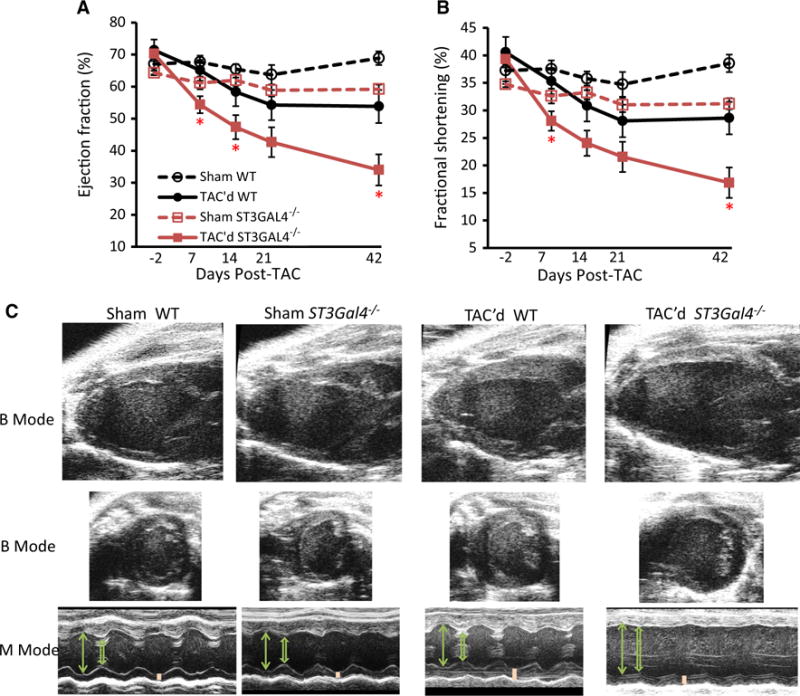
Chronic stress-induced left heart failure presents in younger ST3Gal4−/− mice. a, b Cardiac systolic function of TAC’d ST3Gal4−/− declined rapidly, reaching a significantly reduced EF % (a) and FS % (b) within 7 days post-TAC. The TAC’d ST3Gal4−/− hearts continued deteriorating over time, developing congestive left heart failure by the sixth week post-TAC (EF % = 34 ± 5.2 % and FS % = 16.9 ± 2.9 %). In comparison, TAC’d WT systolic function was only marginally reduced, remaining significantly higher and within the normal range throughout the 6-week experiment (6 weeks after TAC surgery: EF % = 53.8 ± 7.4 % and FS % = 28.6 ± 4.5 %). Cardiac systolic function remained normal and relatively constant in sham WT and sham ST3Gal4−/− mice throughout the experiment (6 weeks after TAC surgery: sham WT EF % = 68.9 ± 2.1 % and FS % = 38.6 ± 1.6 %, sham ST3Gal4−/− EF % = 59.2 ± 1.5 % and FS % = 31.2 ± 1 %). TAC’d WT (n = 7), black solid circles/lines; TAC’d ST3Gal4−/− (n = 12), red solid squares/lines; Sham WT (n = 8), black open circles/dashed lines; Sham ST3Gal4−/− (n = 10), red open squares/dashed lines. *p < 0.05, **p < 0.01; ***p < 0.001, NS (p > 0.05; not statistical significant) versus TAC’d WT. c Representative two-dimensional B-mode and M-mode echocardiographs measured 6 weeks after TAC or sham surgery. TAC’d ST3Gal4−/− hearts showed significantly thinner LV walls, enlarged LVID (systolic and diastolic), and reduced contractility when compared to TAC’d WT. Arrow LVID;d, Open arrow LVID;s, Bar LVPW;d
Insufficient LV adaptation and progressive ventricular dilation in TAC’d ST3Gal4−/− mice
The mean LV wall thickness of TAC’d WT hearts was nearly doubled after TAC due to substantial LV hypertrophy (Fig. 7). However, TAC’d ST3Gal4−/− hearts presented only a limited increase in wall thickness, with progressive left ventricular dilation observed as thinner LVPW;d (Fig. 7b), increased EDV (Fig. 7c), and enlarged LVID;d (Fig. 7d); all parameters showed significant differences from TAC’d WT hearts starting 1 week post-surgery. No significant differences or changes were observed in LVPW;d, EDV and LVID;d of sham WT or sham ST3Gal4−/− mice throughout the 6-week experiment. Table 2 compares all parameters for sham and TAC’d WT and ST3Gal4−/− hearts measured at 6 weeks post-surgery.
Fig. 7.
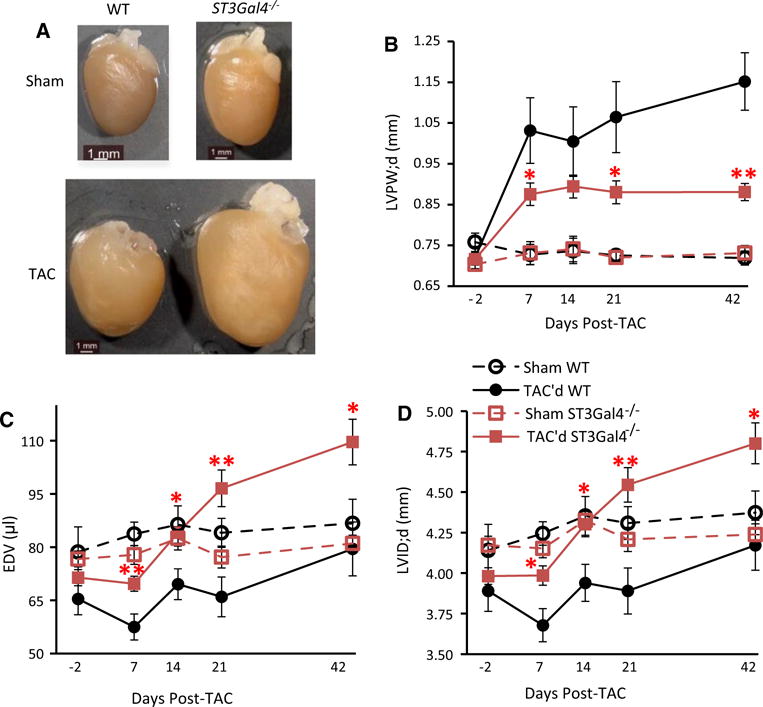
Insufficient cardiac adaptation and progressive LV dilation in ST3Gal4−/− hearts following TAC surgery. a Gross morphology of the WT and ST3Gal4−/− hearts 6 weeks after TAC or Sham surgery. Scale bar 1 mm. b The LVPW;d of TAC’d WT is significantly thicker than the LVPW;d of the TAC’d ST3Gal4−/−, starting 1 week post-surgery. EDV (c) and LVID;d (d) were significantly enlarged in TAC’d ST3Gal4−/− mice compared to TAC’d WT mice, also starting 1 week post-surgery and progressively increasing throughout the 6-week experiment. b–d Sham-operated animals showed no significant changes in LVPW;d, EDV and LVID;d before or after sham surgery. The mean ± SEM parameter values measured 6 weeks post-TAC: LVPW;d—TAC’d WT = 1.15 ± 0.1 mm; TAC’d ST3Gal4−/− = 0.88 ± 0.03 mm; p < 0.01; LVID;d: TAC’d WT = 4.17 ± 0.18 mm; TAC’d ST3Gal4−/− = 4.8 ± 0.16 mm; p < 0.05; EDV: TAC’d WT = 79.5 ± 8.31 μl; TAC’d ST3Gal4−/− = 109.63 ± 8.19 μl; p < 0.05. *p < 0.05, **p < 0.01 versus TAC’d WT. TAC’d WT (n = 7), black solid circles/lines; TAC’d ST3Gal4−/− (n = 12), red solid squares/lines; Sham WT (n = 8), black open circles/dashed lines; Sham ST3Gal4−/− (n = 10), red open squares/dashed lines
Heart weight, left atrial weight, right atrial weight, left ventricle weight, right ventricle weight, lung weight, liver weight, and tibia length were compared among all four groups. Both TAC’d WT and TAC’d ST3Gal4−/− mice demonstrated significantly heavier LV weight and heart weight when compared to sham WT and sham ST3Gal4−/− mice, respectively (p < 0.001) (data not shown). However, TAC’d ST3Gal4−/− mice exhibited much heavier left atrial weight/body weight (LA/BW), lung weight/body weight (Lung/BW) and right ventricle weight/body weight (RV/BW) when compared to TAC’d WT mice. Together, the data suggest that TAC’d ST3Gal4−/− hearts lack sufficient cardiac adaptation and, therefore, decompensate rapidly, deteriorating into left side heart failure under chronic stress conditions, while TAC’d WT heart function remained within the normal range throughout the 6-week period of chronic stress.
Discussion
Here, we report that the partial reduction of N-linked α2,3-sialylation achieved through ST3Gal4 gene deletion leads to the late-onset of a mild dilated cardiomyopathy (DCM) and the rapid development of stress-induced heart failure (HF) at a younger age. Thus, these data show for the first time that glycoprotein sialylation is relevant to cardiac adaptation and contractile function; we also report the first cardiac phenotype for ST3Gal4−/− mice under stressed conditions.
The DCM symptoms progressed gradually and presented with thinner LV walls and dilation of all four chambers (Figs. 1, 2; Tables 1), but with mostly preserved systolic function. The lack of increased fibrosis in ST3Gal4−/− hearts (Fig. 2) renders fibrosis-induced arrhythmogenesis or cardiac remodeling as unlikely mechanisms. However, under chronic stress conditions (pressure-overload), younger ST3Gal4−/− mice demonstrated insufficient LV adaptation and rapid cardiac decompensation, presenting with significantly (compared to WT) reduced systolic function by the end of week 1 that deteriorated into HF by the end of week six post-surgery (Figs. 4, 6, 7; Table 2). In contrast to the negative impact of TAC surgery on the ST3Gal4−/− heart, TAC’d WT hearts showed the classic characteristics of stress-induced adaptation, observed as substantial thickening of the LV wall, and relatively maintained LVID;d, EDV, and contractile functions throughout the 6-week experiment. That is, TAC’d ST3Gal4−/− hearts developed eccentric cardiac hypertrophy as indicated by the significantly increased LVID and the rapid cardiac decompensation (severely reduced systolic function). On the other hand, TAC’d WT hearts presented with classical concentric hypertrophy manifested as significantly increased LV wall thickness, normal systolic function, and a stable LVID. The reduced calcineurin levels observed in younger ST3Gal4−/− hearts may contribute to reduced myocardial contractile reserve and suggest that the responsible mechanism involves altered hypertrophic gene expression in myocytes rather than an extracellular matrix (ECM)-specific mechanism.
Extensive studies have demonstrated that activation of the calcineurin/NFAT pathway acts as one of the primary mechanisms inducing pathological cardiac hypertrophy in response to stimuli such as pressure overload and adrenergic agonist infusions (e.g., isoproterenol), while inhibition of this pathway prevents cardiac hypertrophy [10, 30, 44, 46]. For example, administration of the calcineurin inhibitors, cyclosporine A or FK506, reduced the hypertrophic response [10, 30, 43]. Cardiac-specific expression of non-competitive calcineurin inhibitory domains from Cabin-1or A-kinase anchoring protein 79 blocked calcineurin activity and attenuated cardiac hypertrophy [15, 30, 43]. In addition, calcineurin dominant negative mutant mice and CnAβ−/− mice show reduced cardiac hypertrophy in response to pathologic stimuli such as pressure-overload, isoproterenol or angiotensin II infusion; overexpression of calcineurin recaptured the hypertrophic phenotype [11, 30, 43, 44, 70]. Further reductions in systolic function (contractility) following ischemia/reperfusion injury were observed in CnAβ−/− mice [9]. Together, these and other examples strongly suggest that reduced calcineurin expression/activity prevents or limits the ability of the stressed heart to respond to (compensate for) pathologic stimuli through the hypertrophic pathway and can contribute to reduced systolic function.
Previous reports have also shown that reduced calcineurin expression can contribute to cardiac dysfunction resulting in cardiomyopathy, even in the absence of a pro-hypertrophic insult [9, 31, 52]. As one example, cardiac dysfunction and remodeling was enhanced in the MLP−/−/CnAβ−/− double knockout compared to the MLP−/− heart. Further, the MLP−/− heart was partially protected following mild overexpression of activated calcineurin [31].
In summary, as discussed above and in a commentary by Wolska [67], data strongly suggest that altered calcineurin levels adversely affect heart function, with increased expression/activity promoting a pathological hypertrophic response and reduced calcineurin expression/activity limiting the hypertrophic response and potentially leading to reduced contractile function that can contribute to cardiomyopathy. Our data here are consistent with the previous reports describing the impact of reduced calcineurin levels/activity on cardiac function. That is, in younger ST3Gal4−/− mice, we observed significantly reduced basal cardiac calcineurin expression and significantly reduced hypertrophy in response to pressure overload that rapidly deteriorated into HF (Figs. 4, 6, 7; Table 2). Further, older ST3Gal4−/− mice, in the absence of a pathologic stimulus, presented with late-onset mild DCM (Figs. 1, 2; Table 1).
Our data also suggested a link between partial reductions in cardiac protein sialylation and DCM. Previous studies described a correlation between altered glycosylation and aberrant cardiac function. For example, each of >45 CDG subtypes results in modest but variable (among subtypes) reduced glycoprotein glycosylation, with nearly all subtypes resulting in reduced sialylation and dramatic multi-system effects including cardiomyopathy with related arrhythmias and a high infant mortality rate [1, 14, 23, 27, 38]. We showed here and previously [18, 19, 45, 56] that a mild perturbation in the glycosylation pathway (e.g., gene deletion of ST3Gal4) resulting in a modest reduction in sialylation of only some glycoproteins, is sufficient to induce rather strong cardiac phenotypes.
In addition, altered glycosylation occurs with environmental/metabolic risk factors for DCM including smoking, excessive drinking, and diabetes [36, 64]. Interestingly, non-congenital changes in glycosylation occur in humans with DCM and heart failure [2, 33, 69], but whether altered glycosylation is pathogenic or subsequent to DCM onset has not been determined. Relevant examples showed mRNA levels for the galactotransferase, β4galT1 (acts to attach penultimate galactose to N-/O-glycans, required for the addition of the terminal sialic acids), a putative sialyltransferase, and the CMP sialic acid transporter, were lower in end-stage human DCM [2, 33, 69]. These studies, in agreement with the current work, suggest a correlation between congenital and non-congenital changes in sialylation and reduced cardiac function that may contribute to DCM and stress-induced HF.
The data presented here combined with our previous efforts provide a possible mechanistic link among reduced sialylation, aberrant electrical signaling, reduced calcineurin levels, DCM, and stress-induced HF [18, 19]. That is, we previously showed that 12- to 20-week-old ST3Gal4−/− mice showed aberrant voltage-gated ion channel sialylation and gating that contribute to a pro-arrhythmogenic phenotype. Here, we show that older ST3Gal4−/− animals present with a mild late-onset DCM. Potential cellular mechanisms responsible for human congenital arrhythmic DCM as described by others (for e.g., see [5, 25, 26, 48, 54, 59]) include: (1) disruption or weakening of cytoskeletal components or of interacting voltage-gated ion channel auxiliary proteins [25, 59], and (2) altered Ca2+ homeostasis secondary to aberrant electrical signaling [5, 26, 48]. While we cannot definitively rule out involvement of cytoskeletal components, we see no evidence to support a mechanism that involves disruption of proteins interacting with voltage-gated ion channel alpha subunits, as no change in current densities of any cardiomyocyte voltage-gated ionic current was observed [18, 19]. Thus, aberrant Ca2+ homeostasis secondary to aberrant electrical signaling remains a potential mechanism; calcineurin activity might be affected by a sustained change in cytosolic Ca2+ levels through one of many possible mechanisms as described by others [5, 13, 26, 46, 48, 63]; future studies will investigate the viability of such a mechanism.
We showed previously that reduced ST3Gal4−/− cardiomyocyte NaV sialylation resulted in altered NaV gating and arrhythmic activities similar to that described for the MLP−/−mouse model for HF and for SCN5A mutations correlated to DCM in humans [19, 40–42, 48, 60, 65]. Using statistical in silico modeling, we recently verified that these sialic acid-dependent NaV gating changes directly contribute to arrhythmic activity [16]. In addition, we reported for ST3Gal4−/− ventricular myocytes and hearts, reduced sialylation and gating of KV isoforms, reduced cellular and ventricular refractory periods, and AP/QT segment prolongations, all of which likely also contribute to the arrhythmic behavior that is similar to that described previously for these DCM-correlated human SCN5A mutations and the MLP−/− HF model [18, 19, 40–42, 48, 60, 65]. The exact mechanism(s) by which a variety of congenital changes (SCN5A mutations, MLP deletion, and now, ST3Gal4 deletion) that result in similar arrhythmic behavior and also contribute to DCM and/or HF is not yet known. Determining whether and how such sialo-dependent chronic arrhythmias contribute to DCM onset and stress-induced HF in this ST3Gal4 model will be the focus of future experimental and in silico studies.
Conclusions
In summary, the partial reduction of cardiac protein α2,3-sialylation is sufficient to cause a moderate DCM in older mice and chronic stress-induced HF in younger mice. This is the first study to report the cardiac phenotype occurring with reduced sialylation and specifically, in ST3Gal4−/− mice under normal and stressed conditions. Further, aberrant cardiomyocyte electrical signaling and reduced calcineurin levels precede the onset of DCM and stress-induced HF, suggesting a potential novel mechanistic link among reduced sialylation, arrhythmias, regulation of calcineurin expression, DCM, and stress-induced HF. These findings not only further our understandings of the causes of and potential novel mechanisms for DCM and HF, but also provide potential therapeutic targets for disease treatment.
Acknowledgments
We would like to thank Dr. Jamey Marth for generously providing the ST3Gal4+/− mouse strain. This work was supported, in part, by two Grants from the National Science Foundation [IOS-1146882 and CMMI-1266331]; an American Heart Association, Greater Southeast Affiliate Grant-In-Aid [14GRNT20450148]; an American Heart Association, Greater Southeast Affiliate Postdoctoral Fellowship [15POST25710010]; and a Grant from the National Heart, Lung, Blood Institute [1R01HL102171-03].
Footnotes
Conflict of interest On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standards All animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Specific national laws have been observed. The manuscript does not contain clinical studies or patient data.
References
- 1.Al-Owain M, Mohamed S, Kaya N, Zagal A, Matthijs G, Jaeken J. A novel mutation and first report of dilated cardiomyopathy in ALG6-CDG (CDG-Ic): a case report. Orphanet J Rare Dis. 2010;5:7. doi: 10.1186/1750-1172-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrans JD, Allen PD, Stamatiou D, Dzau VJ, Liew CC. Global gene expression profiling of end-stage dilated cardiomyopathy using a human cardiovascular-based cDNA microarray. Am J Pathol. 2002;160:2035–2043. doi: 10.1016/S0002-9440(10)61153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett E, Urcan MS, Tinkle SS, Koszowski AG, Levinson SR. Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism. J Gen Physiol. 1997;109:327–343. doi: 10.1085/jgp.109.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett ES. Isoform-specific effects of sialic acid on voltage-dependent Na+ channel gating: functional sialic acids are localized to the S5–S6 loop of domain I. J Physiol. 2002;538:675–690. doi: 10.1113/jphysiol.2001.013285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.RES.87.4.275. [DOI] [PubMed] [Google Scholar]
- 6.Born GV, Palinski W. Unusually high concentrations of sialic acids on the surface of vascular endothelia. Br J Exp Pathol. 1985;66:543–549. [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks S, Hall DS. Lectin histochemistry to detect altered glycosylation in cells and tissues. In: Dwek M, Brooks SA, Schumacher U, editors. Metastasis research protocols. Humana Press; Totowa: 2012. pp. 31–50. [Google Scholar]
- 8.Brooks SA, Leathem AJC, Schumacher U, Society RM. Lectin histochemistry: a concise practical handbook BIOS Scientific. Oxford; 1997. [Google Scholar]
- 9.Bueno OF, Lips DJ, Kaiser RA, Wilkins BJ, Dai YS, Glascock BJ, Klevitsky R, Hewett TE, Kimball TR, Aronow BJ, Doevendans PA, Molkentin JD. Calcineurin Abeta gene targeting predisposes the myocardium to acute ischemia-induced apoptosis and dysfunction. Circ Res. 2004;94:91–99. doi: 10.1161/01.RES.0000107197.99679.77. [DOI] [PubMed] [Google Scholar]
- 10.Bueno OF, van Rooij E, Molkentin JD, Doevendans PA, De Windt LJ. Calcineurin and hypertrophic heart disease: novel insights and remaining questions. Cardiovasc Res. 2002;53:806–821. doi: 10.1016/S0008-6363(01)00493-X806-821. [DOI] [PubMed] [Google Scholar]
- 11.Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Impaired cardiac hypertrophic response in Calcineurin Abeta -deficient mice. Proc Natl Acad Sci USA. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. doi: 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camacho Londono JE, Tian Q, Hammer K, Schroder L, Camacho Londono J, Reil JC, He T, Oberhofer M, Mannebach S, Mathar I, Philipp SE, Tabellion W, Schweda F, Dietrich A, Kaestner L, Laufs U, Birnbaumer L, Flockerzi V, Freichel M, Lipp P. A background Ca2 + entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J. 2015;36:2257–2266. doi: 10.1093/eurheartj/ehv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, McKew JC, Gahl WA, Carrillo-Carrasco N, Huizing M. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. 2014;35:915–926. doi: 10.1002/humu.22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, Glascock BJ, Kimball TF, del Monte F, Hajjar RJ, Molkentin JD. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3322–3327. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du D, Yang H, Yang H, Ednie A, Bennett E. Statistical Metamodeling and Sequential Design of Computer Experiments to Model Glyco-altered Gating of Sodium Channels in Cardiac Myocytes. IEEE J Biomed Health Inform. 2015:1. doi: 10.1109/JBHI.2015.2458791. [DOI] [PubMed] [Google Scholar]
- 17.Ednie AR, Bennett ES. Modulation of voltage-gated ion channels by sialylation. Compr Physiol. 2012;2:1269–1301. doi: 10.1002/cphy.c110044. [DOI] [PubMed] [Google Scholar]
- 18.Ednie AR, Bennett ES. Reduced sialylation impacts ventricular repolarization by modulating specific K+ channel isoforms distinctly. J Biol Chem. 2015;290:2769–2783. doi: 10.1074/jbc.M114.605139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ednie AR, Horton KK, Wu J, Bennett ES. Expression of the sialyltransferase, ST3Gal4, impacts cardiac voltage-gated sodium channel activity, refractory period and ventricular conduction. J Mol Cell Cardiol. 2013;59:117–127. doi: 10.1016/j.yjmcc.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, Le DT, Marth JD. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc Natl Acad Sci USA. 2002;99:10042–10047. doi: 10.1073/pnas.142005099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 22.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 23.Footitt EJ, Karimova A, Burch M, Yayeh T, Dupre T, Vuillaumier-Barrot S, Chantret I, Moore SE, Seta N, Grunewald S. Cardiomyopathy in the congenital disorders of glycosylation (CDG): a case of late presentation and literature review. J Inherit Metab Dis. 2009;32(Suppl 1):S313–S319. doi: 10.1007/s10545-009-1262-1. [DOI] [PubMed] [Google Scholar]
- 24.Francis GS, Desai MY. Contractile reserve: are we beginning to understand it? JACC Cardiovasc Imaging. 2008;1:727–728. doi: 10.1016/j.jcmg.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–414. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 26.Ge J, Sun A, Paajanen V, Wang S, Su C, Yang Z, Li Y, Wang S, Jia J, Wang K, Zou Y, Gao L, Wang K, Fan Z. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 2008;1:83–92. doi: 10.1161/CIRCEP.107.750752. [DOI] [PubMed] [Google Scholar]
- 27.Gehrmann J, Sohlbach K, Linnebank M, Bohles HJ, Buderus S, Kehl HG, Vogt J, Harms E, Marquardt T. Cardiomyopathy in congenital disorders of glycosylation. Cardiol Young. 2003;13:345–351. doi: 10.1017/S1047951103000702. [DOI] [PubMed] [Google Scholar]
- 28.Geisler C, Jarvis DL. Effective glycoanalysis with Maackia amurensis lectins requires a clear understanding of their binding specificities. Glycobiology. 2011;21:988–993. doi: 10.1093/glycob/cwr080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics C, Stroke Statistics S Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 31.Heineke J, Wollert KC, Osinska H, Sargent MA, York AJ, Robbins J, Molkentin JD. Calcineurin protects the heart in a murine model of dilated cardiomyopathy. J Mol Cell Cardiol. 2010;48:1080–1087. doi: 10.1016/j.yjmcc.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 33.Hwang JJ, Allen PD, Tseng GC, Lam CW, Fananapazir L, Dzau VJ, Liew CC. Microarray gene expression profiles in dilated and hypertrophic cardiomyopathic end-stage heart failure. Physiol Genomics. 2002;10:31–44. doi: 10.1152/physiolgenomics.00122.2001. [DOI] [PubMed] [Google Scholar]
- 34.Johnson D, Bennett ES. Isoform-specific effects of the beta2 subunit on voltage-gated sodium channel gating. J Biol Chem. 2006;281:25875–25881. doi: 10.1074/jbc.M605060200. [DOI] [PubMed] [Google Scholar]
- 35.Johnson D, Montpetit ML, Stocker PJ, Bennett ES. The sialic acid component of the beta1 subunit modulates voltage-gated sodium channel function. J Biol Chem. 2004;279:44303–44310. doi: 10.1074/jbc.M408900200. [DOI] [PubMed] [Google Scholar]
- 36.Knezevic A, Polasek O, Gornik O, Rudan I, Campbell H, Hayward C, Wright A, Kolcic I, O’Donoghue N, Bones J, Rudd PM, Lauc G. Variability, heritability and environmental determinants of human plasma N-glycome. J Proteome Res. 2009;8:694–701. doi: 10.1021/pr800737u. [DOI] [PubMed] [Google Scholar]
- 37.Kono M, Ohyama Y, Lee YC, Hamamoto T, Kojima N, Tsuji S. Mouse beta-galactoside alpha 2,3-sialyltransferases: comparison of in vitro substrate specificities and tissue specific expression. Glycobiology. 1997;7:469–479. doi: 10.1093/glycob/7.4.469. [DOI] [PubMed] [Google Scholar]
- 38.Kranz C, Basinger AA, Gucsavas-Calikoglu M, Sun L, Powell CM, Henderson FW, Aylsworth AS, Freeze HH. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am J Med Genet A. 2007;143A:1371–1378. doi: 10.1002/ajmg.a.31791. [DOI] [PubMed] [Google Scholar]
- 39.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mann SA, Castro ML, Ohanian M, Guo G, Zodgekar P, Sheu A, Stockhammer K, Thompson T, Playford D, Subbiah R, Kuchar D, Aggarwal A, Vandenberg JI, Fatkin D. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol. 2012;60:1566–1573. doi: 10.1016/j.jacc.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 41.McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L, Familial Cardiomyopathy Registry Research G SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 42.McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L, Familial Cardiomyopathy Registry Research G SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011;57:2160–2168. doi: 10.1016/j.jacc.2010.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/S0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montpetit ML, Stocker PJ, Schwetz TA, Harper JM, Norring SA, Schaffer L, North SJ, Jang-Lee J, Gilmartin T, Head SR, Haslam SM, Dell A, Marth JD, Bennett ES. Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc Natl Acad Sci USA. 2009;106:16517–16522. doi: 10.1073/pnas.0905414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J: Off Public Fed Am Soc Exp Biol. 2006;20:1660–1670. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nanka O, Peumans WJ, Van Damme EJ, Pfuller U, Valasek P, Halata Z, Schumacher U, Grim M. Lectin histochemistry of microvascular endothelium in chick and quail musculature. Anat Embryol (Berl) 2001;204:407–411. doi: 10.1007/s004290100212. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen TP, Wang DW, Rhodes TH, George AL., Jr Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res. 2008;102:364–371. doi: 10.1161/CIRCRESAHA.107.164673. [DOI] [PubMed] [Google Scholar]
- 49.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 50.Okumura T, Murohara T. Contractile reserve in dilated cardiomyopathy. In: José Milei GA, editor. Cardiomyopathies InTech. 2013. pp. 47–59. [DOI] [Google Scholar]
- 51.Ram R, Mickelsen DM, Theodoropoulos C, Blaxall BC. New approaches in small animal echocardiography: imaging the sounds of silence. Am J Physiol Heart Circ Physiol. 2011;301:H1765–H1780. doi: 10.1152/ajpheart.00559.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaeffer PJ, Desantiago J, Yang J, Flagg TP, Kovacs A, Weinheimer CJ, Courtois M, Leone TC, Nichols CG, Bers DM, Kelly DP. Impaired contractile function and calcium handling in hearts of cardiac-specific calcineurin b1-deficient mice. Am J Physiol Heart Circ Physiol. 2009;297:H1263–H1273. doi: 10.1152/ajpheart.00152.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwetz TA, Norring SA, Bennett ES. N-glycans modulate K(v)1.5 gating but have no effect on K(v)1.4 gating. Biochim Biophys Acta. 2010;1798:367–375. doi: 10.1016/j.bbamem.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 54.Simantirakis EN, Koutalas EP, Vardas PE. Arrhythmia-induced cardiomyopathies: the riddle of the chicken and the egg still unanswered? Europace. 2012;14:466–473. doi: 10.1093/europace/eur348. [DOI] [PubMed] [Google Scholar]
- 55.Sperandio M, Frommhold D, Babushkina I, Ellies LG, Olson TS, Smith ML, Fritzsching B, Pauly E, Smith DF, Nobiling R, Linderkamp O, Marth JD, Ley K. Alpha 2,3-sialyltransferase-IV is essential for L-selectin ligand function in inflammation. Eur J Immunol. 2006;36:3207–3215. doi: 10.1002/eji.200636157. [DOI] [PubMed] [Google Scholar]
- 56.Stocker PJ, Bennett ES. Differential sialylation modulates voltage-gated Na+ channel gating throughout the developing myocardium. J Gen Physiol. 2006;127:253–265. doi: 10.1085/jgp.200509423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takashima S. Characterization of mouse sialyltransferase genes: their evolution and diversity. Biosci Biotechnol Biochem. 2008;72:1155–1167. doi: 10.1271/bbb.80025. [DOI] [PubMed] [Google Scholar]
- 58.Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, Horak KM, Molkentin JD, Wang X. Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res. 2010;88:424–433. doi: 10.1093/cvr/cvq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Towbin JA, Lorts A. Arrhythmias and dilated cardiomyopathy common pathogenetic pathways? J Am Coll Cardiol. 2011;57:2169–2171. doi: 10.1016/j.jacc.2010.11.061. [DOI] [PubMed] [Google Scholar]
- 60.Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. 2001;276:28197–28203. doi: 10.1074/jbc.M102548200. [DOI] [PubMed] [Google Scholar]
- 61.Verma SK, Krishnamurthy P, Kishore R. Transverse aortic constriction: a model to study heart failure in small animals. In: Ardehali H, Bolli R, Losordo DW, editors. Manual of research techniques in cardiovascular medicine. Wiley; Oxford, UK: 2014. [DOI] [Google Scholar]
- 62.Vinhas M, Araujo AC, Ribeiro S, Rosario LB, Belo JA. Transthoracic echocardiography reference values in juvenile and adult 129/Sv mice. Cardiovasc Ultrasound. 2013;11:12. doi: 10.1186/1476-7120-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest. 2016;126:112–122. doi: 10.1172/JCI84669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waszkiewicz N, Szajda SD, Zalewska A, Szulc A, Kepka A, Minarowska A, Wojewodzka-Zelezniakowicz M, Konarzewska B, Chojnowska S, Ladny JR, Zwierz K. Alcohol abuse and glycoconjugate metabolism. Folia Histochem Cytobiol. 2012;50:1–11. doi: 10.2478/18690. [DOI] [PubMed] [Google Scholar]
- 65.Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, Wang DW, Hipkens SB, Leake B, Hall L, Kupershmidt S, Chopra N, Magnuson MA, Tanabe N, Knollmann BC, George AL, Jr, Roden DM. Striking In vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124:1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilkins BJ, Molkentin JD. Calcineurin and cardiac hypertrophy: where have we been? Where are we going? J Physiol. 2002;541:1–8. doi: 10.1113/jphysiol.2002.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolska BM. Calcineurin and cardiac function: is more or less better for the heart? Am J Physiol Heart Circ Physiol. 2009;297:H1576–H1577. doi: 10.1152/ajpheart.00833.2009. [DOI] [PubMed] [Google Scholar]
- 68.Wu AM, Wu JH, Tsai MS, Yang Z, Sharon N, Herp A. Differential affinities of Erythrina cristagalli lectin (ECL) toward monosaccharides and polyvalent mammalian structural units. Glycoconj J. 2007;24:591–604. doi: 10.1007/s10719-007-9063-y. [DOI] [PubMed] [Google Scholar]
- 69.Yung CK, Halperin VL, Tomaselli GF, Winslow RL. Gene expression profiles in end-stage human idiopathic dilated cardiomyopathy: altered expression of apoptotic and cytoskeletal genes. Genomics. 2004;83:281–297. doi: 10.1016/j.ygeno.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 70.Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, Nagai R, Yazaki Y, Komuro I. Calcineurin plays a critical role in the development of pressure overload-induced cardiac hypertrophy. Circulation. 2001;104:97–101. doi: 10.1161/01.CIR.104.1.97. [DOI] [PubMed] [Google Scholar]