

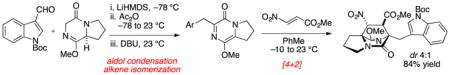
Abstract
A stereoselective intermolecular Diels–Alder cycloaddition of an intermediate pyrazinone with both achiral and chiral acrylate-derived dienophiles provides rapid access to the bicyclo[2.2.2]diazaoctane core shared among several prenylated indole alkaloids. The product derived from cycloaddition with 2-nitroacrylate required an additional five to six synthetic operations to intercept established precursors to premalbrancheamide and brevianamide B. The chemistry detailed in this manuscript constitutes a formal total synthesis (12 steps each) of these [2.2.2]diazabicyclic natural products from proline methyl ester.
Graphical abstract
INTRODUCTION
Isolated by Birch and Wright in 1969, the brevianamides were the first metabolites discovered that possess the bicyclo[2.2.2]-diazaoctane architecture.1 The [2.2.2]diazabicyclic alkaloid family has since grown and now numbers nearly 100 distinct fungal-derived prenylated indole natural products.2 Intriguing biological activity is found across the family, and the alkaloids exhibit antitumor, antihelmintic, insecticidal, and selective phosphodiesterase (PDE1) inhibition and neuroprotective properties.2,3 As a result, the [2.2.2]diazabicyclic alkaloids are lead compounds in the treatment of several diseases and cancer.
Within the [2.2.2]diazabicyclic alkaloid family, metabolites can possess either the syn or anti stereochemical configuration at C19 (brevianamide numbering).4 Continued isolation efforts have revealed enantiomeric5 and epimeric6 metabolites from different fungal organisms (e.g., (+)-, (−)-stephacidin A, and 6-epi-stephacidin A, Figure 1), a discovery that only increases the allure of these alkaloids.
Figure 1.
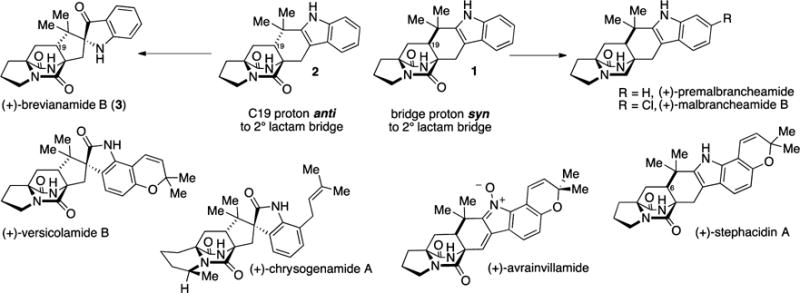
[2.2.2]Diazabicyclic natural products with syn and anti fusion.
Because of potent and diverse bioactivities as well as appealing molecular structure, the entire alkaloid group has attracted the attention of the synthetic community. Efforts directed to the synthesis of various congeners have revealed several methods for the construction of the [2.2.2]diazabicyclic core: (1) SN2′ cyclization, (2) biomimetic intramolecular Diels–Alder cycloaddition (IMDA), (3) N-acyliminium-initiated cyclization, (4) oxidative enolate coupling, (5) radical cyclization, and (6) Dieckmann condensation with an isocyanate component.7,8
The epimeric [2.2.2]diazabicycles 1 and 2 are the most simple structures that contain all rings apparent within these natural products. The syn-configured 1 can be converted to premalbrancheamide, the biogenic precursor to more highly halogenated compounds (e.g., malbrancheamide A and B).9 The anti-configured 2 is not a natural product but has served as the penultimate intermediate in several syntheses of brevianamide B (3).10,8a The synthesis of compounds 1 or 2 have been a proving ground for methods to construct the [2.2.2]-diazabicyclic ring system, and the chemistry revealed in these studies is often applied to more elaborate targets within the natural product family.
The development of the biomimetic IMDA cycloaddition by Williams has resulted in the total synthesis of several [2.2.2]diazabicyclic alkaloids and led to important insights into the biogenesis of these alkaloids.11 As an example of this chemistry, diketopiperazine 4 can be isomerized to the reactive pyrazinone intermediate 7, which undergoes cycloaddition to give the diastereomers 5 and 6 (dr 2:1, Figure 2, entry 1). Because the pyrazinone Diels–Alder precursor 7 is achiral, the observed cycloadducts are racemic. Production of asymmetric [2.2.2]diazabicyclic alkaloids via the IMDA has been realized using a chiral aminal auxiliary precursor 88d (e.g., entry 3) as well as two other nonracemic substrates.11a,12 Additionally, Johnston and co-workers recently extended the IMDA chemistry using a hydrogen bonding catalyst to modulate the diastereo- and enantioselectivity of the reaction (entry 2).13 In this way, syn-fused 5 can be produced in up to 4:1 dr and up to 44% ee depending on the catalyst used. In many ways, this represents a normal evolution of reaction development, where an initial reaction is rendered diastereoselective first with auxiliaries and later made to be a catalytic enantioselective process.
Figure 2.
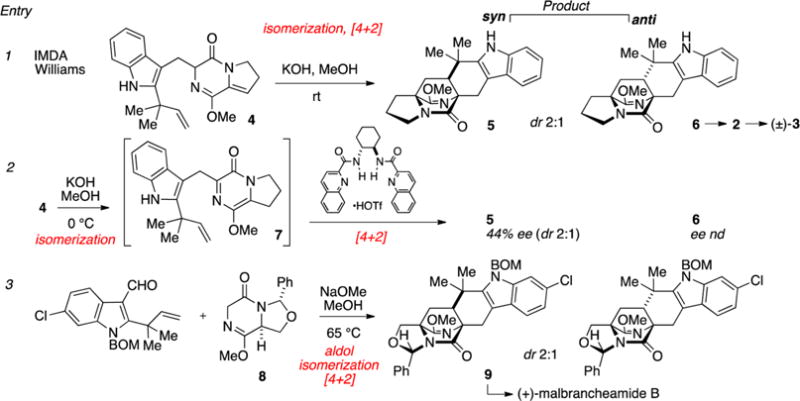
Intramolecular Diels–Alder approaches to the formation of [2.2.2]diazabicyclic structures.
RESULTS AND DISCUSSION
This manuscript describes our efforts toward 1 and 2 that focus on an intermolecular Diels–Alder cycloaddition for the construction of the [2.2.2]diazabicyclic system.
The Diels–Alder reaction is naturally efficient (forming two bonds in concerted fashion), and the intermolecular variant is intrinsically convergent. We believed that a strategy based on intermolecular cycloaddition might offer improvements in efficiency over several known routes and potentially permit selective formation of either diastereomer 1 or 2 based on modulation of the reaction conditions. Most importantly, the fundamentals of the pyrazinone intermolecular Diels–Alder cycloaddition are not fully understood, and we viewed a synthesis of 1 and 2 as an instructive target to advance this chemistry in more detail.
We previously established that pyrazinone Diels–Alder precursors can be prepared in a single step by a domino aldol condensation/alkene isomerization sequence.14 Accordingly, our proposed synthesis toward 1 and 2 planned to engage the requisite pyrazinone 10 prepared in this manner with a suitable acrylate-derived dienophile to afford cycloadduct 3 (Scheme 1). Subsequent addition of an organometallic reagent (e.g., MeMgBr) to the ester to afford an intermediate tertiary alcohol would then permit Friedel–Crafts cyclization to produce the quaternary carbon and complete the ring system. Because both 1 and 2 are known, we anticipated that verification of stereochemical outcome from the intermolecular cycloaddition could be quickly determined by comparing to published spectroscopic data or authentic standards.
Scheme 1.
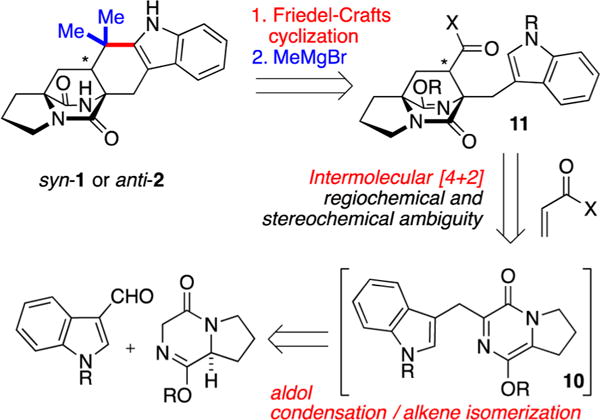
Retrosynthesis Featuring an Intermolecular [4 + 2] Reaction
Although this synthesis plan appeared concise, there were significant regiochemical and stereochemical ambiguities with regard to the intermolecular cycloaddition as well as fundamental questions regarding reactivity. We predicted that a modest preference for the endo cycloadduct might be enforced (leading to the anti-bicyclo fusion).
In order to evaluate the intermolecular cycloaddition, pyrazinone 14 was prepared in one reaction vessel starting from the proline-derived diketopiperazine (DKP) 12 (Scheme 2).15 The reaction sequence is initiated by enolization (LiHMDS, 1.1 equiv) followed by aldol addition to the indole carboxaldehyde. The intermediate β-alkoxy adduct was acylated (Ac2O, 1.1 equiv) and treated with base (DBU, 1.4 equiv) in order to effect elimination to the exocyclic diene in 13 and subsequent isomerization to the DKP azadiene 14. Based on experimental evidence, the exocyclic diene in 13 is short-lived and isomerizes rapidly to the endocyclic azadiene 14, the apparent thermodynamic alkene isomer and only identified product from the reaction.
Scheme 2.
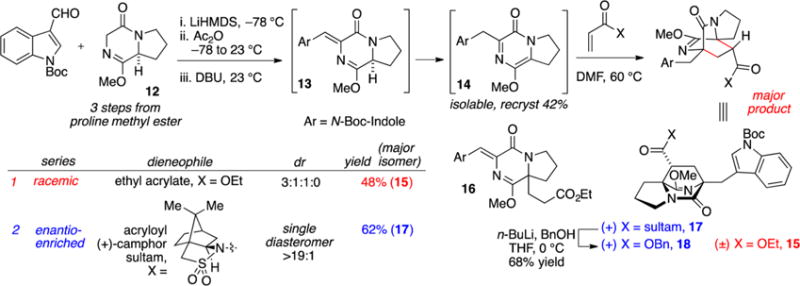
Intermolecular Diels–Alder Cycloaddition with Acrylate Dienophiles
Although isolable, azadiene 14 proved somewhat difficult to manipulate. The pyrazinone decomposed on exposure to silica gel and was reactive with air, undergoing decomposition within hours at room temperature.16 Nonetheless, we became experienced at handling 14 as the unpurified product mixture (obtained in 94% yield from the β-acetoxyaldol adduct, 90–95% pure by 1H NMR) or as a homogeneous material obtained by recrystallization, albeit in diminished yield (42%). In practice, we also found it convenient to isolate and purify the intermediate β-acetoxy adduct and advance material through the elimination and isomerization to form 14 as needed.
We first attempted Diels–Alder cycloaddition of 14 with ethyl acrylate and observed three products in a 3:1:1 ratio.17 The major product 15 was obtained in 48% isolated yield and corresponds to cycloaddition from the endo transition state. Due to the inconvenience of handling metastable pyrazinone 14, we also attempted a “one pot” reaction in order to circumvent isolation of the reactive intermediate. Addition of ethyl acrylate to the reaction vessel following completion of the aldol condensation and alkene isomerization sequence produced a low yield (15%) of desired cycloadduct 15. The observed major product 16 (50% yield) was the result of Michael addition. Formation of the Michael adduct could be almost completely avoided with addition of a stoichiometric amount of acetic acid (in order to buffer the basicity of residual DBU) prior to addition of acrylate. In this way, the desired major cycloadduct 15 was obtained in a commensurate amount (40% isolated yield) and identical stereoselection (3:1:1) as compared to the two-step sequence. Overall, this intermolecular Diels–Alder reaction sequence required five to six steps from commercially available proline methyl ester to create the [2.2.2]diazabicyclic structure; however, the dominant product possessed the undesired regiochemistry and would not permit synthesis of 2 in a straightforward manner.
In the course of preparing suitable acrylate precursors, we observed that the acrylamide derivative of Oppolzer’s sultam was more selective in engaging pyrazinone substrate 14 and could effectively introduce asymmetry to and improve diastereoselection in the cycloaddition.18 The thermal cyclo-addition of 14 with acryloyl camphor sultam afforded cycloadduct 17 in 62% yield as an apparent single diastereomer (>19:1) as determined by 1H NMR on the unpurified reaction product mixture (Scheme 2, blue highlight).19 The acyl sultam in 17 could be converted with lithio benzyl alcohol into the corresponding ester 18.20
In order to obtain the desired regiochemistry whereby the carboxyl moiety is positioned proximal to the indole nucleus, we attempted cycloaddition with methyl 2-nitroacrylate (19), a dienophile substrate that can be effective in reversing cycloaddition regioselectivity.21 In the event, nitroacrylate 19 was more reactive than ethyl acrylate and cycloaddition with pyrazinone 14 was achieved near ambient temperatures. Two regioisomeric cycloadducts 20 and 21 (ratio 1:4) were produced in 84% combined yield (Scheme 3). Both cycloadducts arise from orientation of the nitro function in the endo transition state. The dominant cycloadduct 21 possessed the desired regiochemistry and we turned our attention to the three remaining tasks to assemble the complete ring system relevant to the [2.2.2]diazabicyclic alkaloid family: (1) removal of the nitro group, (2) methylation of the ester (MeMgBr), and (3) Friedel–Crafts cyclization.
Scheme 3.
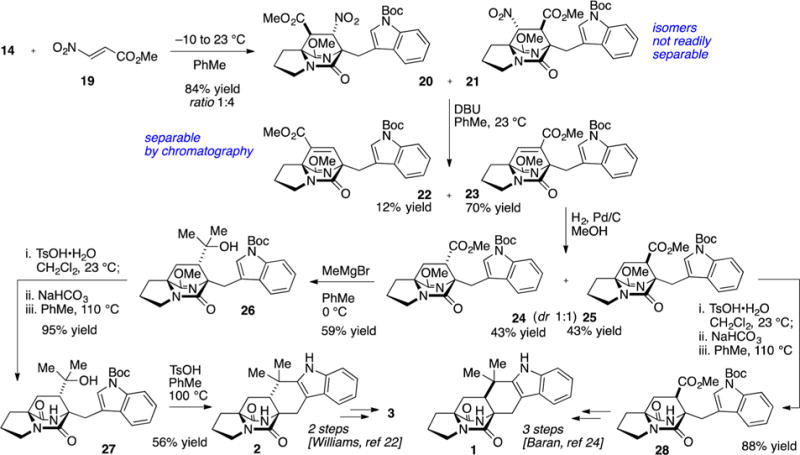
Cycloaddition with Nitroacrylate and Formal Synthesis of Brevianamide B and Premalbrancheamide
Direct radical reduction of the secondary nitro group in 21 was attempted (Bu3SnH, AIBN, PhMe, 110 °C), although only the product resulting from elimination (23) was observed. Performing the reduction at lower temperatures (70–80 °C) afforded mostly elimination product 23 but also a limited quantity (ca. 10% yield) of 25, thereby validating the stereochemistry of the cycloaddition. Due to the strong propensity for elimination, we found it operationally most convenient to expose the 1:4 mixture of cycloadducts 20 and 21 to DBU, which promoted elimination to the two isomeric unsaturated ester products. The major isomer 23 was easily separated by chromatography from the regioisomeric product 22. Hydrogenation of the unsaturation in 23 gave a nearly equal mixture of syn- and anti-fused products 24 and 25.
Both diastereomers 24 and 25 were advanced in a short number of operations to intercept known compounds. Product 24 was converted to anti-fused 2, a well-established intermediate toward brevianamide B.22 Toward this end, the ester in 24 was consumed with excess MeMgBr to afford the derived tertiary alcohol. Acidic hydrolysis23 of the lactim ether functionality to give the lactam 26 was required prior to Friedel–Crafts cyclization of the indole on the derived tertiary carbocation. Intermediate 27, which features the tertiary alcohol and lactam structures, cleanly underwent indole BOC deprotection and cyclization to give the desired annulated product 2 in 83% yield. The spectral data for product 2 thus obtained was identical to all properties of an authentic sample provided by Williams.
The analogous lactim hydrolysis on syn-fused intermediate 25 intercepted the known lactam 28, an intermediate produced by Baran and co-workers in the course of their synthesis of 1 and more complex natural products.24
The synthesis of 2 and 28 constitute 12-step formal total syntheses of both brevianamide B (3) and premalbrancheamide, representative anti- and syn-fused [2.2.2]diazabicyclic alkaloid natural products. The synthetic strategy based on intermolecular pyrazinone cycloaddition is convergent and provides a direct entry to the [2.2.2]diazabicyclic feature (four to five steps from proline methyl ester). Further insight into the pyrazinone Diels–Alder was obtained in this study. In particular, we demonstrated that the use of a 2-nitroacrylate dienophile was required to obtain the desired regiochemistry. Extension of this intermolecular cycloaddition with chiral nitroacrylate derivatives25 could enable construction of structures in either enantiomeric series. Additionally, we plan to continue to explore other possible stereoselective operations, such as directed or diastereoselective reduction, in the application of this synthetic strategy to more intricate and bioactive natural products in the [2.2.2]diazabicyclic prenylated indole alkaloid family.
EXPERIMENTAL SECTION
General Information
All reactions were carried out under an atmosphere of nitrogen in flame-dried or oven-dried glassware with magnetic stirring unless otherwise indicated. Acetonitrile, THF, toluene, and Et2O were degassed with argon and purified by passage through a column of molecular sieves and a bed of activated alumina. Dichloromethane was distilled from CaH2 prior of use. All reagents were used as received unless otherwise noted. Flash column chromatography was performed using silica gel (230–400 mesh). Analytical thin-layer chromatography was performed on 60 Å glass plates. Visualization was accomplished with UV light, anisaldehyde, ceric ammonium molybdate (CAM), potassium permanganate, or ninhydrin, followed by heating. 1H NMR spectra were recorded on a 400 MHz spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm) or tetramethylsilane (0.00 ppm). Proton-decoupled 13C NMR spectra were recorded on a 400 MHz spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 77.0 ppm). All compounds were judged to be homogeneous (>95% purity) by 1H and 13C NMR spectroscopy unless otherwise noted as mixtures. Mass spectra data analysis was obtained through positive electrospray ionization (ICR-MS w/NaCl).
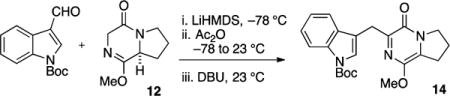
tert-Butyl 3-((1-Methoxy-4-oxo-4,6,7,8-tetrahydropyrrolo-[1,2-a]pyrazin-3-yl)methyl)-1H-indole-1-carboxylate (14)
To diketopiperazine (DKP) 128d (384 mg, 2.29 mmol) in PhMe (8 mL) and DMF (8 mL) at −78 °C was added LiHMDS (2.50 mL, 1.0 M in THF, 1.1 equiv) dropwise over 5 min by syringe. After the mixture was stirred for 15 min at −78 °C, N-Boc-indole carboxaldehyde (0.56 g, 1.0 equiv) in PhMe (8 mL) was added, and the reaction was stirred for an additional 15 min at −78 °C. Ac2O (0.28 mL, 3.0 mmol, 1.3 equiv) was added, and the cooling bath was removed. The reaction mixture was stirred at rt for 3 h, at which time DBU (0.48 mL, 3.2 mmol, 1.4 equiv) was added and the reaction was stirred at rt for 18 h. The reaction was then diluted with 0.1 M HCl (50 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with satd aqueous NaHCO3 and brine, dried (Na2SO4), and concentrated in vacuo. The resulting product (0.91 g, quant mass recovery, ca. 95% pure by 1H NMR) was recrystallized (hexanes/EtOAc) to afford analytically pure 14 (0.38 g, 42% yield) as a yellow amorphous solid. Pyrazinone 14 can be stored (in the dark) for several weeks under inert gas at −15 °C without appreciable degradation: TLC (100% EtOAc) Rf 0.40 (UV/CAM); IR (film) 2976, 2939, 1730, 1653, 1584, 1579, 1452, 1369, 1349, 1256, 1157, 1080, 1046, 1016, 855, 750 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.75 (d, J = 7.3 Hz, 1H), 7.61 (s, 1H), 7.29–7.19 (m, 2H), 4.19 (s, 2H), 4.14 (t, J = 7.0 Hz, 2H), 3.82 (s, 3H), 3.04 (t, J = 7.4 Hz, 2H), 2.21 (q, J = 7.4 Hz, 2H), 1.65 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 154.0, 149.8, 149.6, 142.5, 135.5, 130.8, 125.4, 124.2, 124.1, 122.2, 120.0, 116.7, 115.0, 83.3, 54.8, 49.5, 28.4, 28.2, 28.1, 21.6; exact mass calcd for C22H25N3O4 [M + Na]+ 418.1743, found 418.1735.
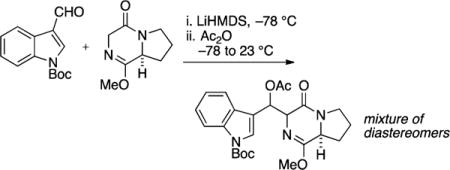
tert-Butyl 3-(Acetoxy((8aS)-1-methoxy-4-oxo-3,4,6,7,8,8a-hexahydropyrrolo[1,2-a]pyrazin-3-yl)methyl)-1H-indole-1-carboxylate
To DKP 12 (2.16 g, 12.9 mmol) in THF (71 mL) at −78 °C was added LiHMDS (14.1 mL, 1 M in THF) dropwise and the mixture stirred for 30 min at −78 °C. N-Boc-indole carboxaldehyde (3.31 g, 13.5 mmol) was added as a solution in THF (10 mL) dropwise over 5 min, and the reaction was stirred for an additional 30 min at −78 °C. Ac2O (1.82 mL, 19.3 mmol) and pyridine (1.55 mL, 19.3 mmol) were each added dropwise over 5 min, and the cold bath was removed. The reaction was allowed to warm to rt and stir for 16 h and was then diluted with HCl (20 mL, 0.1 M) and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with satd aqueous NaHCO3 (50 mL), saturated NaCl (50 mL), dried (Na2SO4), filtered, and concentrated in vacuo to afford the β-acetoxy product as a mixture of four diastereomers. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 30% → 100% EtOAc in hexanes). The four diastereomers were collected together to afford a pale yellow amorphous solid (5.32 g, 91% yield): mp 58.0–59.1 °C; TLC (80% EtOAc in hexanes) Rf 0.25–0.35 (UV/CAM); IR (film) 2980, 2945, 2886, 1732, 1676, 1657, 1451, 1368, 1338, 1310, 1252, 1219, 1152, 1080, 1017, 947, 853, 839, 764, 747, 700, 631, 600, 592 cm−1; 1H NMR (400 MHz, CDCl3) 8.13–8.01 (m, 1H), 7.96–7.91 (m, 1H), 7.74–7.52 (m, 1H), 7.32–7.18 (m, 2H), 6.89–6.41 (m, 1H), 4.77–4.28 (m, 1H), 4.05–3.94 (m, 1H), 3.79–3.72 (m, 3H), 3.70–3.63 (m, 1H), 3.50–3.31 (m, 1H), 2.31–2.22 (m, 1H), 2.16–2.01 (m, 3H), 1.93–1.69 (m, 2H), 1.62–1.29 (m, 1H), 1.66–1.64 (m, 9H); 13C NMR (100 MHz, CDCl3): δ 170.2, 169.8, 169.4, 165.9, 165.5, 165.2, 165.1, 163.4, 163.1, 162.5, 162.4, 149.6, 149.7, 149.5, 135.3, 135.1, 129.8, 129.5, 128.9, 128.6, 125.6, 125.4, 125.0, 124.7, 124.6, 124.4, 124.3, 124.1, 122.7, 122.6, 122.5, 122.3, 121.8, 120.8, 120.4, 120.0, 118.6, 116.9, 116.8, 116.7, 115.2, 115.1, 114.8, 84.1, 83.6, 83.6, 71.1, 71.0, 70.4, 66.0, 65.8, 63.7, 63.4, 56.9, 56.8, 56.6, 56.3, 53.8, 53.7, 53.6, 44.6, 44.4, 44.3, 44.2, 29.6, 29.4, 29.1, 28.7, 28.3, 28.2, 28.1, 22.6, 22.4, 21.9, 21.7, 21.3, 21.2, 21.1; exact mass calcd for C24H29N3O6Na [M + Na]+ 478.1949, found 478.1946.
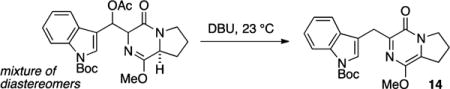
tert-Butyl 3-((1-Methoxy-4-oxo-4,6,7,8-tetrahydropyrrolo-[1,2-a]pyrazin-3-yl)methyl)-1H-indole-1-carboxylate (14)
A flame-dried flask was charged with β-acetoxy aldol addition product (1.37 g, 3.00 mmol), wrapped in aluminum foil, flushed with N2 gas for 10 min, and dissolved in PhMe (15 mL) and DMF (3 mL). To the reaction flask was added DBU (0.89 mL, 6.00 mmol), and the reaction was stirred for 16 h, diluted with HCl (20 mL, 0.2 M), and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with satd aqueous NaHCO3 (50 mL) and saturated NaCl (50 mL), dried (Na2SO4), filtered, and concentrated in vacuo to afford pyrazinone 14 as a yellow amorphous solid (1.12 g, 94% yield). Vide infra for spectral information.
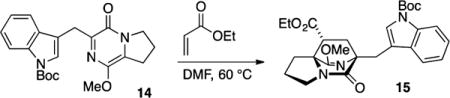
Ethyl (6R*,8R*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-5-oxo-2,3,5,6,7,8-hexahydro-1H-6,8a-(azenometheno)indolizine-8-carboxylate (15)
A flame-dried flask was charged with pyrazinone 14 (227 mg, 0.575 mmol) and fitted with a reflux condenser. The starting material was dissolved in DMF (3.8 mL, 0.15 M), and the reaction vessel was degassed (evacuated and backfilled with N2 × 5). Ethyl acrylate (0.12 mL, 114 mg, 1.14 mmol) was added, and the reaction vessel was heated at 60 °C for 24 h, cooled to rt, and concentrated in vacuo to afford a brown oil. Analysis of the unpurified reaction mixture reveals a 3:1:1 mixture of diastereomers. Purification by flash column chromatography on silica gel (gradient elution: 30% →100% EtOAc in hexanes) afforded product 15 (138 mg, 48% yield) as a colorless oil: TLC (60% EtOAc in hexane) Rf 0.30 (CAM); IR (film) 2985, 2945, 2876, 1736, 1683, 1642, 1456, 1416, 1308, 1260, 1156, 1085, 1016, 852, 768, 745 cm−1; 1H NMR (400 MHz, CDCl3) 8.10 (br s, 1H), 7.78 (d, J = 7.5 Hz, 1H), 7.68 (s, 1H), 7.24 (m, 2H), 4.05 (m, 2H), 3.75 (s, 3H), 3.50–3.30 (m, 2H), 3.44 (d, J = 14.8 Hz, 1H), 3.34 (d, J = 14.8 Hz, 1H), 2.78 (dd, J = 9.8, 5.5 Hz, 1H), 2.61 (m, 1H), 2.05–1.85 (m, 4H), 1.80 (dd, J = 13.2, 5.5 Hz, 1H), 1.64 (s, 9H), 1.18 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.3, 163.0, 160.8, 149.5, 134.9, 132.1, 130.3, 129.0, 128.4, 128.2, 125.3, 124.7, 123.0, 118.9, 116.1, 155.5, 115.1, 104.8, 83.7, 65.6, 60.7, 54.3, 43.8, 35.0, 32.4, 29.0, 28.2, 28.1, 20.2, 14.0; exact mass calcd for C27H33N3O6 [M + Na]+ 518.2261, found 518.2259.
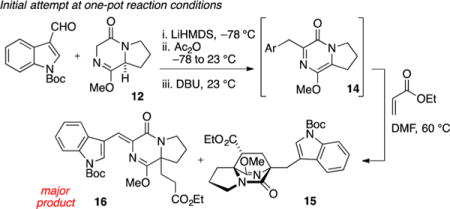
tert-Butyl (Z)-3-((8a-(3-Ethoxy-3-oxopropyl)-1-methoxy-4-oxo-6,7,8,8a-tetrahydropyrrolo[1,2-a]pyrazin-3(4H)-ylidene)-methyl)-1H-indole-1-carboxylate (16)
The reaction was executed according to the conditions previously described for the preparation of 14. Prior to aqueous workup of 14 (0.38 mmol, assuming quantitative formation), ethyl acrylate (0.080 mL, 0.76 mmol, 2 equiv) was added, and the reaction was warmed to 60 °C and stirred for an additional 24 h. The reaction mixture was diluted with 0.2 M HCl (30 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with satd aqueous NaHCO3 and brine, dried (Na2SO4), and concentrated in vacuo. The resulting residue was purified by flash chromatography on silica gel (gradient elution: 40 → 100% EtOAc in hexane) to afford 15 (28 mg, 15% yield) and 16 (94 mg, 50% yield) as a yellow oil. Data for compound 16: TLC (60% EtOAc in hexane) Rf 0.50 (UV, CAM); IR (film) 2980, 2880, 1732, 1680, 1646, 1456, 1411, 1300, 1270, 1154, 1018, 740 cm−1; 1H NMR (400 MHz, CDCl3); 8.60 (s, 1H), 8.18 (d, J = 7.4 Hz, 1H), 7.84 (dd, J = 7.4, 1.6 Hz, 1H), 7.34 (m, 2H), 4.10–3.92 (m, 2H), 4.01 (s, 3H), 2.22–2.00 (m, 8H), 3.40 (m, 4H), 1.67 (s, 9H), 1.14 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.3, 162.9, 160.8, 149.5, 132.0, 130.2, 128.3, 124.6, 123.0, 118.8, 116.0, 115.4, 115.1, 83.6, 65.6, 60.7, 54.3, 43.8, 35.0, 32.3, 28.9, 28.2, 28.0, 20.0, 14.0; exact mass calcd for C27H33N3O6 [M + Na]+, 518.2261, found 518.2257.
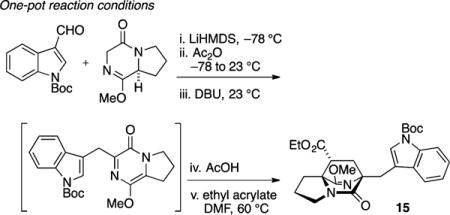
The reaction was executed according to the conditions previously described conditions used for the preparation of 14. Prior to aqueous workup of 14 (0.1 mmol, assuming quantitative formation), AcOH (6 μL, 0.1 mmol, 1 equiv) and ethyl acrylate (20 μL, 0.2 mmol, 2 equiv) were added, and the reaction was warmed to 60 °C and stirred for an additional 24 h. The reaction mixture was diluted with 0.2 M HCl (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with satd aqueous NaHCO3 and brine, dried (Na2SO4), and concentrated in vacuo. The resulting residue was purified by flash chromatography on silica gel (gradient elution: 40 → 100% EtOAc in hexane) to afford 15 (20 mg, 40% yield). Vide infra for spectral information.
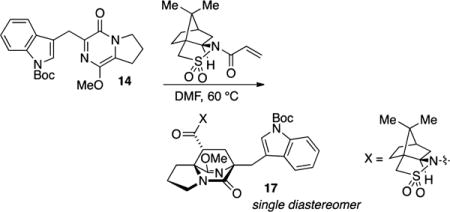
tert-Butyl 3-(((6R,8R,8aS)-8-((7aR)-8,8-Dimethyl-2,2-dioxidohexahydro-3H-3a,6-methanobenzo[c]isothiazole-1-carbonyl)-9 – methoxy – 5 – oxo – 2, 3, 7, 8 – tetrahydro – 1 H – 6, 8 a -(azenometheno)indolizin-6(5H)-yl)methyl)-1H-indole-1-carboxylate (17)
To a flame-dried flask were added acryloyl (+)-camphorsultam (135 mg, 0.51 mmol) and pyrazinone 14 (188 mg, 0.48 mmol). The flask was fitted with a condenser and flushed with N2. The reactants were dissolved in DMF (3.2 mL, 0.15 M), and the vessel was evacuated and backfilled with N2 (×5) before heating to 60 °C. After being heated for 24 h, the reaction mixture was cooled to rt and concentrated in vacuo to afford a brown oil. Analysis of the unpurified reaction mixture by 1H NMR reveals a single cycloadduct as well as some unreacted sultam dienophile. Purification by flash column chromatography on silica gel (gradient elution: 30% → 70% EtOAc in hexanes) afforded cycloadduct 17 (260 mg, 62% yield) as a colorless oil: TLC (80% EtOAc in hexane) Rf 0.3 (CAM); [α]25D = +59 (c 1.0, CH2Cl2); IR (film) 3854, 3744, 3061, 2986, 2882, 2360, 2337, 1730 1683, 1648, 1635, 1452, 1363, 1351, 1308, 1298, 1268, 1258, 1218, 1164, 1135, 1083, 1015, 987, 768, 735, 667, 533 cm−1; 1H NMR (400 MHz, CDCl3) 8.1 (s, 1H), 7.77 (d, J = 1.14 Hz, 1H), 7.66 (s, 1H), 7.23 (m, 2H), 3.77 (s, 3H), 3.75 (m, 1H), 3.52–3.42 (m, 4H), 3.38 (d, J = 13.7 Hz, 1H), 3.30 (d, J = 14.8 Hz, 1H), 2.47 (dd, J = 11.7, 6.6 Hz, 1H), 2.15–1.81 (m, 10H), 1.68 (dd, J = 13.3, 5.5 Hz, 1H), 1.66 (s, 9H), 1.30 (m, 2H), 1.15 (s, 3H), 0.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.2, 170.9, 169.5, 149.8, 135.1, 132.1, 125.5, 123.6, 121.9, 120.4, 116.5, 114.8, 83.1, 66.3, 66.2, 65.4, 54.5, 53.1, 49.0 48.2, 47.7, 44.6, 43.2, 38.5, 34.9, 32.9, 28.8, 28.2, 27.2, 26.3, 24.4, 20.9, 19.8; exact mass calcd for C35H44N4O7S [M + Na]+ 687.2823, found 687.2813.
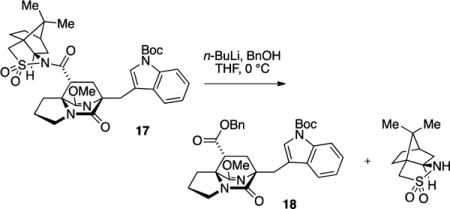
Benzyl (6R,8R,8aS)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-5-oxo-2,3,5,6,7,8-hexahydro-1H-6,8a-(azenometheno)indolizine-8-carboxylate (18)
To a flame-dried reaction flask under N2 were added BnOH (13 μL, 0.12 mmol) and THF (0.6 mL), the vessel was cooled to −78 °C, and a solution of n-BuLi (3.0 M in pentane, 33 μL, 0.01 mmol) was added. In a separate flask, sultam adduct 17 (20 mg, 0.03 mmol) was dissolved in THF (0.25 mL), and the solution was transferred via cannula to the reaction flask (which contained the LiOBn solution). After the cannula wire was rinsed with an additional 0.15 mL of THF, the −78 °C cooling bath was exchanged for an ice/water bath (0 °C). The reaction mixture was stirred at 0 °C for 8 h, diluted with satd aq NH4Cl (15 mL), and extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with satd aq NaHCO3 and brine, dried (Na2SO4), and concentrated in vacuo. The resulting oil was purified by flash column chromatography on silica gel (gradient elution: 30% → 70% EtOAc in hexanes) to afford the benzyl ester 18 as a light yellow oil (13 mg, 75% yield): TLC (60% EtOAc in hexane) Rf 0.6 (CAM); [α]D25 = +25 (c 0.3, CH2Cl2); IR (film) 2985, 2942, 2882, 2358, 2330, 1730, 1686, 1639, 1452, 1370, 1308, 1257, 1160, 1085, 1015, 912, 857, 746, 668, 664 cm−1; 1H NMR (400 MHz, CDCl3) 8.11 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.66 (s, 1H), 7.34–7.28 (m, 4H), 7.24–7.18 (m, 3H) 5.06 (d, J = 12.1 Hz, 1H), 4.98 (d, J = 12.1 Hz, 1H), 3.65 (s, 3H), 3.50 (m, 1H), 3.45 (d, J = 14.8 Hz, 1H), 3.35 (m, 1H), 3.34 (d, J = 14.8 Hz, 1H), 2.84 (dd, J = 10.2, 5.5 Hz, 1H), 2.57 (dd, J = 13.3, 5.1 Hz, 1H), 2.10–1.85 (m, 4H), 1.84 (dd, J = 13.3, 5.5, 1H), 1.66 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 172.4, 171.4, 169.6, 135.3, 132.3, 128.8, 125.8, 124.1, 122.3, 120.7, 116.7, 115.1, 83.4, 67.2, 66.6, 65.9, 54.6, 49.3, 43.6, 34.2, 30.6, 29.1, 28.5, 28.4, 24.9, 13.8; exact mass calcd for C32H35N3O6 [M + Na]+ 580.2418, found 580.2405.
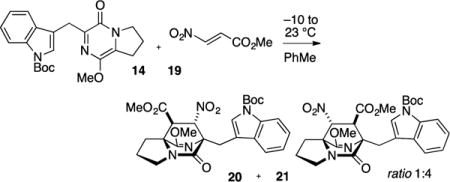
Methyl (6S*,7S*,8S*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-7-nitro-5-oxo-2,3,5,6,7,8-hexa-hydro-1H-6,8a-(azenometheno)indolizine-8-carboxylate (20) and Methyl (6S*,7R*,8R*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-8-nitro-5-oxo-2,3,5,6,7,8-hexahydro-1H-6,8a-(azenometheno)indolizine-7-carboxylate (21)
A dry flask was charged with pyrazinone 14 (531 mg, 1.34 mmol) and flushed with N2 gas for 30 min and dissolved in toluene (7 mL). A separate flask was charged with nitroacrylate 19 (158 mg, 1.21 mmol) and flushed with N2 gas for 30 min and dissolved in toluene (2 mL). The reaction flask containing pyrazinone 14 was cooled to −10 °C (ice/salt bath). The nitroacrylate solution was dropwise to the reaction vessel over 10 min. The reaction was allowed to stir and slowly warm to rt overnight (16 h). The solution was concentrated in vacuo to afford a brown oil, which contained the two regioisomeric cycloadducts 20 and 21 in a 1:4 ratio (as judged by 1H NMR on the unpurified reaction mixture). The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 10% → 80% EtOAc in hexanes). A small amount of pure 21 (40 mg) was obtained for analytical purposes and the bulk of the material was obtained as a yellow oil that was comprised of a 1:4 mixture of 20 and 21 (593 mg, 84% yield): TLC (80% EtOAc in hexanes) Rf 0.30, 0.35 (CAM/UV). IR (film) 3052, 2980, 2950, 1733, 1704, 1641, 1608, 1559, 1477, 1452, 1410, 1370, 1309, 1219, 1197, 1158, 1086, 1016, 979, 937, 897, 857, 811, 768, 747, 703, 656 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 5.9 Hz, 1H), 7.76 (d, J = 1.6, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.29 (dt, J = 8.2, 1.2 Hz, 1H) 7.21 (dt, J = 7.8, 0.8 Hz, 1H), 5.05 (d, J = 4.3 Hz, 1H), 3.77 (s, 3H) 3.76 (s, 3H), 3.62–3.52 (m, 3H), 3.34 (d, J = 4.3 Hz, 1H), 3.30 (d, J = 15.7 Hz, 1H), 2.72–2.66 (m, 1H), 2.25–2.00 (m, 3H), 1.68 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 169.6, 169.1, 168.7, 149.7, 135.1, 132.0, 125.5, 124.0, 121.9, 120.5, 115.3, 114.9, 88.9, 83.3, 68.6, 65.3, 55.4, 53.1, 51.0, 44.0, 28.2, 27.2, 26.8, 24.3; exact mass calcd for C26H30N4O8Na [M + Na]+ 549.1953, found 549.1956.
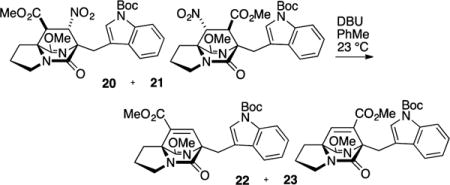
Methyl (6S*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-5-oxo-2,3,5,6-tetrahydro-1H-6,8a-(azenometheno)indolizine-8-carboxylate (22) and Methyl (6S*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-5-oxo-2,3,5,6-tetrahydro-1H-6,8a-(azenometheno)indolizine-7-carboxylate (23)
A flask was charged with cycloadducts 20 and 21 (ratio 1:4, 588 mg, 1.12 mmol), flushed with N2 gas, and dissolved in PhMe (18 mL). To the reaction vessel was added DBU (277 μL, 1.86 mmol), and the reaction mixture was stirred for 16 h at rt, diluted with HCl (0.1 M, 10 mL), and extracted with EtOAc (3 × 15 mL). The combined organic portions were washed with saturated NaHCO3 (20 mL) and saturated NaCl (20 mL), dried (Na2SO4), filtered, and concentrated in vacuo to give the α,β-unsaturated methyl ester regioisomers 22 and 23 in a 1:4 ratio as judged by 1H NMR on the unpurified reaction mixture. The residue was purified by flash column chromatography on silica gel (gradient elution: 20% → 80% EtOAc in hexanes) to afford the minor regioisomer 22 as a light yellow oil (62.1 mg, 12% yield) and 23 as a pale yellow powder (376 mg, 70% yield). Spectroscopic data reported for 22: TLC (40% EtOAc in hexanes) Rf 0.45 (CAM/UV); IR (film) 2890, 2884, 1721, 1688, 1634, 1607, 1451, 1369, 1339, 1327, 1308, 1267, 1256, 1244, 1157, 1086, 1044, 1003, 984, 910, 856, 746, 732, 665 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.0 Hz, 1H), 7.83 (d, J = 7.5 Hz, 1H), 7.68 (s, 1H), 7.50 (s, 1H) 7.33–7.29 (m, 1H), 7.27–7.23 (m, 1H), 3.80 (s, 3H), 3.73 (d, J = 12.1 Hz, 3H), 3.59 (dd, J = 15.3, 1.2 Hz, 2H), 3.49–3.44 (m, 1H) 3.19–3.12 (m, 1H) 2.87–2.76 (m, 2H) 2.14–2.04 (m, 1H), 1.98–1.87 (m, 1H) 1.66 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 175.4, 169.6, 162.8, 152.8, 149.8, 138.2, 135.1, 131.7, 124.9, 124.1, 122.3, 120.0, 116.1, 115.0, 83.4, 74.0, 69.4, 56.3, 51.8, 43.0, 28.2, 26.7, 25.6, 25.4; exact mass calcd for C26H29N3O6 [M + Na]+ 502.1949, found 502.1944.
Spectroscopic data reported for α,β-unsaturated methyl ester 23: mp 94.0–95.0 °C; TLC (40% EtOAc in hexanes) Rf 0.28 (CAM/UV); IR (film) 2980, 2949, 2882, 1722, 1690, 1640, 1607, 1451, 1370, 1339, 1304, 1256, 1221, 1155, 1080, 1017, 995, 978, 930, 907, 856, 835, 764, 739, 619 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.81 (dd, J = 7.1, 1.3 Hz, 1H), 7.62 (s, 1H), 7.28- 7.20 (m, 2H), 7.13 (s, 1H), 4.07 (dd, J = 15.6, 0.8 Hz, 1H), 3.79 (s, 3H), 3.64 (dd, J = 15.6, 0.8 Hz, 1H), 3.40 (s, 3H), 3.40–3.35 (m, 1H), 3.25–3.19 (m, 1H), 2.74 (m, 1H), 2.25–2.18 (m, 1H), 2.10–2.00 (m, 2H), 1.63 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 174.4, 169.7, 164.6, 150.0, 147.5, 144.4, 135.1, 132.3, 124.5, 124.0, 122.1, 120.7, 117.6, 114.9, 83.1, 75.7, 67.6, 56.3, 52.1, 42.7, 28.4, 26.8, 25.5, 25.3; exact mass calcd for C26H29N3O6 [M + Na]+ 502.1949, found 502.1944.
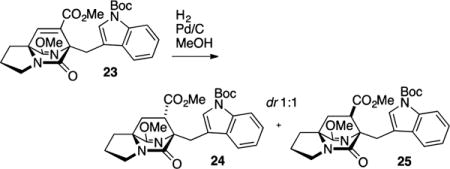
Methyl (6S*,7S*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-9-methoxy-5-oxo-2,3,5,6,7,8-hexahydro-1H-6,8a-(azenometheno)indolizine-7-carboxylate (24) and Methyl (6S*,7R*,8aS*)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)-methyl)-9-methoxy-5-oxo-2,3,5,6,7,8-hexahydro-1H-6,8a-(azenometheno)indolizine-7-carboxylate (25)
Unsaturated ester 23 (182 mg, 0.378 mmol) was dissolved in MeOH (4 mL) at rt, and 10% Pd/C (25 mg) was added. One balloon of H2 gas was used to sparge the reaction mixture (10 min); a second balloon was maintained above the reaction mixture during the course of the reaction to provide an atmosphere of H2. When TLC indicated consumption of starting material (1 h), the reaction mixture was filtered through Celite, and the filter pad was washed with MeOH. After removal of the solvent in vacuo, a white powder was obtained that contained an equal mixture of diastereomers as judged by 1H NMR on the unpurified reaction mixture. The mixture was purified by flash chromatography on silica gel (20% to 100% EtOAc in hexanes) to afford 24 (77 mg, 43% yield) and 25 (78 mg, 43% yield), both obtained as a colorless amorphous solids. Spectroscopic data for 24: TLC (60% EtOAC in hexanes) Rf 0.36 (CAM/UV); IR (film) 2980, 2948, 2883, 1732, 1730, 1695, 1687, 1641, 1476, 1457, 1452, 1416, 1371, 1360, 1308, 1257, 1213, 1185, 1161, 1084, 1016, 913, 857, 768, 745, 701, 672 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 5.8 Hz, 1H), 7.76 (s, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.24–7.19 (m, 2H), 3.78 (s, 3H), 3.54 (d, J = 14.8 Hz, 1H), 3.47 (s, 3H), 3.44–3.31 (m, 2H), 3.22 (d, J = 14.9 Hz, 1H), 3.06 (q, J = 4.7 Hz, 1H), 2.65–2.58 (m, 1H), 2.09–1.87 (m, 5H), 1.63 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 172.4, 171.0, 169.9, 149.9, 134.9, 132.3, 125.7, 123.6, 121.9, 120.3, 116.7, 114.8, 82.8, 69.3, 64.1, 54.7, 51.8, 45.8, 43.4, 37.3, 28.6, 28.2, 27.5, 24.4; exact mass calcd for C26H31N3O6Na [M + Na]+ 504.2105, found 504.2102.
Spectroscopic data for 25: TLC (60% EtOAC in hexanes) Rf 0.43, (CAM/UV); IR (film) 2979, 2948, 2881, 1731, 1693, 1690, 1689, 1452, 1437, 1419, 1370, 1343, 1301, 1258, 1218, 1162, 1120, 1084, 1045, 1013, 990, 856, 767, 747, 666 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.09 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.68 (s, 1H), 7.25 (t, J = 6.2 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 3.75 (s, 1H), 3.71 (s, 3H), 3.68 (s, 3H), 3.29 (d, J = 15.3 Hz, 1H), 2.76 (q, J = 5.1 Hz, 1H), 2.60–2.54 (m, 1H), 2.08–1.83 (m, 5H), 1.66 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 172.9, 172.4, 169.8, 149.9, 135.2, 132.3, 125.5, 123.8, 121.8, 121.1, 116.5, 114.8, 83.1, 68.9, 63.3, 54.7, 52.1, 45.2, 43.5, 36.3, 28.7, 28.2, 27.2, 24.6; exact mass calcd for C26H31N3O6Na [M + Na]+ 504.2105, found 504.2102.
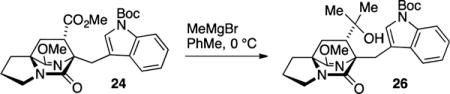
tert-Butyl 3-(((6S*,7S*,8aS*)-7-(2-Hydroxypropan-2-yl)-9-methoxy-5-oxo-2,3,7,8-tetrahydro-1H-6,8a-(azenometheno)-indolizin-6(5H)-yl)methyl)-1H-indole-1-carboxylate (26)
A flame-dried flask was charged with 24 (12.5 mg, 0.026 mmol) and flushed with N2 gas for 10 min, dissolved in toluene (0.52 mL), and cooled to 0 °C. To this solution was added MeMgBr (74 μL, 0.104 mmol, 1.4 M in THF). The reaction was stirred for 1 h at 0 °C, diluted with NH4Cl (10 mL), and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated NaCl (40 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The resulting product was purified by flash chromatography on silica gel (50% → 100% EtOAc in hexanes) to afford the derived lactim tertiary alcohol 26 as a white foam (7.4 mg, 59% yield): TLC (80% EtOAc in hexanes) Rf 0.35 (CAM/UV); IR (film) 3406, 2976, 2945, 2880, 1726, 1661, 1451, 1424, 1368, 1354, 1256, 1207, 1155, 1103, 1080, 1011, 955, 924, 924, 856, 820, 743 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.05 (br s, 1H), 7.84–7.82 (m, 1H), 7.69 (s, 1H), 7.24–7.18 (m, 2H), 3.75 (d, J = 14.8 Hz, 1H), 3.69 (s, 3H), 3.51 (d, J = 14.9 Hz, 1H), 3.47–3.41 (m, 1H), 3.28–3.22 (m, 1H), 2.50 (dt, J = 12.9, 7.4 Hz, 1H), 2.35 (dd, J = 9.8, 6.2 Hz, 1H), 2.04 (dd, J = 12.9, 9.8, 1H), 1.97–1.82 (m, 3H), 1.76 (br s, 1H), 1.63 (s, 9H), 1.41 (dd, J = 12.9, 6.2 Hz, 1H), 1.32 (s, 3H), 1.22 (s, 3H); 13C (100 MHz, CDCl3) δ 171.5, 169.8, 150.0, 134.8, 132.6, 125.5, 123.3, 121.7, 120.7, 118.8, 114.7, 82.7, 74.2, 71.7, 63.4, 54.1, 51.9, 43.2, 37.9, 31.2, 28.6, 28.2, 25.9, 24.2; exact mass calcd for C27H35N3O5Na [M + Na]+ 504.2469, found 504.2468.
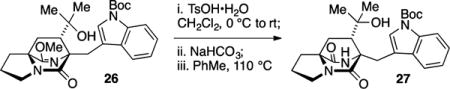
tert-Butyl 3-(((6S,7S,8aS)-7-(2-Hydroxypropan-2-yl)-5,9-diox-otetrahydro-1H-6,8a-(epiminomethano)indolizin-6(5H)-yl)-methyl)-1H-indole-1-carboxylate (27)
A reaction flask was charged with lactim tertiary alcohol 26 (15.8 mg, 0.033 mmol) and flushed with N2 gas for 10 min, dissolved in CH2Cl2 (3.5 mL), and cooled to 0 °C. To the reaction flask was added p-TsOH·H2O (7.3 mg, 0.038 mmol). The reaction was stirred and slowly warmed to ambient temperature over 6 h. The reaction mixture was diluted with satd aq NaHCO3 (10 mL), and extracted with EtOAc (4 × 10 mL). The combined organic layers were washed with saturated NaCl (40 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The resulting product (which contained some amino methyl ester) was dissolved in toluene (3 mL) and stirred at 110 °C for 18 h. The solution was concentrated in vacuo to obtain lactam tertiary alcohol 27 (15.5 mg, 95% yield) which was used without further purification: TLC (80% EtOAc in hexanes) Rf 0.32 (CAM/UV); IR (film) 3393, 2928, 1688, 1682, 1674, 1451, 1367, 1339, 1310, 1258, 1221, 1153, 1085, 1022, 961, 934, 856, 802, 745, 694, 665, 592 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 7.4 Hz, 1H), 7.64 (s, 1H), 7.60 (d, J = 7.1 Hz, 1H), 7.33–7.16 (m, 2H), 5.70 (br s, 1H), 4.01 (d, J = 15.6 Hz, 1H), 3.64–3.52 (m, 2H), 3.40 (dt, J = 11.8, 6.6 Hz, 1H), 2.65 (dt, J = 12.9, 7.4 Hz, 1H), 2.36 (s, 1H), 2.31 (dd, J = 10.2, 6.6 Hz, 1H), 2.13 (dd, J = 13.3, 10.2 Hz, 1H), 1.97 (apparent quint, J = 7.0 Hz, 2H), 1.82–1.74 (m, 2H), 1.66 (s, 9H), 1.45 (s, 3H), 1.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.4, 169.3, 149.6, 135.1, 131.5, 126.8, 124.5, 122.9, 118.0, 115.5, 114.6, 83.7, 73.8, 66.8, 65.8, 50.7, 44.2, 36.2, 32.0, 28.2, 24.1, 23.7, 23.5; exact mass calcd for C26H33N3O5Na [M + Na]+ 490.2312, found 490.2312.
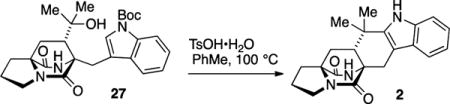
12,12-Dimethyl-2,3,11,12,12a,13-hexahydro-1H,5H,6H-5a,13a-(epiminomethano)indolizino[7,6-b]carbazole-5,14-dione (2)
A reaction flask was charged with lactam tertiary alcohol 27 (11.5 mg, 0.025 mmol), fitted with a reflux condenser, and flushed with N2 gas for 10 min. PhMe (3.7 mL) and p-TsOH·H2O (4.7 mg, 0.025 mmol) were added, and the reaction vessel was heated to 100 °C for 6 h. After being cooled to rt, the reaction was diluted with satd aqueous NaHCO3 (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated NaCl (40 mL), dried (Na2SO4), filtered, and concentrated in vacuo to obtain 2 as a white powder (7.1 mg, 83% yield). The material thus obtained was >95% pure as judged by 1H NMR and further purification was not performed: TLC (100% EtOAc) Rf 0.45 (CAM/UV); IR (film) 3325, 3310, 3233, 3221, 3210, 2990, 2947, 2901, 1656, 1670, 1451, 1404, 1369, 1339, 1300, 1258, 1196, 1161, 1092, 1011, 922, 860, 802, 772, 748, 718, 698, 644, 594; 1H NMR (400 MHz, CDCl3) δ 7.89 (br s, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 7.9 Hz, 1H), 7.18 (td, J = 7.5, 1.3 Hz, 1H), 7.12 (td, J = 7.5, 1.2 Hz, 1H), 5.88 (br s, 1H), 3.93 (d, J = 18 Hz, 1H), 3.54 (t, J = 6.9 Hz, 2H), 2.91 (d, J = 18 Hz, 1H), 2.81 (quint, J = 6.7 Hz, 1H), 2.34 (dd, J = 9.8, 4.3 Hz, 1H), 2.18–1.99 (m, 4H), 1.89–1.82 (m, 1H), 1.33 (s, 3H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.7, 169.0, 139.6, 136.4, 127.2, 122.1, 119.7, 118.3, 110.7, 103.6, 67.1, 61.6, 45.7, 44.2, 34.5, 32.6, 29.1, 29.0, 25.3, 24.5, 23.9. exact mass calcd for spray dimer C42H46N6O4Na [M + M + Na]+ 721.3473, found 721.3476. The spectral data for 2 were identical with an authentic sample provided by Williams (Colorado State University).
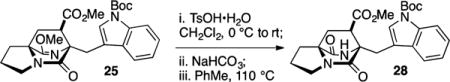
Methyl (6S,7R,8aS)-6-((1-(tert-Butoxycarbonyl)-1H-indol-3-yl)methyl)-5,9-dioxohexahydro-1H-6,8a-(epiminomethano)-indolizine-7-carboxylate (28)
A reaction flask was charged with lactim methyl ester 25 (48 mg, 0.099 mmol) and flushed with N2 gas for 10 min, dissolved in CH2Cl2 (10 mL), and cooled to 0 °C. To the reaction flask was added pTsOH·H2O (24 mg, 0.13 mmol), and the reaction was stirred for 6 h while slowly warming to rt. The reaction mixture was diluted with satd aq NaHCO3 (10 mL) and extracted with EtOAc (4 × 10 mL). The combined organic layers were washed with saturated NaCl (40 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The resulting product (which contained some amino methyl ester) was dissolved in toluene (10 mL) and stirred at 110 °C for 18 h. The solution was concentrated in vacuo to obtain the lactam 28 as a white powder (15.7 mg, 88% yield). The material thus obtained was >95% pure as judged by 1H NMR, and further purification was not performed: TLC (70% EtOAc in hexanes) Rf 0.30 (CAM/UV); IR (film) 3175, 3117, 3082, 3051, 2978, 2936, 2882, 1736, 1724, 1690, 1450, 1385, 1362, 1312, 1254, 1196, 1153, 1092, 1308, 1018, 964, 934, 922, 899, 849, 837, 768, 745, 691, 563; 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.71 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.36 (td, J = 7.6, 1.2 Hz, 1H), 7.29 (td, J = 7.6, 1.2 Hz, 1H), 5.75 (br s, 1H), 3.82 (s, 3H), 3.66–3.53 (m, 3H), 3.30 (dd, J = 9.8, 4.7 Hz, 1H), 3.19 (d, J = 15.6 Hz, 1H), 2.73–2.66 (m, 1H), 2.28- – 2.15 (m, 2H), 2.10–1.98 (m, 2H), 1.85–1.79 (m, 1H), 1.69 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 172.3, 171.6, 166.4, 149.4, 135.4, 130.8, 126.2, 125.0, 123.3, 118.1, 115.7, 112.4, 84.1, 66.1, 63.4, 52.7, 48.2, 44.3, 34.8, 28.9, 28.2, 24.3, 24.0.exact mass calcd for C25H29N3O6Na [M + Na]+ 490.1949, found 490.1948. Spectral data agrees with that reported by Baran.24
Supplementary Material
Acknowledgments
We thank Professor Robert M. Williams (Colorado State University) for authentic samples of 1 and 2. We acknowledge primary support from the National Institutes of Health (R15 GM107702 to J.R.S.). The donors of the ACS Petroleum Research Fund supported early aspects of this work. Support was also provided by the Camille and Henry Dreyfus Foundation (Henry Dreyfus Teacher–Scholar Award to J.R.S.). The NSF MRI provided funds for the acquisition and upgrade of a 400 MHz NMR Spectrometer (Award No. 1337295).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.5b02744.
Spectroscopic data (1H NMR and 13C NMR) for all new compounds as well as known compounds 2 and 28 (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Birch AJ, Wright JJ. J Chem Soc, Chem Commun. 1969:644–645. [Google Scholar]; (b) Birch AJ, Russell RA. Tetrahedron. 1972;28:2999–3008. [Google Scholar]
- 2.Finefield JM, Frisvad JC, Sherman DH, Williams RM. J Nat Prod. 2012;75:812–833. doi: 10.1021/np200954v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Qian-Cutrone J, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. J Am Chem Soc. 2002;124:14556–14557. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]; (b) Martínez-Luis S, Rodríguez R, Acevedo L, González MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817–1822. [Google Scholar]; (c) Lin Z, Wen J, Zhu T, Fang Y, Gu Q, Zhu W. J Antibiot. 2008;61:81–85. doi: 10.1038/ja.2008.114. [DOI] [PubMed] [Google Scholar]
- 4.The syn/anti nomenclature was introduced by Williams. For a more detailed description see ref 2.
- 5.(a) Tsukamoto S, Kawabata T, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. Org Lett. 2009;11:1297–1300. doi: 10.1021/ol900071c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sunderhaus JD, Sherman DH, Williams RM. Isr J Chem. 2011;51:442–452. doi: 10.1002/ijch.201100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai S, Luan Y, Kong X, Zhu T, Gu Q, Li D. Org Lett. 2013;15:2168–2171. doi: 10.1021/ol400694h. [DOI] [PubMed] [Google Scholar]
- 7.The synthesis of [2.2.2]diazabicycles has been reviewed:; Miller KA, Williams RM. Chem Soc Rev. 2009;38:3160–3174. doi: 10.1039/b816705m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Additional strategies not encompassed in the review:; (a) Frebault FC, Simpkins NS. Tetrahedron. 2010;66:6585–6596. [Google Scholar]; (b) Crick PJ, Simpkins NS, Highton A. Org Lett. 2011;13:6472–6475. doi: 10.1021/ol202769f. [DOI] [PubMed] [Google Scholar]; (c) Simpkins N, Pavlakos I, Male L. Chem Commun. 2012;48:1958–1960. doi: 10.1039/c1cc16510k. [DOI] [PubMed] [Google Scholar]; (d) Laws SW, Scheerer JR. J Org Chem. 2013;78:2422–2429. doi: 10.1021/jo3026059. [DOI] [PubMed] [Google Scholar]; (e) Simpkins NS, Pavlakos I, Weller MD, Male L. Org Biomol Chem. 2013;11:4957–4970. doi: 10.1039/c3ob40979a. [DOI] [PubMed] [Google Scholar]; (f) Mercado-Marin EV, Sarpong R. Chem Sci. 2015;6:5048–5052. doi: 10.1039/c5sc01977j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Amatov T, Pohl R, Cisarova I, Jahn U. Angew Chem, Int Ed. 2015;54:12153–12157. doi: 10.1002/anie.201504883. [DOI] [PubMed] [Google Scholar]
- 9.Ding Y, Greshock TJ, Miller KA, Sherman DH, Williams RM. Org Lett. 2008;10:4863–4866. doi: 10.1021/ol8019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Adams LA, Valente MWN, Williams RM. Tetrahedron. 2006;62:5195–5200. [Google Scholar]; (b) Williams RM, Glinka T, Kwast E. J Am Chem Soc. 1988;110:5927–5929. [Google Scholar]; (c) Williams RM, Glinka T, Kwast E, Coffman H, Stille JK. J Am Chem Soc. 1990;112:808–821. [Google Scholar]
- 11.(a) Miller KA, Tsukamoto S, Williams RM. Nat Chem. 2009;1:63–68. doi: 10.1038/nchem.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Finefield JM, Kato H, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM. Org Lett. 2011;13:3802–3805. doi: 10.1021/ol201284y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Williams RM. J Org Chem. 2011;76:4221–4259. doi: 10.1021/jo2003693. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Williams RM, Cox RJ. Acc Chem Res. 2003;36:127–139. doi: 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]
- 12.Sanz-Cervera JF, Williams RM. J Am Chem Soc. 2002;124:2556–2559. doi: 10.1021/ja017425l. [DOI] [PubMed] [Google Scholar]
- 13.Sprague DJ, Nugent BM, Yoder RA, Vara BA, Johnston JN. Org Lett. 2015;17:880–883. doi: 10.1021/ol503626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margrey KA, Chinn AJ, Laws SW, Pike RD, Scheerer JR. Org Lett. 2012;14:2458–2461. doi: 10.1021/ol3007056. [DOI] [PubMed] [Google Scholar]
- 15.DKP 12 was prepared from proline methyl ester in three steps (1 chromatographic separation) and 80% overall yield. See ref 8d.
- 16.Pyrazinone structures undergo cycloaddition with molecular oxygen (without added sensitizer):; (a) Machin PJ, Porter AEA, Sammes PG. J Chem Soc, Perkin Trans. 1973;1:404–409. [Google Scholar]; (b) Markham JL, Sammes PG. J Chem Soc, Perkin Trans. 1979;1:1885–1888. [Google Scholar]
- 17.Isomeric ratio determined by 1H NMR on the unpurified reaction mixture.
- 18.Acryloyl camphor sultam was prepared as described in:; Thom C, Kocienski P. Synthesis. 1992;1992:582–586. [Google Scholar]
- 19.(a) Oppolzer W, Chapuis C, Bernardinelli G. Helv Chim Acta. 1984;67:1397–1401. [Google Scholar]; (b) Oppolzer W. Pure Appl Chem. 1990;62:1241–1250. [Google Scholar]
- 20.The esterification was accomplished using reaction conditions similar to those initially reported by Evans for removal of oxazolidinone auxiliaries:; Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1988;110:1238–1256. [Google Scholar]
- 21.Danishefsky S, Prisbylla MP, Hiner S. J Am Chem Soc. 1978;100:2918–2920. [Google Scholar]
- 22.(a) Williams RM, Kwast E. Tetrahedron Lett. 1989;30:451–454. [Google Scholar]; (b) Greshock TJ, Williams RM. Org Lett. 2007;9:4255–4258. doi: 10.1021/ol701845t. [DOI] [PubMed] [Google Scholar]
- 23.Hydrolysis of the lactim ether affords an intermediate ammonium methyl ester. On basification of the medium, cyclization re-establishes the [2.2.2]diazabicyclic bridge and reveals the lactam functionality.
- 24.(a) Baran PS, Hafensteiner BD, Ambhaikar NB, Guerrero CA, Gallagher JD. J Am Chem Soc. 2006;128:8678–8693. doi: 10.1021/ja061660s. [DOI] [PubMed] [Google Scholar]; (b) Baran PS, Guerrero CA, Ambhaikar NB, Hafensteiner BD. Angew Chem, Int Ed. 2005;44:606–609. doi: 10.1002/anie.200461864. [DOI] [PubMed] [Google Scholar]
- 25.(a) Clive DLJ, Bo Y, Selvakumar N, McDonald R, Santarsiero BD. Tetrahedron. 1999;55:3277–3290. [Google Scholar]; (b) Calmès M, Escale F, Didierjean C, Cazals G, Martinez J. Tetrahedron: Asymmetry. 2007;18:2491–2496. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.