

Abstract
Purpose
Mutations in the estrogen receptor-alpha (ER) gene, ESR1, have been identified in breast cancer metastases after progression on endocrine therapies. Due to limitations of metastatic biopsies, the reported frequency of ESR1 mutations may be underestimated. Here, we show a high frequency of ESR1 mutations using circulating plasma tumor DNA (ptDNA) from metastatic breast cancer patients.
Experimental Design
We retrospectively obtained plasma samples from eight patients with known ESR1 mutations and three patients with wild type ESR1 identified by next generation sequencing (NGS) of biopsied metastatic tissues. Three common ESR1 mutations were queried for using droplet digital polymerase chain reaction (ddPCR). In a prospective cohort, metastatic tissue and plasma were collected contemporaneously from eight ER-positive and four ER-negative patients. Tissue biopsies were sequenced by NGS and ptDNA ESR1 mutations were analyzed by ddPCR.
Results
In the retrospective cohort, all corresponding mutations were detected in ptDNA, with two patients harboring additional ESR1 mutations not present in their metastatic tissues. In the prospective cohort, three ER-positive patients did not have adequate tissue for NGS, and no ESR1 mutations were identified in tissue biopsies from the other nine patients. In contrast, ddPCR detected seven ptDNA ESR1 mutations in six of twelve patients (50%).
Conclusions
We show that ESR1 mutations can occur at a high frequency and suggest that blood can be used to identify additional mutations not found by sequencing of a single metastatic lesion.
Keywords: ESR1, mutation, estrogen receptor, breast cancer, circulating tumor DNA, plasma
Introduction
Estrogen receptor-alpha (ER) is a part of the nuclear hormone receptor family and is expressed in ∼70% of breast cancers (1). Drugs that target ER and estrogen production have become effective standard of care therapies (2). Notably, selective estrogen receptor modulators (SERMs), selective estrogen receptor down regulators (SERDs) and aromatase inhibitors (AIs) have significantly improved overall survival of ER-positive breast cancer patients (3). Nevertheless, de novo and acquired resistance may arise after prolonged exposure to these therapies (4). Recently, next generation sequencing (NGS) studies of metastatic ER-positive breast cancer patients have revealed genetic alterations that may account for acquired resistance to endocrine therapy (5-9). These studies collectively report mutations in the ligand binding domain (LBD) of ESR1 in approximately 20% of these patients, and presumably these mutations act as a driver of endocrine therapy resistance. Interestingly, these mutations were predicted in mutagenesis models and identified in patient xenograft studies reported almost two decades ago (10, 11). Molecular modeling and preclinical studies characterizing ESR1 LBD mutations reveal a conformational change that leads to constitutive activation of ER signaling in the absence of ligand (6-8). However, these studies also suggest that cells with ESR1 LBD mutations may still be sensitive to SERM and SERD therapy, albeit at higher doses compared to cells with wild type ESR1 (7, 8). The identification of ESR1 mutations that are responsible for endocrine therapy resistance in ER-positive breast cancers opens the door for developing new diagnostic tools and novel targeted therapies. However, given the problem of tumor heterogeneity, the true frequency of ESR1 mutations may be underestimated, since mutational profiles can vary between different sites of metastatic disease (12). Most studies heretofore have employed NGS of a single metastatic site, and indeed, one study demonstrated an ESR1 mutation in a liver metastatic biopsy but not a lung metastasis obtained from the same patient (6). Further, in many cases, fresh biopsies of metastatic disease cannot be safely obtained and/or archival tissues are inadequate or unavailable. Finally, these mutations appear to evolve during endocrine treatment, and therefore a non-invasive method of monitoring patients might provide an opportunity to alter therapy as these mutations emerge. Thus, there is need to develop non-invasive methods to quickly assess mutational profiles across multiple metastases from an individual patient.
Recently, we and others have examined the use of circulating cell-free plasma tumor DNA (ptDNA) as a biomarker for cancer detection (13-20). It is known that DNA molecules from both normal and cancer cells are shed or released into the circulation (21, 22). Because DNA from cancer cells harbor somatic mutations and rearrangements, these can serve as specific genetic biomarkers for the presence of cancer. Further, the quantity of ptDNA directly correlates with tumor burden and response to therapies (23). Additionally, several groups have demonstrated the ability to detect the presence of acquired drug resistance mutations in ptDNA (24, 25), which opens the possibility for earlier therapeutic intervention. More recently, our group has shown that a next generation digital PCR platform, termed droplet digital PCR (ddPCR) has exquisite sensitivity and specificity for detecting cancer mutations in early stage breast cancer patients (19). We hypothesized that ddPCR could be a more sensitive platform for ESR1 mutation detection in metastatic breast cancer patients and may show a more accurate frequency of these mutations in ER-positive disease. To test this hypothesis, we performed ddPCR for ESR1 mutations on cell-free plasma samples from patients with metastatic breast cancer and compared ESR1 mutations in ptDNA with NGS of metastatic tumor tissue from the same patients.
Materials and Methods
Patient and sample collection
We conducted this clinical study at the University of Michigan Comprehensive Cancer Center (UMCCC) and the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center (JHSKCCC). Men and women with metastatic (stage IV) breast cancer were eligible. All patients signed informed consent. For the UMCCC cohort, patients were recruited from breast cancer patients undergoing a research tumor biopsy of metastatic disease for whole exome sequencing through UMCCC's MiONCOSEQ program (7, 26). In particular, these patients were recruited in a companion trial to MiONCOSEQ, designated MiCTC-ONCOSEQ approved by the University of Michigan Health System IRB. Under this protocol, any metastatic breast cancer patients previously enrolled or enrolling in the parent MiONCOSEQ protocol were asked to provide blood samples for ptDNA collected in BCT DNA tubes (Streck) and CTC analyses (data not reported in this publication). For the JHSKCCC cohort, patients were consented and enrolled in an ongoing longitudinal tissue and blood repository protocol, allowing for research use of human tissues and bodily fluids from patients with breast disease. An IRB subprotocol approved for genomic analyses of tumor tissues and blood from breast cancer patients of any stage was used to obtain metastatic tumor biopsies and subsequent blood samples from ER-positive metastatic patients. Metastatic tumor samples obtained as FFPE blocks and slides were sent for NGS DNA analysis using a commercial source (Foundation Medicine). In this cohort, blood samples of 30ml were collected in EDTA tubes or BCT DNA tubes after patients with ESR1 mutations were identified. Prospective enrollment is also allowed for this protocol.
Isolation and Quantification of ptDNA for ddPCR
Blood samples and plasma DNA preparation were performed as previously described (19). Briefly, plasma was obtained by a double spin centrifuge protocol of whole blood to remove cellular contaminants and DNA extracted using the Qiagen Circulating Nucleic Acid kit (Qiagen) per the manufacturer's protocol. Blood was centrifuged within 1 hour if collected in EDTA tubes and within 7 days if collected in DNA BCT tubes (Streck). The Bio-Rad QX200 platform was then used for ddPCR per the manufacturer's protocol, with results reported as a percentage or fractional abundance of mutant DNA alleles to total (mutant plus wild type) DNA alleles as previously described (19). Further details are provided in Supplemental Methods.
Statistical Analysis
In order to quantify the percent ptDNA containing mutant ESR1 in plasma samples, a fractional abundance calculation using the QuantaSoft program (Bio-Rad Technologies) was employed, using the total number of droplets (with and without DNA) to calculate the number of DNA molecules as copies/μl, and then dividing the number of mutant DNA molecules by the number of total DNA molecules (mutant plus wild type), multiplied by 100 to yield a percentage of mutant DNA molecules in a sample taking into account a Poisson distribution of occupied to unoccupied droplets. For cohort 2, Fisher's exact two-tailed test (GraphPad) was used to calculate differences in ESR1 mutation status (mutant vs. wild type) between tissue and blood using a 2×2 contingency table with 9 samples for tissue and 12 samples for blood.
Results
We enrolled a total of 23 patients in two separate cohorts (Figure 1) from UMCCC and JHSKCCC. Systemic endocrine therapies are shown (Table 1), though many patients also received prior chemotherapies. To determine if we could identify circulating ESR1 mutations in patients with known tissue ESR1 status, we initially performed a retrospective analysis by obtaining plasma samples from eleven patients who had previously undergone NGS of a metastatic lesion (Table 2). Plasma DNA was obtained from these patients less than 1 year after their tissue biopsy (median = 145 days, range 54 to 344 days). Eight of these patients (patients 1-8) had ESR1 mutations identified via NGS in their metastatic biopsies, and three patients (patients 9-11) had wild type ESR1. Patients 1- 3 had previously been reported as having an ESR1 tissue mutation (7) identified via the MiONCOSEQ program at UMCCC (26), while patients 4-8 had ESR1 mutations identified at JHSKCCC using a commercial platform (Foundation Medicine). All patients had documented ER-positive disease, and NGS was performed on samples representing diverse metastatic sites (Table 1). The plasma specimens were interrogated for all three ESR1 hotspot mutations: Y537S, Y537N and D538G, using mutation specific probes and ddPCR as previously reported (19). These three mutations were chosen as they collectively represent the most frequent ESR1 mutations in metastatic disease (9). As demonstrated in Figure S1, each probe was specific for its respective mutation using Y537S, Y537N and D538G mutant and wild type templates. As shown in Table 2, ddPCR successfully detected all mutations in ptDNA that were detected in the metastatic biopsy, confirming the ability to detect mutations present within the tumor sample. The majority of patients had significant tumor burden with multiple metastatic sites of disease (Table S1), though patient 5 had no evidence of disease after removal of her metastatic lesion. Indeed, although she did have a circulating ESR1 mutation (D538G), it was detected at a relatively low fractional abundance (0.03%) in her plasma.
Figure 1.
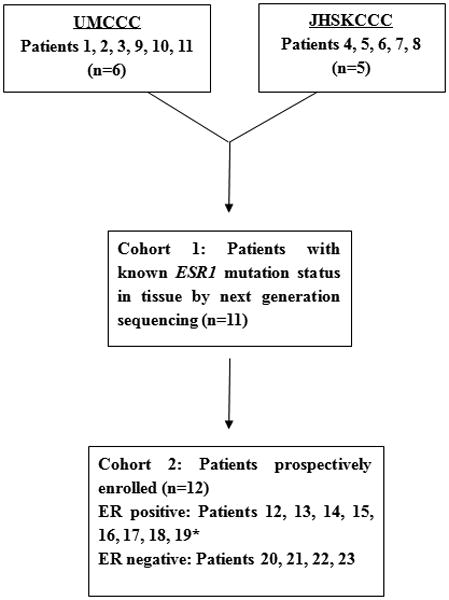
Patient enrollment and distribution. Cohort 1: Patients were enrolled retrospectively after next generation sequencing of a metastatic biopsy at UMCCC and JHSKCCC (cohort 1) confirming the presence of an ESR1 mutation (patients 1, 2, 3, 4, 5, 6, 7, 8, 9), or wild type ESR1 (patients 10, 11, 12), with blood obtained after tissue sequencing was performed for ddPCR. Cohort 2: Patients were enrolled prospectively, with tissue and blood obtained contemporaneously. Tissue was subjected to next generation sequencing and blood was analyzed by ddPCR, with sequencing results blinded to ddPCR investigators. *All cohort 2 patients were enrolled at UMCCC except patient 19.
Table 1. Patient characteristics.
| Patient | Age at study entry (median = 58) | Site of tissue biopsy | Primary ER/PR/HER2 | Metastatic ER/PR/HER2 | Treatment |
|---|---|---|---|---|---|
| Cohort 1 | |||||
| 1 | 58 | Peritoneal fluid | +/+/- | +/+/- | Tamoxifen × 5 years (adjuvant), letrozole (metastatic), tamoxifen (metastatic), fulvestrant, exemestane +everolimus |
| 2 | 45 | Liver | +/+/- | +/+/- | Tamoxifen × 4 years (adjuvant), anastrozole (metastatic), fulvestrant, estrace, exemestane |
| 3 | 60 | Liver | +/+/- | +/+/+ | Tamoxifen × 5 years (adjuvant), letrozole (metastatic) |
| 4 | 56 | Liver | NA | +/+/- | Tamoxifen and zolendronic acid × 4 years, anastrozole × 6 weeks, fulvestrant × l year, exemestane × 2 months |
| 5 | 37 | Liver | +/-/- | +/-/- | Tamoxifen × 2 year (adjuvant), letrozole (+sorafenib on trial) × 4 months, anastrozole × lyear, fulvestrant × 4months |
| 6 | 63 | Brain | +/+/NA | +/+/- | Tamoxifen × l year, letrozole × l year, fulvestrant × 6 months |
| 7 | 70 | Liver | +/+/NA | +/+/- | Tamoxifen (adjuvant), letrozole, exemestane + everolimus, fulvestrant (many years) |
| 8 | 65 | Liver | +/+/NA | +/+/- | Tamoxifen × 5 years (adjuvant), letrozole × 5 years (adjuvant), anastrozole × 1.5 years, fulvestrant × 8 months |
| 9 | 46 | Sternal mass | +/+/- | +/-/- | Tamoxifen (adjuvant), anastrozole (adjuvant), anastrozole (metastatic), fulvestrant + anastrozole (metastatic) |
| 10 | 67 | Skinand subcutaneous tissue | +/+/- | +/+/- | Tamoxifen × 5 years (adjuvant), exemestane, fulvestrant (metastatic) |
| 11 | 58 | Bone | +/+/- | +/-/- | Tamoxifen × 2years (adjuvant), anastrozole (adjuvant), fulvestrant (metastatic) |
| Cohort 2 | |||||
| 12 | 61 | Unable to get tissue for analysis | +/NA/NA | +/+/- | Tamoxifen × 3.5 years (adjuvant), letrozole 1.5 years (adjuvant), anastrozole (adjuvant), fulvestrant + anastrozole (metastatic) |
| 13 | 63 | Periaortic lymp node | +/+/- | +/+/- | Tamoxifen × 3years (adjuvant), exemestane 5 years (adjuvant), anastrozole (metastatic), anastrozole + fulvestrant, exemestane |
| 14 | 48 | Pleural fluid | +/+/+ | NA | Tamoxifen (metastatic), fulvestrant + leuprolide, fulvestrant + anastrozole + euprolide |
| 15 | 56 | Right axillary lymph node | +/-/- | +/NA/- | Letrozole + goserelin (neoadjuvant), letrozole + goserelin (adjuvant), fulvestrant (metastatic), fulvestrant + letrozole |
| 16 | 77 | Liver | +/+/+ | +/-/- | Tamoxifen × 5 years, exemestane (adjuvant), anastrozole (adjuvant) |
| 17 | 63 | Axillary lymph node | +/-/- | NA | Anastrozole (metastatic), fulvestrant, tamoxifen, exemestane + everolimus |
| 18 | 65 | Subcutaneous chest wall nodule | +/-/- | +/-/- | Anastrozole (adjuvant), tamoxifen (adjuvant), fulvestrant (metastatic) |
| 19 | 65 | Unable to get tissue for analysis | +/+/- | NA | Letrozole × 5 years (metastatic), fulvestrant |
| 20 | 63 | Skin | -/-/+ | -/-/+ | None |
| 21 | 41 | Lung, RLL | -/-/+ | -/-/+ | None |
| 22 | 49 | Right anterior chest wall | -/-/- | -/-/- | None |
| 23 | 57 | Liver | -/-/- | -/-/- | None |
NA = not available
Table 2. Cohort 1: ESR1 mutations in metastatic tissues are present in ptDNA from blood within 1 year of biopsy.
Tumor samples from 11 patients with ER-positive, metastatic breast cancer were analyzed for ESR1 mutations using next generation sequencing (Sequencing FFPE tumor tissue) and additionally, blood samples were analyzed by ddPCR for ESR1 Y537S, Y537N and D538G mutations. Percentage reflects the fractional abundance of mutant ESR1 (Y537S, Y537N or D538G) to total ESR1 DNA.
| Patient | Days between tissue biopsy and blood draw | Sequencing FFPE tumor tissue | ddPCR plasma for ESR1 Y537S | ddPCR plasma for ESR1 Y537N | ddPCR plasma for ESR1 D538G |
|---|---|---|---|---|---|
| 1 | 186 | ESR1 Y537S | Y537S (0.87%) | wild type | D538G (0.01%) |
| 2 | 344 | ESR1 Y537S | Y537S (1.69%) | wild type | wild type |
| 3 | 275 | ESR1 D538G | wild type | wild type | D538G (1.55%) |
| 4 | 68 | ESR1 Y537S | Y537S (0.63%) | wild type | wild type |
| 5 | 64 | ESR1 D538G | wild type | wild type | D538G (0.03%) |
| 6 | 165 | ESR1 D538G | wild type | wild type | D538G (4.23%) |
| 7 | 88 | ESR1 D538G | wild type | wild type | D538G (0.01%) |
| 8 | 60 | ESR1 Y537N | wild type | Y537N (0.68%) | wild type |
| 9 | 54 | wild type | wild type | wild type | D538G (0.01%) |
| 10 | 145 | wild type | wild type | wild type | wild type |
| 11 | 270 | wild type | wild type | wild type | wild type |
In addition to harboring the known tissue mutation (Y537S in her circulation), patient 1 also had a low fractional abundance (0.01%) of a second circulating mutation, D538G, which was not detected in the metastatic tissue. It should be noted, however, that her blood was drawn 186 days after biopsy, and thus a new subclonal population within the same metastatic site could have been present at the time of blood draw. Similarly, patient 9 was wild type for ESR1 in the metastatic lesion but showed a D538G mutation at a relatively low fractional abundance in a plasma sample obtained 54 days after biopsy. These results suggest that ddPCR of ptDNA can reliably detect ESR1 mutations first identified in metastatic tissues and may also detect subclonal populations in the metastatic biopsy below the limit of detection by NGS, or mutations from other sites of disease.
The presence of two additional mutations in patients 1 and 9 may have been due to clonal evolution in the interim between tissue biopsy and blood draw for ptDNA analysis. To address this possibility, we prospectively enrolled eight additional ER-positive patients (patients 12-19) to simultaneously collect metastatic tissue biopsies and blood for NGS and ddPCR analysis, respectively. As controls, we also obtained metastatic tissue and blood samples from four ER-negative patients (patients 20-23). All patients were enrolled at UMCCC except patient 19 who was enrolled at JHSKCCC. As shown in Table 3, sufficient tissue could not be obtained for patients 12 and 19, while patient 14 did not have adequate sample for NGS analysis. These patients highlight the fact that metastatic biopsies are not always obtainable and that the amount of tissue can preclude genomic analysis.
Table 3. Cohort 2: ESR1 mutations are present in ptDNA in patients with wild type ESR1 metastatic biopsies when obtained contemporaneously.
Tumor samples from 8 and 4 patients with ER-positive and ER-negative metastatic breast cancer, respectively, were analyzed for ESR1 mutations using next generation sequencing (Sequencing FFPE tumor tissue) and additionally, blood samples were analyzed by ddPCR for ESR1 Y537S, Y537N and D538G mutations. Percentage reflects the fractional abundance of mutant ESR1 (Y537S, Y537N or D538G) to total ESR1 DNA. n/a = not available
| Patient | Days between tissue biopsy and blood draw | Sequencing FFPE tumor tissue | ddPCR plasma for ESR1 Y537S | ddPCR plasma for ESR1 Y537N | ddPCR plasma for ESR1 D538G |
|---|---|---|---|---|---|
| ER-positive | |||||
| 12 | - | n/a | Y537S (0.47%) | wild type | wild type |
| 13 | 0 | wild type | wild type | wild type | D538G (0.01%) |
| 14 | 0 | n/a | Y537S (5.02%) | wild type | D538G (2.62%) |
| 15 | 0 | wild type | wild type | wild type | wild type |
| 16 | 0 | wild type | wild type | wild type | D538G (0.01%) |
| 17 | 0 | wild type | wild type | wild type | wild type |
| 18 | 0 | wild type | wild type | wild type | D538G (0.01%) |
| 19 | - | n/a | wild type | Y537N (0.06%) | wild type |
| ER-negative | |||||
| 20 | 5 | wild type | wild type | wild type | wild type |
| 21 | 3 | wild type | wild type | wild type | wild type |
| 22 | 0 | wild type | wild type | wild type | wild type |
| 23 | 0 | wild type | wild type | wild type | wild type |
After plasma DNA extraction, ddPCR analysis was performed in a blinded fashion. As seen in Table 3, all patients had blood drawn at the time of tissue biopsy, except two patients (patients 20 and 21) had blood drawn 5 and 3 days after biopsy, respectively, for logistical reasons. Of the five ER positive patients for whom tissue NGS results could be obtained, no ESR1 mutations were identified in their metastatic biopsies. However, ESR1 mutations were detected in the ptDNA samples from three of these patients (patients 13, 16, and 18), all of whom had their blood drawn the same day as biopsy. Of note, patient 16 was a known germline BRCA2 mutation carrier and may have had a primary peritoneal (ovarian) carcinoma concurrent with her liver metastases, thus obfuscating the origin of the liver lesion. As expected, the ER-negative patients (patients 20-23) did not have detectable ESR1 mutations in their ptDNA. The difference in mutational status was statistically significant between tissue and blood using two-tailed Fisher's exact test (p<0.0186).
Interestingly, patient 14, who was ER-positive, had a high fractional abundance of two distinct circulating ESR1 mutations (Y537S, 5.02%; D538G, 2.62%). Her only metastatic site amenable to biopsy was a pleural effusion, which was inadequate for NGS. The ptDNA from this patient collected concurrently at the time of biopsy contained two distinct mutations at differing allelic frequencies, suggestive of two separate clonal populations. This was similar to patient 1, and suggestive that the mutations were on separate alleles. To prove this, we developed a dual mutation specific probe and positive control template. As shown in Figure S2, this probe has specificity for a synthetic allele harboring both mutations. Analysis of ptDNA from patients 1 and 14 using this probe showed no positive signals, demonstrating that the two ESR1 mutations are on separate alleles, further supporting that these ESR1 mutations are derived from different clonal populations.
An additional noteworthy case is patient 19, who presented at the time of diagnosis with wide spread, bone only ER-positive metastatic disease. She initiated treatment with the AI letrozole, and after 1 year of therapy restaging scans showed disease stabilization of her bony metastasis, and complete resolution of her breast tumor. She elected to have bilateral mastectomies, which revealed that the affected breast and the contralateral breast had no evidence of disease. She remained on letrozole for 5 years with stable disease. She enrolled in our study while still in remission, although restaging scans continued to demonstrate only prior bony lesions, which were not amenable to biopsy. Nonetheless, her plasma demonstrated the presence of the Y537N mutation. Because of her unusual presentation, this is the only patient in our cohort that had developed an ESR1 mutation after exposure to a single endocrine therapy, letrozole. Subsequently, she had an asymptomatic elevation in her tumor markers and her therapy was changed to fulvestrant. Clinically she remains without evidence of progression and has had stabilization of tumor markers. Although other studies have suggested that AIs may be the class of endocrine therapies that selects for LBD ESR1 mutations (27), most studies have enrolled patients who have received multiple lines of endocrine therapy in both the adjuvant and metastatic settings, which precludes any definitive conclusions. This patient demonstrates that an ESR1 mutation can indeed occur after prolonged exposure to an AI without other endocrine or systemic therapies, and that ESR1 mutations do not necessarily preclude response to a subsequent fulvestrant.
Discussion
There are several important conclusions with potential therapeutic implications derived from our study. First, we have demonstrated that ESR1 mutations can be readily detected using ddPCR on plasma from patients with metastatic ER-positive disease after progression on endocrine therapies. Given challenges that can arise in obtaining a metastatic biopsy as encountered in this study, the use of ptDNA as a “liquid biopsy” holds great promise for future molecular analysis of human cancers. Moreover, monitoring for emergence of mutated clones by repeat sampling can be more easily performed with a simple blood test than with multiple tissue biopsies. Second, we demonstrate that blood can be a more sensitive source for detecting ESR1 mutations. In our study two patients harbored a distinct, second ESR1 mutation not present in the corresponding metastatic biopsies. Perhaps more importantly, one patient in cohort 1 and three patients in cohort 2 had wild type ESR1 in their metastatic biopsies, but had ESR1 mutations detected in their corresponding ptDNA sample. These results support the increasingly recognized problem of tumor heterogeneity and are in agreement with a prior report demonstrating differences in ESR1 mutation status between two metastatic sites within the same patient (6). Third, our results support the previously proposed hypothesis that ESR1 LBD mutations may be selected for after progression on AIs (7). This was particularly striking in patient 19, who was positive for an ESR1 mutation and had received only prolonged exposure to letrozole. Fourth, our study shows that ddPCR of ptDNA is capable of detecting ESR1 mutations even in patients who have no radiographic evidence of disease. Although the clinical validity and utility of this observation remains to be proven, we suggest that detecting drug resistant mutations may afford the opportunity to change therapies earlier or enroll in trials of novel targeted therapies, which may lead to improved outcomes for patients. Finally, the frequency of circulating ESR1 mutations in our study is notably higher than prior reports using a single metastatic biopsy. The majority of studies thus far have detected ESR1 mutations only in patients with metastatic disease after progression on endocrine therapies, though one study did find a low incidence (3%) in primary tumors (8). The largest study to date of ESR1 mutations in metastatic tissue biopsies suggests an overall frequency of 12%, with a frequency of 20% in a subgroup analysis of patients who received an average of 7 lines of therapy (9). However, we found additional mutations not detected by sequencing of metastatic lesions. In cohort 1, two additional mutations were discovered: patient 1 who had an additional ESR1 mutation found in ptDNA compared to her metastatic biopsy, and patient 9 who was wild type for ESR1 on her metastatic tissue sample. Additionally, in cohort 2, we detected seven ESR1 mutations in six of the eight ER-positive patients not detected in metastatic biopsies, although three of these patients did not have adequate tissue for NGS. These results highlight the potential impact of using blood as a more sensitive and accessible source for mutation detection.
The higher frequency of ESR1 mutations in blood compared to biopsied tissues could be due to several non-overlapping reasons. As mentioned, tumor heterogeneity can lead to the detection of mutations in ptDNA that are present in other non-biopsied metastatic sites. It is also conceivable that sampling error of biopsies may miss subclonal populations in a given metastatic lesion, and/or certain clonal populations may have a propensity for releasing ptDNA versus other clonal variants. For example, it is possible that ptDNA shed from CTCs is more abundant than ptDNA derived from other metastatic sites. Further studies are needed to clarify the origins and kinetics of ptDNA as related to sites of metastases, and any underlying biology that may favor the enrichment of clonal populations that shed higher versus lower amounts of ptDNA into the circulation.
There are limitations of our study, most notably the small sample size, which prevents our assessing the true prevalence of ESR1 mutations in plasma from patients with ER-positive breast cancer. Further, we only queried for the three most common ESR1 LBD mutations, and it is likely ptDNA contains other ESR1 mutations associated with endocrine therapy resistance. Although additional ESR1 LBD mutations have been described at lower frequency (5-9), we did not identify these mutations by NGS of tissues in the retrospective cohort, and they were therefore not queried by ddPCR. In addition, because these mutations are all in close proximity to one another, each ESR1 ddPCR mutation probe was run separately due to potential competition for the same template molecule, which could theoretically decrease the sensitivity for any given probe. This can limit the number of mutations that can be assayed due to low amounts of plasma DNA. However, this limitation may have led us to underestimate the prevalence of ESR1 mutations in our study.
In summary, we confirm the feasibility of detecting ESR1 mutations in ptDNA, and that plasma may prove to be a superior source than metastatic biopsies for ESR1 mutation detection. However, the clinical utility of using ddPCR for ESR1 mutations to guide therapy for patients requires careful prospective study before adoption into clinical practice. It is unknown what allelic frequency of ESR1 mutation is associated with symptomatic disease progression, and whether changing endocrine therapies can improve patient outcomes. Nevertheless, the ability to detect ESR1 mutations in the plasma of patients, independent of the tissue mutational status, provides the foundation for future clinical trials to track and monitor the emergence of endocrine therapy resistance.
Supplementary Material
Statement of Translational Relevance.
ESR1 mutations can arise in estrogen receptor-positive breast cancer metastases after progression on endocrine therapies. However, due to tumor heterogeneity and difficulty in obtaining metastatic biopsies, a “liquid biopsy” using circulating plasma tumor DNA (ptDNA) would facilitate assessment of ESR1 mutations. We developed a blood based assay to detect ESR1 mutations using droplet digital PCR (ddPCR) and compared the results with next generation sequencing (NGS) of metastatic tissue biopsies in patients with breast cancer. In a retrospective cohort (n=11), we detected all mutations in blood that were present in tissues by NGS and discovered two additional ESR1 mutations in ptDNA samples. In a prospective cohort (n=12) we identified seven ESR1 mutations in blood and no mutations were detected in metastatic biopsies. These results demonstrate a higher frequency of ESR1 mutations in ptDNA than in corresponding metastatic biopsies and suggest that ddPCR of ptDNA may be preferred for ESR1 mutation detection.
Acknowledgments
We thank Moshe Talpaz, M.D. for assistance with the MiONCOSEQ data.
Financial Support: This work was supported in part by The Breast Cancer Research Foundation (BCRF) grant N003173 (JMR and DFH), The Avon Foundation (B.H.P., J.L.), DOD W81XWH-14-1-0284 (P.J.H.), NIH CA009071 (H.P., K.C., B.H.P.), GM007309 (D.J.Z.), CA168180 (R.L.C.), CA167939 (S.C.) and the Sandy Garcia Charitable Foundation (D.C. and B.E.). We would also like to thank and acknowledge the support of NIH P30 CA006973, the Commonwealth Foundation, the Santa Fe Foundation, the Breast Cancer Research Foundation (B.H.P.), the Health Network Foundation, the ME Foundation, the Augustine Fellowship (W.B.D.) The Walsh Fund (A.M.), The Robin Page/Lebor Foundation and Fashion Footwear Charitable Foundation of New York/QVC Presents Shoes on Sale™ (D.F.H.). None of the funding sources influenced the design, interpretation or submission of this manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest: B.H.P. is a paid consultant for Novartis and is a member of the scientific advisory boards of Horizon Discovery, LTD and Loxo Oncology, and has research contracts with Genomic Health, Inc and Foundation Medicine. Under separate licensing agreements between Horizon Discovery, LTD and The Johns Hopkins University, B.H.P. is entitled to a share of royalties received by the University on sales of products. The terms of this arrangement are being managed by the Johns Hopkins University, in accordance with its conflict of interest policies. D.F.H. has received clinical and laboratory research funding from Janssen, AstraZeneca, and Pfizer. The University of Michigan holds two patents regarding isolation and characterization of circulating tumor cells, one of which has been licensed to Janssen, naming D.F.H. as the inventor. D.F.H. also has stock options in two diagnostic companies: InBiomotion and OncImmune. None of these conflicts pertains to this manuscript. All other authors declare no potential conflicts.
Author Contributions: Conception and design: D.C. C.P. D.F.H., J.R., B.H.P.
Development of methodology: D.C., C.P., C.G., D.A.V., R.L.C., P.V.T., S.C., H.Y.W., D.J.Z., J.C., R.L.C., B.E., K.C., K.K.S., B.B., N.T., H.P., W.B.D, R.G., A.M., P.J.H., J.L., B.H.P.
Acquisition of data: S.C., C.P., D.R., H.Y.W., D.J.Z., D.C., M.M., D.M.R., R.S., J.C., R.L.C., H.P., W.B.D., B.E., B.B., K.C., K.K.S., N.T., K.S.J., K.A., P.J.H., J.L., B.H.P.
Analysis and interpretation of data: All authors
Writing, review, and/or revision of the manuscript: All authors
References
- 1.Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609–18. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- 2.Burstein HJ, Prestrud AA, Seidenfeld J, Anderson H, Buchholz TA, Davidson NE, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol. 2010;28:3784–96. doi: 10.1200/JCO.2009.26.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 4.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports. 2013;4:1116–30. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73:6856–64. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 7.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997;57:1244–9. [PubMed] [Google Scholar]
- 11.Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–98. doi: 10.1210/mend.10.11.8923465. [DOI] [PubMed] [Google Scholar]
- 12.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18:3462–9. doi: 10.1158/1078-0432.CCR-11-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4:162ra54. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra68. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 17.Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A. 2013;110:18761–8. doi: 10.1073/pnas.1313995110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–12. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 19.Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014;20:2643–50. doi: 10.1158/1078-0432.CCR-13-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stroun M, Maurice P, Vasioukhin V, Lyautey J, Lederrey C, Lefort F, et al. The origin and mechanism of circulating DNA. Ann N Y Acad Sci. 2000;906:161–8. doi: 10.1111/j.1749-6632.2000.tb06608.x. [DOI] [PubMed] [Google Scholar]
- 22.Choi JJ, Reich CF, 3rd, Pisetsky DS. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology. 2005;115:55–62. doi: 10.1111/j.1365-2567.2005.02130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra21. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oesterreich S, Davidson NE. The search for ESR1 mutations in breast cancer. Nat Genet. 2013;45:1415–6. doi: 10.1038/ng.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.