

Abstract
The promises of data-independent acquisition (DIA) strategies are a comprehensive and reproducible digital qualitative and quantitative record of the proteins present in a sample. We developed a fast and robust DIA method for comprehensive mapping of the urinary proteome that enables large scale urine proteomics studies. Compared to a data-dependent acquisition (DDA) experiments, our DIA assay doubled the number of identified peptides and proteins per sample at half the coefficients of variation observed for DDA data (DIA = ~8%; DDA = ~16%). We also tested different spectral libraries and their effects on overall protein and peptide identifications and their reproducibilities, which provided clear evidence that sample type-specific spectral libraries are preferred for reliable data analysis. To show applicability for biomarker discovery experiments, we analyzed a sample set of 87 urine samples from children seen in the emergency department with abdominal pain. The whole set was analyzed with high proteome coverage (~1300 proteins/sample) in less than 4 days. The data set revealed excellent biomarker candidates for ovarian cyst and urinary tract infection. The improved throughput and quantitative performance of our optimized DIA workflow allow for the efficient simultaneous discovery and verification of biomarker candidates without the requirement for an early bias toward selected proteins.
Keywords: DIA, QE, urine proteomics, biomarker discovery, spectral library
Graphical abstract
INTRODUCTION
Urine is of particular interest for proteomic biomarker discovery studies as urine has several advantages over, for example, physiologically active blood-derived body fluids such as serum or plasma including (1) superior stability as the easily degradable proteins are proteolyzed during the prolonged “storage” at body temperature; (2) lower biohazard as bloodborne pathogens including HIV or M. tuberculosis have a much lower titer in urine; (3) excellent availability as urine is easily and noninvasively obtainable in large amounts; and (4) smaller analytical challenge as the urinary proteome is characterized by lower complexity and dynamic range. The urinary proteome comprises proteins from the blood that passed the glomerular barrier of the kidney as well as proteins secreted or shed by the kidney and genitourinary organs, which are in direct contact with the urine. Thus, urine is a systemic body fluid reflecting the state of the entire organism as well as a proximal body fluid for the kidney and genitourinary tract with obvious potential as a source for biomarkers of, for example, renal diseases (reviewed in ref 1). The systemic nature of urine has been exploited in studies aiming at identifying urinary biomarkers for a wide range of diseases unrelated to the kidney or the genitourinary tract including (but not limited to) Kawasaki disease,2 coronary artery disease,3 prostate cancer,4 appendicitis,5 tuberculosis,6 and major depressive disorder.7 Attempts to map the urinary proteomes date back to at least 2001 when Spahr and colleagues identified 124 proteins.8 Since then, the field has evolved allowing for the routine identification of 1000+ proteins in a single urine samples and more than 2500 proteins when using prefractionation and combining the proteomic data from several urine specimens.9 This relatively low number of proteins has been consistent across numerous extensive urine proteomics studies, suggesting that the vast majority of the accessible urinary proteome is covered by these ~2500 proteins. Thus, the urinary proteome lends itself to be analyzed without any prefractionation in a single liquid chromatography–mass spectrometry (LC–MS) experiment when using state-of-the-art instrumentation.
Leveraging the unique characteristics of urine as a promising source for clinically relevant biomarkers, we aimed at developing a robust method for the comprehensive mapping of the urinary proteome that enables large scale urine proteomics studies requiring fast, accurate, and reproducible quantitation across many samples. This need for robust quantification of thousands of features raises the question as to whether data-dependent acquisition (DDA) routines are the appropriate choice given the stochastic aspect of the precursor ion selection,10 which is particularly problematic for unfractionated samples of high complexity resulting in noticeable undersampling.11 This undersampling, in turn, leads to a large number of missing data points, which poses a particular problem for the discovery and initial verification phases where it is crucial to distinguish between a peptide signal being truly absent for biological reasons or simply missing due to the limitation in the acquisition method. A typical way to overcome this problem is the use of targeted acquisition methods such as selected reaction monitoring (SRM).12 In such targeted acquisition method, a select set of easily detectable, that is, “proteotypic”,13 peptides of the protein of interest are monitored, which allow very sensitive, accurate, and precise protein quantification in urine samples across several orders of magnitude.14 The drawback is that only the targeted, that is, preselected, set of peptides/proteins can be quantified, potentially missing biomarker candidates that were not identified as such in the initial discovery experiment. Additionally, the development of the SRM assay can be a very time-consuming undertaking and needs to be optimized for each protein individually.
Recent studies showed that data-independent acquisition (DIA) methods can overcome several of the limitations associated with DDA, resulting in SRM-like quantification for thousands of proteins with fewer missing values.15 In contrast to SRM experiments, in which the analytes have to be preselected prior to the data acquisition, a DIA data set is most commonly analyzed with a spectral library comprising a list of peptides from previously identified proteins.15a This strategy also allows for revisiting old data sets to quantify initially missed biomarker candidates with SRM-like precision.
Here, we evaluate the use of DIA methods on the newest generation quadrupole Orbitrap instrument, the Q Exactive HF mass spectrometer,16 for urine protein biomarker discovery, with particular emphasis on effect of spectral library on the data analysis, DIA data reproducibility, and quantitation precision. The optimized DIA workflow was subsequently applied to the analysis of 87 urine samples from pediatric patients visiting the emergency room (ER) because of abdominal pain. In this proof of concept study, we focused on patients that were diagnosed with ovarian cyst (12 patients) and urinary tract infections (UTI, 11) and combined other causes in a symptomatic control group (64), representing the intended use population.
MATERIALS AND METHODS
Urine Sample Collection
Urine samples were collected from consenting patients visiting the ER at Boston Children’s Hospital in Boston, MA, USA. Samples were taken before final diagnosis of the patients. The study was reviewed and approved by Boston Children’s Hospital’s Internal Review Board (Protocol Number: X06–10–0493).
Sample Preparation/Digestion
Samples were prepared using the in-house developed MStern blot protocol.17 In brief, undiluted neat urine (150 μL, i.e., ~15 μg of protein) was added to a mixture of 150 μg of urea and 30 μL of dithiothreitol (DTT) (100 mM in 1 M Tris/HCl pH 8.5). The samples were incubated for 20 min, and the cysteine residues were blocked with 50 mM iodoacetamide for 20 min in the dark. Afterward, samples were transferred into a 96-well plate with a PVDF membrane at the bottom (MSIPS4510, Millipore). Protein digestion was performed with sequencing-grade trypsin (V5111, Promega) at a nominal enzyme to substrate ratio of 1:15. After incubation for 2 h at 37 °C in a humidified incubator, the remaining digestion buffer was evacuated. Resulting peptides were eluted twice with 150 μL of 40% ACN (v/v)/0.1% (v/v) formic acid (FA) each, and the solutions were pooled and subsequently dried in a vacuum concentrator. For DDA experiments, iRT peptides (Biognosys, Schlieren, Switzerland) were spiked into the sample, and for the DIA experiments, HRM calibration peptides (Biognosys) were added to the samples prior to analysis according to manufacturer instructions.
DDA Sample Analysis and Database Search
For the spectral library, all 87 samples were analyzed using a nanoLC system (Eksigent, Dublin, CA) equipped with a LC-chip system (cHiPLC nanoflex, Eksigent, trapping column: Nano cHiPLC Trap column 200 μm × 0.5 mm Reprosil C18 3 μm 120 Å, analytical column: Nano cHiPLC column 75 μm × 15 cm Reprosil C18 3 μm 120 Å) coupled online to a Q Exactive mass spectrometer (Thermo Scientific, Bremen, Germany). Peptides (4 μL of digest) were separated by a linear gradient from 93% buffer A (0.2% FA in water)/7% buffer B (0.2% FA in ACN) to 75% buffer A/25% buffer B within 75 min. The mass spectrometer was operated in data-dependent TOP10 mode with the following settings: mass range 400–1000 Th; resolution for MS1 scan 70 000 @ 200 Th; lock mass: 445.120025 Th; resolution for MS2 scan 17 500 @ 200 Th; isolation width 1.6 Th; NCE 27; underfill ratio 1%; charge state exclusion: unassigned, 1, >6; dynamic exclusion 30 s.
Additionally, a subset of randomly chosen 23 samples was analyzed on Q-TOF mass spectrometer (Sciex, TripleTOF 5600) using the same LC setup and gradient as described earlier of the Q Exactive-based analysis. The mass spectrometer was operated in data-dependent TOP50 mode with following settings: MS1 mass range 400–1000 Th with 250 ms acc. time; MS2 mass range 100–1700 Th with 50 ms accumulation time and following MS2 selection criteria: UNIT resolution, intensity threshold 100 cts; charge states 2–5. Dynamic exclusion was set to 17 s.
The human UNIPROT protein sequence database (only reviewed entries, downloaded on October 31, 2014) was searched with MaxQuant (v1.5.0.0)18 directly using the .RAW and .WIFF files. The protein sequence database was appended with common laboratory contaminants (cRAP, version 2012.01.01) and the iRT fusion protein sequence (Biognosys) resulting in 20 296 entries. The following settings were applied: trypsin with up to two missed cleavages; mass tolerances set to 20 ppm for the first search and 4.5 ppm for the second search for the Q Exactive data and 0.1 Da for the first search and 0.01 for the main search for the TripleTOF 5600 data. Oxidation of M was chosen as dynamic modification (+15.995 Da) and carbamidomethylation of C as static modification (+57.021 Da). False discovery rate (FDR) was set to 1% on peptide and protein level. For the analysis of the DDA replicate data, the matching option was used. For all other search parameters, the default settings were used.
To generate a spectral library for the analysis of the UTI samples, we searched these DDA files against a concatenated human (as described earlier), Escherichia coli, and Staphylococcus saprophylicus database (both downloaded from uniprot.org on December 4, 2014); this combined database featured in total 27 005 entries.
Generation of Spectral Libraries
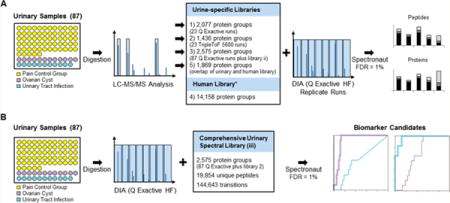
For spectral library testing, two libraries were generated using data from those 23 samples that were analyzed on both the Q Exactive and the TripleToF 5600 Q-TOF instrument. Two spectral libraries were generated in Spectronaut 7.0 (Biognosys) using a Q value cutoff of 0.01 and minimum of three and a maximum of six fragment ions. Proteins were grouped according to the MaxQuant search result. Library 1 was based on the Q Exactive data (Supplementary Table 1A) and library 2 on the TripleToF 5600 data (Supplementary Table 1B). To generate a comprehensive urinary library (library 3), the MaxQuant search results of all 87 Q Exactive files were loaded into Spectronaut and merged with library 2 (Supplementary Table 1C). To merge the libraries, peptide precursors occurring in only one of the two spectral libraries were simply combined. For peptide precursors occurring in both spectral libraries, a weighted average of the iRT was taken based on the number of observations of the peptide in the DDA data. The relative fragment ion intensities were averaged between the two spectral libraries without weighting. The comprehensive human library (library 419) was downloaded from SWATHAtlas (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/GetDIALibs) and directly used in Spectronaut. The subset library was generated by extracting the identified proteins from comprehensive urinary library 3 from publicly available human library 4 (library 5, Supplementary Table 1D). An overview of all libraries can be found in Table 1. For data analysis of the biomarker study, the comprehensive urinary library 3 was applied. Additionally, we generated an UTI-library in Spectronaut based on the search results against the concatenated human and bacterial database (Supplementary Table 1E).
Table 1.
Overview of Spectral Librariesa
| library | protein groups | proteins | peptides | fragment ions | instrument | samples | source |
|---|---|---|---|---|---|---|---|
| 1 | 2077 | 1862 | 14 832 | 109 064 | Q Exactive | 23 | urine |
| 2 | 1463 | 1315 | 9132 | 65 880 | TripleTOF 5600 | 23 | urine |
| 3 | 2510 | 2183 | 18 631 | 144 643 | Q Exactive/TripleTOF 5600 | 87 + 23 | urine |
| 4 | 14 158 | 10 346 | 149 420 | 2 832 306 | TripleTOF 5600 | Rosenberger et al., 2014 | |
| 5 | 1869 | 1869 | 40 902 | 925 156 | TripleTOF 5600 | subset of 4 |
For libraries 1 and 2, a randomly selected subset of 23 samples was analyzed by a DDA method on a Q Exactive or a TripleTOF 5600 mass spectrometer. For library 3, the remaining 64 samples were analyzed on a Q Exactive mass spectrometer. Data were searched with MaxQuant and filtered with a FDR of 1% on peptide and protein level. The spectral libraries were generated in Spectronaut, and protein grouping of the MaxQuant search results was applied. Additionally, the number of “single group proteins” is given. For library 3, the TripleTOF 5600 and Q Exactive libraries were merged in Spectronaut. Library 4 was published in Rosenberger et al., 2014. This library comprises ~10 000 single protein entries and ~14 000 protein group entries. Library 5 comprised the subset of proteins from library 4 that was identified in library 3. Supplementary Figure 3 shows an overview of the overlap of peptides and proteins between the libraries.
DIA Sample Acquisition on Q Exactive HF
The samples were analyzed on an EASY-nLC 1000 nanoLC system (Thermo Scientific) equipped with a trapping column (PepMap100, 75 μm × 2 cm, C18, 3 μm, 100 Å) and an analytical column (PepMapRSLC, 75 μm × 25 cm, C18, 2 μm, 100 Å) coupled online to a Q Exactive HF mass spectrometer (Thermo Scientific) equipped with an EASY-Spray nano-electrospray ion source (Thermo Scientific).
Peptides (2 μL of digest) were separated by a linear gradient from 93% buffer A (0.2% FA in water)/7% buffer B (0.2% FA in ACN) to 75% buffer A/25% buffer B within 30 min. The total run time with loading and washing steps was 50 min. Column oven was set to 40 °C.
For the DIA method on Q Exactive HF, each DIA cycle contains one full scan and 24 DIA scans covering a mass range of 400–1000 Th covering 97% of peptides in a urinary sample (Supplementary Figure 1), full scan with a resolution of 30 000 @ 200 Th; AGC target −3e6, maximal IT, 50 ms; mass range 400–1000 Th; followed by DIA scans with resolution 30 000 @ 200 Th; isolation width 20 Th for the first 20 DIA scans, 40 Th for the following two DIA scans, and 60 Th for the last two DIA scans; NCE −b30; target value −1e6, maximal injection time, auto, which automatically calculates the maximal injection time based on the resolution settings. This setting ensured that the mass spectrometer was always working in parallel ion filling and scanning mode. The cycle time was 2 s, which resulting in more than eight scans across the LC peak (8 s @ fwhm).
All mass spectrometric data are available at PeptideAtlas.20 The identifier is PASS00706.
DIA Data Analysis
All DIA data were directly analyzed in Spectronaut 7.0 (Biognosys)15b without any file conversion. The following settings were applied in Spectronaut 7.0: peak detection, dynamic iRT; correction factor 1; dynamic score refinement and MS1 scoring, enabled; interference correction and cross run normalization (total peak area), enabled; peptides were grouped according to the protein grouping. The number of fragment ions was defined in the spectral library (at least 3 and up to 6), and all were required for identification and quantification. Spectronaut utilizes the spiked-in HRM peptides for m/z and retention time calibration. For our data set, the m/z tolerance was in the range of 4 ppm and the median retention time extraction window 8 min. All results were filtered by a Q value of 0.01 (equals a FDR of 1% on peptide level). All other settings were set to default.
Protein intensity was calculated by summing the peptide peak areas (sum of fragment ion peak areas as calculated by Spectronaut) of each protein from the Spectronaut output file. For testing of the spectral libraries, one urinary sample was measured three times on the Q Exactive HF and afterward analyzed in Spectronaut using the spectral libraries 1–5 (Table 1). The results were benchmarked based on the number of detected peptides and proteins as well as the reproducibility of the peptide and protein detection. To enable comprehensive urinary proteome analysis, library 3 (Table 1) was applied to analyze the complete DIA data set of 87 urinary samples. All quantitative data are summarized in Supplementary Table 3.
For statistical analysis, the data were imported into Perseus 1.5.1.6 (http://141.61.102.17/perseus_doku/doku.php?id=start), and missing values were imputed using the lowest intensity of each individual protein. Significance of protein abundance changes was calculated using the nonparametric Mann–Whitney u-test, and Bonferroni multiple testing correction was applied.
RESULTS AND DISCUSSION
The objective of this study was to establish a robust DIA-based method for comprehensively mapping of urinary proteomes to facilitate the discovery of disease-relevant biomarker candidates. To this end, we optimized a workflow based on different parameters including protein identification, data reproducibility, quantification precision, and applied the optimized workflow to a urine proteomics study comprising 87 samples from pediatrics ER patients that were seen because of abdominal pain.
Finding the Right Balance for the DIA Method
For setting up a DIA experiment, it is crucial to cover the mass range containing all analytes of interest. This can be achieved by selecting the entire mass range for simultaneous fragmentation or by fragmenting windows 20–50 Th. The latter increases the specificity by reducing the precursor range at the expense of increased cycle time. An appropriate implementation of such DIA method has to ensure that the cycle time allows for at least 8–10 points per LC peak. To account for this minimum number of data points per LC peak, precursor window size, fragment ion resolution, and LC peak profile have to be considered for the method optimization.
According to our shotgun experiment on the urine sample, >97% peptides fall into the m/z range 400–1000 (Supplementary Figure 1). Typically, we defined 30 windows with 20 Th width using the first generation Q Exactive mass spectrometer to cover this mass range. The DIA scans were acquired with a resolution of 17 500 resulting in a cycle time of 2.6 s; since this cycle time could not be significantly reduced, the LC conditions had to be chosen to ensure LC peak widths in the 25–30 s range, resulting a minimum gradient time of 60 min on the Q Exactive mass spectrometer. The ultrahigh-field Orbitrap mass analyzer of the Q Exactive HF instrument almost doubles the resolution at the same transient times, allowing us to keep the similar cycle time but acquiring all scans at 30 000 resolution settings, which improves the separation of the analyte of interest from interferences. In addition, a rectangular isolation window shape was generated by the segmented quadruple design on Q Exactive HF instrument,16 enabling the accurate isolation of the peptides on the isolation edges. After slightly adopting the DIA method (for details see Materials and Methods), we were able to achieve a duty cycle 2 s to cover the mass range 400–1000 Th, including one full scan and 24 DIA MS/MS scans acquired at 30 000 resolution. Thus, moving to the Q Exactive HF enabled us to shorten gradient time to 30 min at twice the resolution without compromising the number of data points across the LC peaks. This shortened gradient length, which led to a reduced peptide elution peak width of about 40% (shown for three examples in Supplementary Figure 2), resulted in an increased sample throughput, which is important for the analysis of a large number of urinary samples.
Selecting the Most Appropriate Spectral Library
The most common way of analyzing DIA data is an assay-driven approach. By using this approach, DDA data are acquired of some or all of samples of interest and used to generate a spectral library containing all spectral information for the detectable peptides and proteins present in the sample. The spectral library is then the basis of for qualitatively and quantitatively analyzing the DIA data.15a The Aebersold group has recently started to publish spectral libraries for DIA data, namely for M. tuberculosis,21 S. cerevisiae (http://www.swathatlas.org/), and H. sapiens.19
To investigate the influence of the spectral library on the data analysis, we generated and tested five different spectral libraries (workflow in Figure 1A) based on the following inputs: (1) Q Exactive-based DDA data from a random subset of 23 urine samples searched with MaxQuant; (2) DDA data from the same set of 23 samples acquired on a quadrupole TOF type instrument (Sciex TripleToF 5600) searched with MaxQuant; (3) a comprehensive urinary spectral library based on all 87 Q Exactive samples combined with the input from 2; (4) the publicly available spectral library for samples of human origin;19 and (5) a subset of the publically available H. sapiens library featuring 1900 of the 2600 proteins identified in the comprehensive library (see library 3). More details about the fragments, peptides, and proteins covered in these five spectral libraries can be found in Table 1 and an overview of the overlap between the libraries in Supplementary Figure 3A (complete libraries: Supplementary Tables 1A–D).
Figure 1.
Workflow. (A) Generation of spectral library. Eighty-seven urinary samples from kids with abdominal pain, diagnosed with ovarian cyst (purple, 11), urinary tract infection (blue, 11), as well as a pain control group (yellow) were processed in a 96-well plate format. Twenty-three randomly chosen samples were analyzed by LC–MS/MS on a Q Exactive (Thermo Scientific) and TripleToF 5600 mass spectrometer. The resulting data were searched with MaxQuant (FDR 1%), and a spectral library of each search result was generated in Spectronaut (libraries 1 and 2). Additionally, all remaining samples were run on the Q Exactive and combined with the other two libraries to create a comprehensive urinary library (library 3). Library 4 was a publically available spectral library from Rosenberg et al., and library 5 featured those proteins from this publicly available library, which were also identified in library 3. All libraries (Table 1) were used in Spectronaut to analyze the DIA data of an unrelated urinary sample, acquired three times on a Q Exactive HF mass spectrometer. (B) DIA sample acquisition. All 87 samples were analyzed by a 30 min LC gradient on a Q Exactive HF mass spectrometer in DIA mode. We applied the comprehensive urinary spectral library (library 3) to analyze the data in Spectronaut (1% FDR).
We assessed the different spectral library using two different criteria: (1) number of identified peptides and proteins, and (2) the reproducibility of peptide/protein detection, which are measures of the relevance of the database since an irrelevant increase in search space will result in irreproducible hits across, for example, technical repeats. Additionally, the reproducibility can be reduced by variability of the fragment ion intensities and the retention time. Within our workflow, the retention times are normalized by application of the indexed retention time concept (iRT), in which the retention times are converted into a dimensionless space to make them comparable across runs/different gradients.22 By application of this concept, we could correct for the variability within the retention time space. All libraries contained iRT values, and the data analysis was based on iRT rather than the peptide retention times in the library. To this end, we analyzed an unrelated urine samples in triplicate using our optimized DIA routine and searched the data against the five spectral libraries (Table 1). We identified the largest number of peptides with the comprehensive urinary spectral library (library 3: 6061 peptides), followed by the Q Exactive library with 5429 peptides (library 1). Using the other libraries, we identified in total only between 3259 and 3721 peptides (Figure 2A, left panel). For the in-house generated project specific spectral libraries (1–3), the overlap in identified peptides was high; more than 80% of the peptides were detected in more than one library (Supplementary Figure 3B), and it resembled the differences in the overlap of the spectral libraries (Supplementary Figure 3A). The overlap with the libraries based on the publicly available human library (libraries 4 and 5) was comparably low, for example, 54% of the peptides were unique to comprehensive urinary library 3, and 36% of the peptides unique to entire human library 4 (Supplementary Figure 3B).
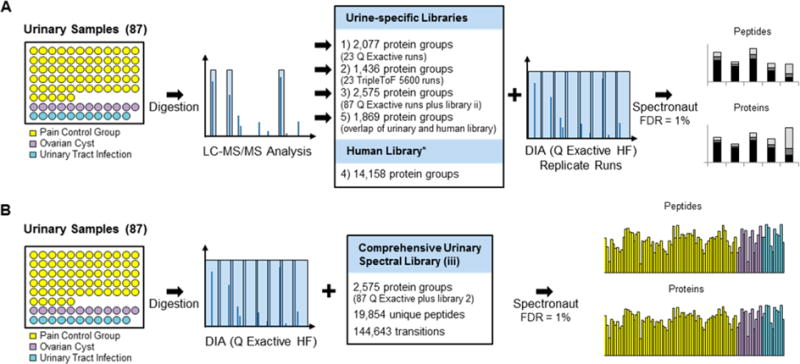
Figure 2.
Validation of workflow. (A) Influence of spectral library. A urinary sample was analyzed in triplicate with a DIA method on a Q Exactive HF mass spectrometer using five different spectral libraries (overview in Table 1). Plotted are the total numbers of identified peptides and proteins; each bar is divided into peptides/proteins that were identified in three of three replicates (dark green), two of three (green), and one of three (light green) as well as the percentages. (B) Number of peptide and protein identifications in replicate runs. A urinary sample was analyzed three times by a DDA and DIA methods on a Q Exactive mass spectrometer and analyzed using the comprehensive urinary library (library 3). The DDA data were analyzed in MaxQuant with and without the ID matching. We plotted the number of identified peptides and proteins (DIA, orange; DDA without matching, blue; DDA with matching, blue/white stripes) as well as the increase in identifications with each of the replicates (DIA, light orange; DDA, light blue). (C) Quantification precision. An independent urine sample was analyzed in triplicate with a DDA and DIA method on a Q Exactive HF mass spectrometer. DDA were quantified in MaxQuant either based on peptide peak areas (DDA, peptide peak areas) or spectral counting (DDA, spectral counting). DIA data were quantified in Spectronaut (DIA, fragment ion peak areas). For both peak area based quantification methods, protein values were calculated by summation of the peptide peak areas. The %CV of the quantification was calculated, plotted against the peptide/protein intensity, and the point density was color coded (Perseus: light blue, highest density; green, lowest density). The table gives an overview of the quantified peptides/proteins as well of number of peptides/proteins with a %CV below 10% and 20%. (D) Protein %CV in relation to protein abundance. The quantified proteins have been binned according to their intensity into 20 bins. The median %CV of each bin was plotted for three quantification methods (right panel, based on peptide peak areas of DDA data; middle panel, spectral counting; left panel, based on fragment peak areas of DIA data). The horizontal bars show the protein intensity spread of each bin.
There was a clear correlation between the number of identified peptides and identified proteins for the in-house generated project specific spectral libraries 1, 2, and 3, which resulted in 1191, 894, and 1393 protein identifications, respectively. In contrast, the two spectral libraries based on the publicly available H. sapiens spectral library were clear outliers (library 4 and 5). These searches resulted in relatively large numbers of identified proteins (1660 and 1122, respectively) despite the small number of identified peptides (3259 and 3396). This problem was particularly noticeable when using the entire H. sapiens spectral library featuring 14 000+ proteins (library 4, Figure 2A, right panel). The overlap between the protein identification results was comparable to the peptide results. We observed a high overlap for libraries 1–3 and a large number of unique proteins comparing the results to libraries 4 and 5, for example, 659 proteins (47% of all identified proteins using library 5) and the majority of the proteins identified by the application of library 4 were unique (908 proteins, 55%, Supplementary Figure 3B).
To assess the reproducibility, we calculated the percentage of peptides and proteins that were detected in all three replicates, in two out of three replicates, or in only one single replicate. With the project specific spectral libraries (library 1–3), between 69% and 77% of the peptide and between 75% and 81% of the proteins were detected in all three replicates. In contrast, only 26% of the peptides and 22% of the proteins were detected in all three replicates when using the entire publicly available human spectral library (library 4). Given this low reproducibility, we concluded that the large human spectral library is inadequate for searching urinary samples. Although the use of a urine specific subset of proteins (library 5) improved the reproducibility to 61% and 62% at the peptide and protein level, respectively, these numbers are still inferior to the project specific in-house generated spectral libraries. In general, the reproducibility of the peptide/protein detection is also lowered by variation in fragment intensity and retention times. Although we made use of the iRT concept,22 the accuracy of the iRT in the library will have a small influence on the detection reproducibility. To compare the libraries in this regard, we generated an additional spectral library only containing the peptide overlap of library 3 and 4. The peptide and protein detection reproducibility was comparable to the in-house generated libraries (71% of peptides were detected in three of three replicates and 75% of the proteins, Supplementary Figure 3C). We concluded that the variability of retention time and fragment ion intensity had only a minor influence on the detection reproducibility. Given that the peptide assignment is FDR-controlled, it can be assumed this lack of reproducibility is not due to false positives, that is, wrong assignments, but due to false negatives, that is, the current software discarded a (true) peptide match because of, for example, the presence of too many spurious signals that can be assigned to other peptides. This problem of false negatives is particularly relevant in the case of very large search spaces as in case of the published human spectral library. Data for peptides are extracted that do not exist, and the multiple testing in the data analysis need to correct for it, resulting in the loss of many true signals thereby reducing the confidence of the original peptide assignment. We assume that with instrument and software improvements in the future, the number of false negatives will decrease.
Since the publicly available spectral library is based on quadrupole-TOF data, we also investigated the possibility that the differences in the search results are due to instrument dependent peptide fragmentation. However, our in-house generated spectral library using quadrupole-TOF data showed the same reproducibility as the Q Exactive data-derived spectral libraries, albeit at lower peptide and protein identifications. We concluded that the instrument type or more precise the fragmentation type has only a minor effect on the spectral library quality. It is more important to apply a sample type-specific spectral library. In this context, it was interesting to note that the Q Exactive and the TripleTOF 5600 resulted in complementary peptide fragmentation spectra such that combining both data sets resulted in the largest number of identified peptides. Therefore, we decided to continue with the most comprehensive urine specific spectral library 3, which provides a good basis for a comprehensive and reliable analysis of the urinary proteome.
In summary, the significantly lower reproducibility in combination with much lower numbers of identified peptides and the low number of overlapping peptides and proteins between the urinary libraries (1–3) to the publicly available libraries (4, 5) clearly shows that the use of project specific spectral library are highly recommendable for successful DIA data analyses and that publicly available spectral libraries can be of limited use (Figure 2A), at least with currently produced DIA data.
Highly Reproducible Peptide Detection and Quantitation in DIA Experiments
In DDA experiments, often technical replicates are acquired to increase the number of identified peptides and proteins23 as well as to improve the quantification. To elucidate how the DIA-based quantification performs compared to DDA-based quantification, we analyzed an unrelated urine sample six times: three technical repeats in standard DDA-mode and three technical repeats using our optimized DIA method. A 30 min gradient on a Q Exactive HF was used for these analyses. The DDA samples were analyzed by MaxQuant18 without and with the matching option activated. The matching options employs the accurate mass tag concept24 and transfers confident peptide identifications from one run to another based on accurate precursor mass and retention time irrespective of whether a MS2 spectrum was acquired or not, however, without providing a proper FDR. This strategy increases the peptide and protein identifications and results in much fewer missing quantification values across multiple LC–MS experiments. The DIA results were analyzed using the comprehensive urinary library (library 3).
With the standard DDA routine, we identified in each replicate 2536 ± 34 peptides and 622 ± 14 proteins (Figure 2B, Supplementary Table 2A). Combining the data of the first two replicates increased the identified peptides and proteins by 15.2% and 7.5%, respectively. Combining all three replicates resulted in an overall increase of 24.6% and 13.8% for the peptides and proteins, respectively. By using the matching option in MaxQuant, the number of identified peptides increased to 2972 ± 1 peptides and 683 ± 7 proteins (Figure 2B, Supplementary Table 2B), that is, 14.6% more peptide and 9% more protein identifications when compared to the searches without matching. The peptide and protein identifications were highly reproducible such that only 5.1% more peptides and 3.4% more proteins were identified when all three replicates were searched together. Given the concept behind the matching option, these numbers will decrease with an increasing number of replicate runs.
Using our optimized DIA workflow, we observed a significantly larger number of peptides and proteins per replicate, namely 5219 ± 42 peptides and 1200 ± 12 proteins corresponding to an increase of more than 75%. Combining the searches of all three replicate LC–MS runs led to 15.8% and 9.7% increase in identified peptides and proteins, respectively (Figure 2B, Supplementary Table 2C), placing the DIA method in between the standard DDA workflow and the matching-based DDA workflow with respect to the peptide and protein identification reproducibility.
After we evaluated the identification reproducibility, we also assessed the reproducibility of the protein quantification, that is, its precision. To this end, we calculated the coefficients of variation (CV) for all peptide and protein identifications for the DIA data and the DDA data using label free quantification algorithm in MaxQuant as well as simple spectral counting. Of note: for estimating the protein CV, we simply added the intensities of all observed peptides associated with a particular protein. The median CVs of the DIA-based peptide and protein quantifications were 6.7% and 8.1%, respectively, with 65% of the peptides and 57% of the proteins showing CVs of ≤10% (84% of the peptides and 76% of the proteins showed a CV of ≤20% Figure 2C).
In contrast, the median CVs of the DDA-based quantification were with 15.7% and 16.3% for the peptide and protein quantifications, respectively, more than twice as large as the CVs of the DIA-based quantifications. These large CVs meant that only 30% of the peptide and protein quantifications featured CVs of ≤10% (61% of the peptides and 59% of the proteins showed a CV of ≤20%). While these DDA-derived numbers are based on searches without the matching option in MaxQuant, using matching only resulted in negligible differences in the CV values on peptide level (16.6%), whereas the CV on protein level dropped to 12.6%. In addition, we also assessed the quantification precision of a peptide peak area based quantification (MS1 level) of the DIA data set. These data showed a slightly better median CV on peptide (12.9%) and a slightly worse CV on protein level (18.2%) as the quantification of the DDA data, suggesting that the peptide quantification on MS2 level is superior to quantification on MS1 level (Supplementary Figure 4).
For completeness, we also evaluated the CV for spectral counting-based protein quantification. Interestingly, the median CV for this protein quantification was in a similar range as the CVs determined for the area-based quantification, namely 16.1% (Figure 2C). However, the spectral counting method showed a very strong dependence on the spectral count, that is, the quantification reproducibility decreased with lower protein spectral counts indicating that spectral counting delivers surprisingly reproducible results for abundant proteins. On the basis of our data, a spectral count of ~10 is needed to ensure that half of the protein quantifications feature a CV of ≤10%, providing an estimate for a minimum for reproducible spectral counting-based quantification.
By binning the protein in 20 intensity bins and calculation of median CV for each bin, we were able to assess the quantification precision in relation to the protein intensity (Figure 2D). For the peptide peak area based quantification, it is possible to maintain a median quantification precision of 25% across the whole intensity range; only the top 30% proteins, that is, 30% of the proteins associated with the largest signal intensities, could be precisely quantified with a CV < 10%. Spectral counting based quantification is highly precise for the top 10% of the proteins (median CV < 10%; spectral counts above 10); a good precision (median CV < 20%) was maintained for about half of the proteins. In contrast, for the fragment ion based quantification in our DIA workflow, the overall highest median CV was only 16% even for the 5% proteins with the lowest signal intensity. More than half of the proteins in the intensity bins were quantified with a CV < 10%, the highest intense 20% proteins even with a CV < 5% (quantitative data for all three approaches in Supplementary Tables 2A–C).
Several conclusions can be drawn from these assessments of the peptide and protein identification and quantification. First, a single DIA experiment on the Q Exactive HF enabled to almost double the number of identified peptides and proteins compared to a standard DDA experiment. The in-house generated urinary spectral library (library 3) allows us to comprehensively map the urinary proteome within a 30 min LC gradient. Second, the quantification precision with the DIA method is very good and with median CVs in the 7%-range is significantly smaller than the biological CV, which has been reported to be in the 60–70% range.25 This allows omitting replicate runs and to focus on the analysis of additional biological samples, which greatly improves the sample throughput. Third, all information about the sample is recorded in a DIA experiment. In case a new biomarker candidate becomes interesting, the samples can be easily re analyzed. It is not necessary to reacquire the samples as it would be the case for a SRM-like MS method.
Comprehensive Urinary Proteome Coverage by DIA Workflow
After we established the optimized DIA workflow, we applied it to a urine proteomics study comprising 87 samples from patients seen in the ER for abdominal pain. Abdominal pain can be related to various diseases, and therefore diagnosis in the ER is not always easy. To prove the applicability of our workflow in a biomarker discovery studies, we focused on two diagnoses that could be unambiguously made after examination of the patients in the ER: ovarian cyst (12 patients) and urinary tract infection (UTI, 11). We imagine the use of the biomarker candidates in a simple urinary test to either diagnose an ovarian cyst and UTI or to exclude these causes of abdominal pain and to further focus on other abdominal pain-causing conditions. Therefore, we created an abdominal pain control group that comprised urinary samples of patients with other diagnoses and cases in which no cause could be made (64 samples).
After digestion in a 96-well plate format using our in-house developed MStern blotting approach, all 87 samples were analyzed in less than 4 days using our optimized DIA workflow (whole workflow in Figure 1B). As for now, the acquisition of the spectral library more than doubled the instrument time. However, now that a comprehensive urine-specific library has been created, no major instrument time has to be spent for DDA data acquisition, and it can easily be applied in other urinary studies.
Prior to further analysis, we applied an arbitrary minimum threshold of 3000 peptides or 800 proteins for a sample to be considered. This threshold resulted in the removal of three samples: one ovarian cyst sample and two abdominal pain control group samples.
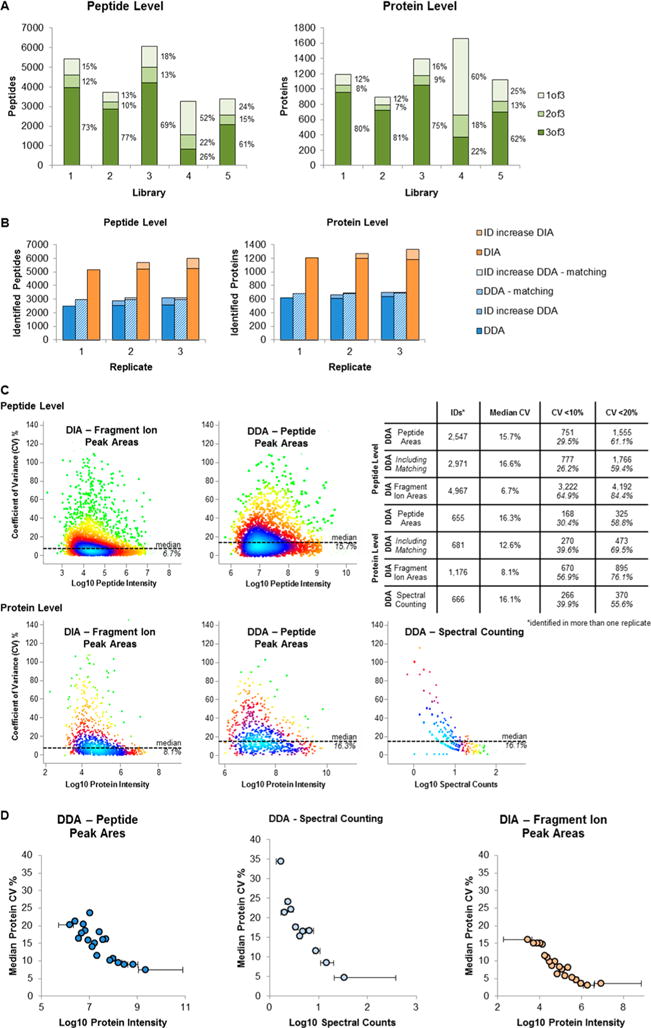
On average, we detected 5714 peptides per sample (3172–8231, Figure 3A) and 1301 protein groups (848–1720, Figure 3B, identification and quantification data in Supplementary Table 3). In total, our DIA workflow resulted in the identification and quantification of 17 303 peptides and 2456 proteins, representing 95% of the proteins in the spectral library. The total number of identified proteins is comparable to recent comprehensive urine proteomics studies.9b,c,26 However, in contrast to these previous studies, we achieved the high coverage of the urinary proteome without any prefractionation and with only 30 min gradient time per sample. Compared to the DDA data, which we acquired to generate the spectral library, the DIA data continuously identified about twice as many proteins in each individual sample: DIA 1301 versus DDA 638, and thus much fewer missing values resulting in more robust protein identification across many samples. Within the DIA data set, 490 proteins were identified in at least 95% of the samples and only 406 proteins in less than 10% of the samples. These numbers are contrasted by the DDA output, in which only 130 proteins were identified in more than 95% of the samples and 838 proteins in less than 10% of the samples (Figure 3C).
Figure 3.
Overview of DIA data set. (A) Overview of peptide identification results. The bar charts (right and left panel) give an overview of the identified peptides/proteins in each of the 87 individual samples using a 30 min gradient on a Q Exactive HF mass spectrometer with a DIA method (yellow, pain control group; purple, ovarian cyst; blue, urinary tract infections). (B) Overview of peptide identification results. (C) Protein sample coverage. We calculated how many proteins were identified in more than 95% of the samples, in 90–95% of the samples, etc. for DIA and DDA data (orange, DIA data; blue, DDA data).
Statistical analysis of the data showed big differences in the composition of the urinary proteome between ovarian cyst, UTI, and pain control group samples. Compared to all other samples, 773 proteins were significantly changed in their amount in the UTI samples (nonparametric Mann–Whitney U test, p < 0.05) and 502 in the ovarian cyst samples. Application of the very conservative Bonferroni multiple testing correction (cut off p = 2.1e–5) reduced the numbers to 55 proteins for the UTI samples and five in the ovarian cyst samples. These numbers reflect the proximal nature of urine for the two conditions: for UTI, urine is the immediate, that is, most proximal body fluid; since ovaries are part of the genitourinary tract, urine might be also considered a proximal body fluid in case of the ovarian cysts.
We also tried to quantify bacterial proteins to detect potential UTI-specific differences. Therefore, we reanalyzed the data set with a new spectral library comprising also bacterial proteins of interest (details in Materials and Methods section, Supplementary Table 1E). Although bacterial proteins were clearly identified in the urine samples, the results were inconclusive (Supplementary Figure 5). We concluded that the limit of detection of the assay may be insufficient to reliably detect sample cohort specific abundance differences of bacterial proteins in the urine samples. Alternatively, the sample handling/processing resulted either in a loss of the majority of the bacteria or in a contamination of the non-UTI urine samples. Nevertheless this analysis showed that the DIA data can be easily reanalyzed once new hypotheses are formulated.
Discovering Disease-Specific Biomarker
The main objective of establishing an optimized DIA workflow was the fast and efficient analysis of a large number of urine samples for biomarker discovery studies. Within this proof of concept study, we were interested in finding urinary biomarker candidates for diagnosing children with UTI or ovarian cyst and to clearly differentiate them from pediatric patients with other abdominal pain causing conditions. For both conditions, excellent urinary biomarker candidates could be identified. The performance of the most significant biomarker candidates was assessed by calculating the area under the receiver-operating characteristic (AUROC) against the other conditions (Figure 4).
Figure 4.
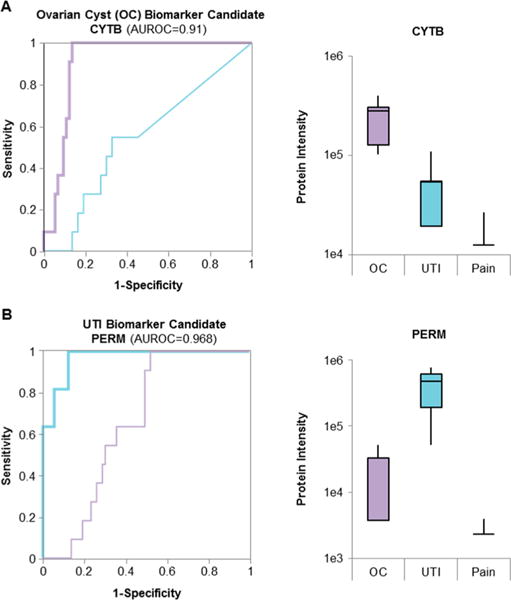
Biomarker candidates. The proteins with the largest area under the ROC (AUROC) were considered the best biomarker candidates. (A) The ROC of the best candidate for ovarian cyst (CYTB, purple) and UTI (PERM, blue) on the ovarian cyst cohort. Diagonal segments are produced by ties. Additionally, the figure shows the intensity of each protein in all conditions as a boxplot. (B) The ROC for CYTB (purple) and PERM (blue) on the UTI sample cohort as well as the protein intensity in all conditions as boxplot.
For ovarian cyst, cystatin-B (CYTB) showed the best performance in separating the ovarian cyst samples from the other samples. Its level was increased by 5.8-fold in the ovarian cyst cohort (p = 1.3e–5, AUROC = 0.91, Figure 4A). Even after the very conservative Bonferroni multiple testing correction was applied, the change in protein level was still statistically significant (q = 0.027). Interestingly, cystatin-B, which is intracellular thiol proteinase inhibitor, has also been described as potential urinary biomarker for bladder cancer27 and as potential biomarker candidate in ovarian clear cell cancer.28 Given the pediatric origin of our urine samples, bladder or ovary cancer is extremely rare, and an ovary cyst diagnosis can be easily confirmed by imaging techniques. Thus, cystatin-B might be a promising biomarker candidate for excluding other potential diagnoses in the context of pediatric patients with abdominal pain.
For the UTI samples with a large number of proteins with significant urinary abundance differences, the protein group PERE/PERM (eosinophil peroxidase/myeloperoxidase) showed the best performance for diagnosing UTI (Figure 4B, AUROC = 0.968). The average observed abundance increase was 122-fold in the UTI samples when compared to the all other non-UTI samples (p = 6.6e–8, q = 1.6e–4). We also identified and quantified peptides unique to PERE and PERM, which allowed us to conclude that the observed abundance increase is caused by PERM. On the basis of the unique PERM peptides, the protein amount is 97-fold increased (p = 1.1e–6, q = 2.8e–3), whereas the PERE-specific peptides did not show a significant abundance difference (p = 0.51). Myeloperoxidase (PERM) has been described as inflammatory marker and is believed to have bactericidal activity in the case of infection.29 A recent study on more than 500 people to identify early stage urinary biomarkers of UTI found that an increased activity of myeloperoxidase in the urine indicates a UTI with high specificity.30 With our study, we were able to explain the increased myeloperoxidase activity by a highly elevated level of the enzyme within in the urine and therefore confirm the myeloperoxidase as potential biomarker for an UTI.
CONCLUSIONS
Here, we present a fast and robust DIA workflow for the efficient, reproducible, and comprehensive quantitative mapping of urinary proteomes, which are ideal for DIA experiments due to their limited complexity in comparison to, for example, whole cell lysates. The established DIA workflow allowed us to analyze 87 urine samples in less than 4 days, that is, 30 min gradients per sample. Without any prefractionation, we identified ~1300 proteins per sample, which is almost twice as many proteins per sample as comparative DDA analyses. This large number significantly reduces the number of missing values, thereby increasing the confidence in identifying relevant biomarker candidates. Interestingly, despite the doubled number of identified proteins, the quantification CV halved, that is, improved from ~16% to ~8%. In summary, this DIA workflow for urine proteomics allows for a sufficient throughput to perform biomarker discovery studies that combine discovery and verification31 as all identified proteins can be precisely quantified in hundreds of samples; thus, the typical focusing on a few selected biomarker candidates during the verification is not necessary any longer (Figure 5).
Figure 5.
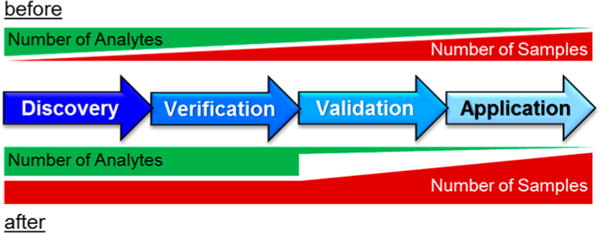
Advanced biomarker research scheme. In a conventional biomarker experiment, the biomarker discovery with high proteome coverage is performed on a small subset of samples (before). Toward the further verification and validation of the candidates, the number of samples is increased, whereas through the application of targeted methods, fewer and fewer analytes are monitored. Focusing on a small number of candidates in the verification phase can result in missed biomarkers. Our optimized DIA workflow enables to keep a high number of analytes throughout the whole discovery and verification phase, increasing the robustness of biomarker discovery in the future (after).
To show the applicability of the DIA workflow in a biomarker discovery study, we analyzed urine samples from children seen in the ER for abdominal pain and identified biomarker candidates for UTI and ovarian cysts. An extension of the study to other diseases will bring us a step closer to quickly stratifying children with abdominal pain simply based on their urine composition.
Supplementary Material
Acknowledgments
We thank Judith Steen for insightful discussions and critically reading the manuscript. Furthermore, we acknowledge the following funding. The TripleTOF 5600 used for some of the DDA experiments was funded by a U.S. National Institute of Health Shared Instrumentation Grant No. S10OD010706 to H.S. Part of this work was made possible by a grant from the Harvard Institute of Translational Immunology and through support from the Leona M. and Harry B. Helmsley Charitable Trust. Part of A.K.’s efforts was funded by the U.S. National Institute of Health Grant No. K08CA160660.
ABBREVIATIONS
- AUROC
area under the receiver-operating characteristics
- DDA
data-dependent acquisition
- DIA
data-independent acquisition
- FDR
false discovery rate
- iRT
indexed retention times
- IT
injection time
- ROC
receiver-operating characteristics
- UTI
urinary tract infection
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.5b00826.
Histogram of precursor m/z of a typical urine sample; peak Width Comparison for 30 and 75 min LC–MS runs; overlap of peptides and protein groups between the spectral libraries and the identification results; quantification precision for MS1 signal intensity based quantification of DIA data set; bacterial protein analysis (PDF)
Spectral library based on 23 Q exactive LC–MS/MS runs (XLSX)
Spectral library based on 23 tripleTOF 5600 LC–MS/MS runs (XLSX)
Comprehensive urinary spectral library (XLSX)
Spectral library based on the protein overlap of the comprehensive spectral library and the human spectral library (Rosenberger et al., 2014) (XLSX)
Spectral library applied in the bacterial protein analysis (XLS)
Identification and quantification result from DIA triplicate runs without using the matching option in MaxQuant (XLSX)
Identification and quantification result from DDA triplicate runs using the matching option in MaxQuant (XLSX)
Identification and quantification result from DDA triplicate runs (XLSX)
Biomarker discovery study, peptide identification, and quantification data (XLSX)
Biomarker discovery study, protein identification, and quantification data (XLSX)
Author Contributions
J.M. carried out the DDA experiments on the Q Exactive/TripleToF 5600 and performed the data analysis. Y.X. carried out all LC–MS experiments on the Q Exactive HF and contributed in writing the manuscript. S.T.B. processed the samples. L.R. contributed to the data analysis. R.B. and A.K. designed the sample cohorts and collected the samples. H.S., Y.X., and J.M. designed the experiments. J.M. and H.S. interpreted the results and wrote the manuscript. All authors critically read and revised the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Wu J, Chen YD, Gu W. Urinary proteomics as a novel tool for biomarker discovery in kidney diseases. J Zhejiang Univ Sci B. 2010;11:227–37. doi: 10.1631/jzus.B0900327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lopez-Giacoman S, Madero M. Biomarkers in chronic kidney disease from kidney function to kidney damage. World J Nephrol. 2015;4:57–73. doi: 10.5527/wjn.v4.i1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pedroza-Diaz J, Rothlisberger S. Advances in urinary protein biomarkers for urogenital non-urogenital pathologies. Biochem Med (Zagreb) 2015;25:22–35. doi: 10.11613/BM.2015.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mischak H, Delles C, Vlahou A, Vanholder R. Proteomic biomarkers in kidney disease: issues in development implementation. Nat Rev Nephrol. 2015;11:221–32. doi: 10.1038/nrneph.2014.247. [DOI] [PubMed] [Google Scholar]
- 2.Kentsis A, Shulman A, Ahmed S, Brennan E, Monuteaux MC, Lee YH, Lipsett S, Paulo JA, Dedeoglu F, Fuhlbrigge R, Bachur R, Bradwin G, Arditi M, Sundel RP, Newburger JW, Steen H, Kim S. Urine proteomics for discovery of improved diagnostic markers of Kawasaki disease. EMBO molecular medicine. 2013;5:210–20. doi: 10.1002/emmm.201201494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zimmerli LU, Schiffer E, Zurbig P, Good DM, Kellmann M, Mouls L, Pitt AR, Coon JJ, Schmieder RE, Peter KH, Mischak H, Kolch W, Delles C, Dominiczak AF. Urinary proteomic biomarkers in coronary artery disease. Mol Cell Proteomics. 2007;7:290–8. doi: 10.1074/mcp.M700394-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Adeola HA, Soares NC, Paccez JD, Kaestner L, Blackburn JM, Zerbini LF. Discovery of novel candidate urinary protein biomarkers for prostate cancer in a multi-ethnic cohort of South African patients via label-free mass spectrometry. Proteomics: Clin Appl. 2015;9:597–609. doi: 10.1002/prca.201400197. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kentsis A, Lin YY, Kurek K, Calicchio M, Wang YY, Monigatti F, Campagne F, Lee R, Horwitz B, Steen H, Bachur R. Discovery validation of urine markers of acute pediatric appendicitis using high-accuracy mass spectrometry. Annals of emergency medicine. 2010;55:62–70. doi: 10.1016/j.annemergmed.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kentsis A, Ahmed S, Kurek K, Brennan E, Bradwin G, Steen H, Bachur R. Detection diagnostic value of urine leucine-rich alpha-2-glycoprotein in children with suspected acute appendicitis. Annals of emergency medicine. 2012;60:78–83. doi: 10.1016/j.annemergmed.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young BL, Mlamla Z, Gqamana PP, Smit S, Roberts T, Peter J, Theron G, Govender U, Dheda K, Blackburn J. The identification of tuberculosis biomarkers in human urine samples. Eur Respir J. 2014;43:1719–29. doi: 10.1183/09031936.00175113. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Chen J, Chen L, Zheng P, Xu HB, Lu J, Zhong J, Lei Y, Zhou C, Ma Q, Li Y, Xie P. Urinary peptidomics identifies potential biomarkers for major depressive disorder. Psychiatry Res. 2014;217:25–33. doi: 10.1016/j.psychres.2014.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Spahr CS, Davis MT, McGinley MD, Robinson JH, Bures EJ, Beierle J, Mort J, Courchesne PL, Chen K, Wahl RC, Yu W, Luethy R, Patterson SD. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I. Profiling an unfractionated tryptic digest. Proteomics. 2001;1:93–107. doi: 10.1002/1615-9861(200101)1:1<93::AID-PROT93>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.(a) Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human urinary proteome contains more than 1500 proteins including a large proportion of membrane proteins. Genome Biol. 2006;7:R80. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kentsis A, Monigatti F, Dorff K, Campagne F, Bachur R, Steen H. Urine proteomics for profiling of human disease using high accuracy mass spectrometry. Proteomics: Clin Appl. 2009;3:1052–1061. doi: 10.1002/prca.200900008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zheng J, Liu L, Wang J, Jin Q. Urinary proteomic non-prefractionation quantitative phosphoproteomic analysis during pregnancy non-pregnancy. BMC Genomics. 2013;14:777. doi: 10.1186/1471-2164-14-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 11.Michalski A, Cox J, Mann M. More than 100 000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res. 2011;10:1785–93. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 12.(a) Zweigenbaum J, Henion J. Bioanalytical high-throughput selected reaction monitoring-LC/MS determination of selected estrogen receptor modulators in human plasma: 2000 samples/day. Anal Chem. 2000;72:2446–54. doi: 10.1021/ac991413p. [DOI] [PubMed] [Google Scholar]; (b) Method of the Year 2012. Nat Methods. 2013;10 doi: 10.1038/nmeth.2329. 1–2.10.1038/nmeth.2329. [DOI] [PubMed] [Google Scholar]
- 13.Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27:190–8. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huttenhain R, Soste M, Selevsek N, Rost H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Nimeus-Malmstrom E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012;4:142–94. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent accurate proteome analysis. Mol Cell Proteomics. 2012;11:1–17. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bruderer R, Bernhardt OM, Gandhi T, Miladinovic SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, Vitek O, Rinner O, Reiter L. Extending the limits of quantitative proteome profiling with data-independent acquisition application to acetaminophen treated 3D liver microtissues. Mol Cell Proteomics. 2015;14:1400–10. doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M. The Q Exactive HF a Benchtop mass spectrometer with a pre-filter high-performance quadrupole an ultra-high-field Orbitrap analyzer. Mol Cell Proteomics. 2014;13:3698–708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger ST, Ahmed S, Muntel J, Cuevas Polo N, Bachur R, Kentsis A, Steen J, Steen H. MStern blotting - high throughput PVDF membrane-based proteomic sample preparation for 96-well plates. Mol Cell Proteomics. 2015;14:1–32. doi: 10.1074/mcp.O115.049650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox J, Mann M. MaxQuant enables high peptide identification rates individualized p.p.b.-range mass accuracies proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberger G, Koh CC, Guo T, Röst HL, Kouvonen P, Collins BC, Heusel M, Liu Y, Caron E, Vichalkovski A, Faini M, Schubert OT, Faridi P, Ebhardt HA, Matondo M, Lam H, Bader SL, Campbell DS, Deutsch EW, Moritz RL, Tate S, Aebersold R. A repository of assays to quantify 10 000 human proteins by SWATH-MS. Sci Data. 2014;1:1–15. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–8. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schubert OT, Mouritsen J, Ludwig C, Rost HL, Rosenberger G, Arthur PK, Claassen M, Campbell DS, Sun Z, Farrah T, Gengenbacher M, Maiolica A, Kaufmann SH, Moritz RL, Aebersold R. The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe. 2013;13:602–12. doi: 10.1016/j.chom.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O. Using iRT a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–21. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muntel J, Hecker M, Becher D. An exclusion list based label-free proteome quantification approach using an LTQ Orbitrap. Rapid Commun Mass Spectrom. 2012;26:701–9. doi: 10.1002/rcm.6147. [DOI] [PubMed] [Google Scholar]
- 24.Smith RD, Anderson GA, Lipton MS, Pasa-Tolic L, Shen Y, Conrads TP, Veenstra TD, Udseth HR. An accurate mass tag strategy for quantitative high-throughput proteome measurements. Proteomics. 2002;2:513–23. doi: 10.1002/1615-9861(200205)2:5<513::AID-PROT513>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 25.Nagaraj N, Mann M. Quantitative analysis of the intra- inter-individual variability of the normal urinary proteome. J Proteome Res. 2011;10:637–45. doi: 10.1021/pr100835s. [DOI] [PubMed] [Google Scholar]
- 26.Marimuthu A, O’Meally RN, Chaerkady R, Subbannayya Y, Nanjappa V, Kumar P, Kelkar DS, Pinto SM, Sharma R, Renuse S, Goel R, Christopher R, Delanghe B, Cole RN, Harsha HC, Pandey A. A comprehensive map of the human urinary proteome. J Proteome Res. 2011;10:2734–43. doi: 10.1021/pr2003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman AS, Banyard J, Wu CL, McDougal WS, Zetter BR. Cystatin B as a tissue urinary biomarker of bladder cancer recurrence disease progression. Clin Cancer Res. 2009;15:1024–31. doi: 10.1158/1078-0432.CCR-08-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takaya A, Peng WX, Ishino K, Kudo M, Yamamoto T, Wada R, Takeshita T, Naito Z. Cystatin B as a potential diagnostic biomarker in ovarian clear cell carcinoma. Int J Oncol. 2015;46:1573–81. doi: 10.3892/ijo.2015.2858. [DOI] [PubMed] [Google Scholar]
- 29.Podrez EA, Abu-Soud HM, Hazen SL. Myeloperoxidase-generated oxidants atherosclerosis. Free Radical Biol Med. 2000;28:1717–25. doi: 10.1016/s0891-5849(00)00229-x. [DOI] [PubMed] [Google Scholar]
- 30.Ciragil P, Kurutas EB, Miraloglu M. New markers: urine xanthine oxidase myeloperoxidase in the early detection of urinary tract infection. Dis Markers. 2014;2014:1–5. doi: 10.1155/2014/269362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery validation: the long uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–83. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.