

Abstract
Cryo-scanning electron microscopy (SEM) of freeze-fractured samples allows investigation of biological structures at near native conditions. Here, we describe a technique for studying the supramolecular organization of photosynthetic (thylakoid) membranes within leaf samples. This is achieved by high-pressure freezing of leaf tissues, freeze-fracturing, double-layer coating and finally cryo-SEM imaging. Use of the double-layer coating method allows acquiring high magnification (>100,000X) images with minimal beam damage to the frozen-hydrated samples as well as minimal charging effects. Using the described procedures we investigated the alterations in supramolecular distribution of photosystem and light-harvesting antenna protein complexes that take place during dehydration of the resurrection plant Craterostigma pumilum, in situ.
Keywords: Plant Biology, Issue 112, High-pressure freezing, freeze-fracture, double-layer coating, cryo-scanning electron microscopy, resurrection plants, chloroplast, thylakoid membrane, supramolecular organization, exoplasmic fracture face, protoplasmic fracture face, photosystem
Introduction
Oxygenic photosynthesis, originating in ancient cyanobacteria, was inherited by algae and land plants by endosymbiotic events that led to development of the chloroplast organelle. In all modern-day oxygenic phototrophs, photosynthetic electron transport and the generation of proton-motive force and reducing power are carried out within flattened sac-like vesicles termed 'thylakoid' membranes. These membranes house the protein complexes that carry out the light-driven reactions of photosynthesis and provide a medium for energy transduction. The thylakoid membranes of plants and (some) algae are differentiated into two distinct morphological domains: tightly appressed membrane regions called 'grana' and unstacked membranes that interconnect the grana, called 'stroma lamellae'1. Various freeze-fracture studies of plant and algal thylakoid membranes have been conducted, starting in the early 1970s. When freeze-fractured, membranes split along their hydrophobic core2, generating an exoplasmic face (EF) and a protoplasmic face (PF), depending on the cellular compartment which the half-membrane borders, as originally coined by Branton et al. in 19753. Plant and algal thylakoids have four different fracture faces: EFs, EFu, PFs and PFu, with 's' and 'u' denoting 'stacked' and 'unstacked' membrane regions, respectively. The membrane protein complexes, which are not split or broken, have the tendency to remain with either the E or P side of the membrane. The initial observations that the different fracture faces of the thylakoids contain particles of different sizes and densities4, and the numerous investigations that followed, led to identification and correlation between the observed particles and the membrane protein complexes that carry out the light reactions5-13 (see also reviews14,15).
Freeze-fracture experiments of thylakoid membranes are typically carried out on preparations of chloroplasts or isolated thylakoid membranes (but see16,17), at the risk of any alteration in structural and/or supramolecular organization that may occur during the isolation procedure. Following fracture, replicas are prepared by evaporation of platinum/carbon (Pt/C), then by a thick layer of carbon (C), and finally digestion of the biological material18. Replicas are visualized by transmission electron microscopy (TEM). The traditional freeze-fracture-replica technique continues to serve as an important tool for studying the supramolecular organization of photosynthetic membranes and their adaption to different, e.g., light, conditions19-23.
In our recent study of the homoiochlorophyllous resurrection plant Craterostigma pumilum24, we aimed to investigate the changes in the supramolecular organization of thylakoid membranes, as well as in overall cellular organization, during dehydration and rehydration. The uniqueness of homoiochlorophyllous resurrection species is that they are able to survive conditions of desiccation in their vegetative tissues (leaves), while retaining their photosynthetic apparatus. Once water is available, these plants recover and resume photosynthetic activity within hours to a few days25. For this study, cryo-scanning EM (SEM) imaging of freeze-fractured leaf samples was combined with high-pressure freezing for sample cryo-immobilization. These procedures provide a means to visualize frozen-hydrated biological samples at a state close to their native state26. One main benefit is that samples are examined directly after freeze-fracture and coating with no successive steps. This is particularly relevant to the investigation of plants at different relative water contents (RWC), as their hydration state is maintained during preparation. However, one critical disadvantage is that frozen-hydrated samples may suffer from beam damage during imaging, especially when scanned at high magnifications, required for accurate measurement of the size of photosynthetic complexes. To overcome this, a method called 'double-layer coating' (DLC)27,28 combined with specific cryo-SEM imaging conditions were utilized. These resulted in samples that are significantly less beam-sensitive and allowed for the elucidation of valuable information on photosynthetic protein supramolecular organization and other cellular constituents of the resurrection plant C. pumilum at high magnifications in situ.
Protocol
1. Cryo-fixation of Leaf Tissues by High-pressure Freezing
Note: This section describes how to carry out high-pressure freezing of leaf tissues for a freeze-fracture experiment. For considerations related to plant samples see29. This can be adapted for other types of tissues or samples with some modification.
- Using the corner of a razor blade, scratch the bottom of the 0.1 mm cavity of a 0.1/0.2 mm aluminum platelet (Figure 1). Prepare at least a dozen platelets (6 samples). Take caution not to create knife marks on the circumference of the disc.
- After scratching, wash discs in absolute ethanol, let them dry and keep in a clean dish.
Prepare the high pressure freezing machine according to the manufacturer's instructions. Fill the high pressure freezing machine's alcohol chamber with isopropanol.
- Prepare a home-made vacuum-assisted infiltration device to replace gas found in the leaf tissue with liquid.
- Tightly connect a 200 µl pipette tip to the end of a 10 ml syringe. Cut off ~1 cm of the tip. Make a hole in the cap of a 1.5 ml microcentrifuge tube to tightly fit the cut tip. Vacuum can be created inside the tube by pulling the plunger of the syringe.
Cut a small piece of leaf from the plant using a razor blade and with tweezers place the leaf inside a microcentrifuge tube half-filled with 1-hexadecene. Infiltrate the intercellular spaces of the leaf with the liquid by gently pulling the plunger to create vacuum, hold for about 30 sec, and then slowly release the plunger.
Remove the leaf piece from the tube using tweezers. Trim the leaf with a razor blade in case it is thicker than 0.2 mm and cut a small piece that will fit into a platelet.
Place a clean platelet with the scratched side up into the freezing machine's holder and then place the cut piece of leaf into the platelet. Fill the remaining space surrounding the leaf with 1-hexadecene. Close the sandwich using another platelet, with the scratches facing the leaf.
Close and tighten the holder and insert it into the machine's chamber. Then press the 'Jet' button to freeze (~2,100 bars for 300 - 500 msec), and quickly transfer the holder into liquid nitrogen. Carefully remove the frozen sandwich from the holder using pre-cooled tweezers, taking care not to fracture the sandwich. Note: Closed frozen sandwiches can be freeze fractured immediately or kept in liquid nitrogen for later use. Use protective clothing and eyewear when handling liquid nitrogen.
2. Freeze-fracture and Double-layer Coating27,28
- Prepare the freeze fracture machine for use, including both guns for evaporation of platinum/carbon (Pt/C) and carbon, according to the manufacturer's instructions. Place the Pt/C gun at 45 degrees and the carbon gun at 90 degrees.
- Pre-program a coating scheme of a 2-nm layer of Pt/C (coating rate 0.02 - 0.04 nm/sec) and a 6-nm layer of carbon (at ~0.1 nm/sec).
- Enter the setup on the freeze-fracture machine's screen. Select a user number where the coating scheme will be saved.
- Choose Pt/C for 'Layer 1'. Select a time frame, e.g., 5 min (to exceed the necessary time needed to complete the coating). Use the up-down arrows to define a thickness of 2 nm.
- Press the 'Layer' button to move to Layer 2. Repeat step 2.1.1.2, except select carbon and define a thickness of 6 nm.
- Test and adjust gun operation.
- Select 'Layer 1'. Select the Pt/C gun by pressing the 'GUN1' button and press the 'ON' button on the evaporation control unit of the freeze-fracture machine. Monitor the evaporation rate and adjust it using the voltage and emission knobs until reaching a rate of 0.02 - 0.04 nm/sec. Press 'OFF' to turn off the Pt/C gun.
- Select 'Layer 2'. Repeat 2.1.2.1, except select 'GUN2' and adjust for an evaporation rate of ~0.1 nm/sec.
Cool the freeze fracture system stage to -140 ˚C or -160 ˚C (for hydrated or dehydrated leaves, respectively). Attach the vacuum cryo transfer shuttle to the machine and fill its chamber with liquid nitrogen. Pressure in the cooled freeze-fracture chamber is usually between 1 - 8 x 10-7 mbar.
Insert a freeze fracturing specimen holder (suitable for 3-mm platelets) into the loading station. Flood the loading station chamber with liquid nitrogen.
Load two frozen sandwiches onto the holder one by one using high-precision grade tweezers. Tighten the first sandwich into the holder and then flip the holder over to check that it fits securely in place. Repeat for the second sandwich.
Detach the cooled shuttle from the freeze fracture machine and connect it to the loading station. Open the shuttle valve, introduce the retracting arm into the holder and lock it into place. Re-introduce the arm with the holder into the shuttle and close the shuttle valve using the loading station knob. Attach the shuttle to the freeze-fracture machine and introduce the holder into the chamber with the retracting arm. Detach the shuttle from the machine.
Align the freeze fracture microtome knife to a height such that it will knock off the top platelets of the sandwiches.
With the microtome knife, quickly break the sandwiches, and then immediately raise it up to prevent contamination of the samples by debris clinging to the knife, and turn the cooled shutter above the newly exposed plane to shield the samples from possible contamination by water molecules in the chamber.
- Coat the samples with platinum and carbon.
- Enter 'Layer 1' on the screen. Turn on the Pt/C gun by pressing the 'GUN1' button followed by the 'ON' button. Examine the evaporation rate and once it reaches 0.02 - 0.04 nm/sec, simultaneously expose the samples and press the 'Measure' button to monitor the thickness of the deposited layer. Note: Gun will turn off automatically when the thickness has reached the pre-selected value.
- Cover the samples with the shutter when Pt/C evaporation is completed.
- Press the 'Layer' button to move to 'Layer 2'. Repeat steps 2.8.1 and 2.8.2, with the exception of selecting 'GUN2' (for carbon) and an evaporation rate of ~0.1 nm/sec.
Warm the freeze fracture system stage to -120 ˚C.
3. Cryo-scanning Electron Microscopy
- Prepare the scanning electron microscope for cryo work.
- Select Stage/Stage Initialise from the main menu and confirm.
- Select Tools/Goto Panel, and open the 'Airlock' panel by selecting it from the list. Click the 'Close Column Chamber Valve' button. Vent the microscope chamber by clicking the Vent button under the 'Vacuum' tab.
- Select Tools/Goto Panel, and open the 'Stage Points List' panel. Select the cryo-exchange position to move the stage to the proper coordinates for accepting the sample holder. Note: The 'cryo-exchange position' is preset to be in alignment with the position of the vacuum transfer unit.
- Open the specimen chamber with a clean pair of gloves and insert the cryo-stage onto the main stage of the microscope. Close the chamber and click the Pump button under the 'Vacuum tab'.
- Cool the microscope to -120 ˚C. Fill the microscope Dewar with liquid nitrogen. Activate the heat function on the cryo-transfer control unit to -120 ˚C once the temperature of the stage drops below -120 ˚C.
Attach the cryo-transfer shuttle to the microscope and transfer the sample holder using the retracting arm into the microscope cryo stage (as in step 2.5). Detach the shuttle from the microscope.
Click on the 'Open Column Chamber Valve' button in the Airlock Panel.
Carefully move the specimen holder close to the objective lens (working distance 1 - 2 mm) using the joystick. Select a 10-µm aperture in the 'Apertures' Tab. Double-click the EHT field in the 'Gun' tab. Enter 10 (kV) for the EHT target and click OK. Click on the bottom EHT tab and select 'EHT On'.
Select 'InLens' from the Detectors drop-down list in the 'Detectors' tab. Optimize imaging conditions (focus, brightness and contrast, beam alignment, astigmatism) as appropriate.
Click on the 'Scanning' tab. Choose a fast scan ('Scan Speed = 1', corresponding to a dwell time of 100 nsec per pixel) to minimize beam damage, and a 'Store resolution' of 1,024 x 768 pixels. Select 'Line Avg' in the 'Noise Reduction' drop-down list. Adjust the value of N to 20 - 30 lines for searching. Move the sample using the 'Centre Point' function (by pressing control-tab on the keyboard) to search for regions with fractured cells and chloroplasts (Figures 2A and 2B).
- Acquire images of fractured chloroplasts at low magnifications (50 - 70 kX) (Figure 2C and 2D). Use the 'Centre Feature' function (by pressing control-shift-tab on the keyboard) to zoom into interesting chloroplast fracture faces and acquire images at high magnifications (100 - 200 kX, Figures 3 and 4). Use the same parameters as in step 3.6 for image acquisition, but change the Line Avg value to N >100 for noise reduction30.
- Press Freeze on the main microscope control unit or in the 'Scanning' tab to stop the scanning and save the image by pressing the 'Save TIFF' button.
4. Image Analysis
Note: This section describes a short procedure for segmentation of membrane particles from freeze-fracture SEM images using the Fiji31 open-source package. Similar results can be obtained with other image analysis software.
Open image with Fiji by dragging image file into the main program window. Convert the image to 8-bit by selecting Image/Type/8-bit. Select Image/Adjust/Brightness/Contrast and adjust as necessary (Figure 5A).
Using the Straight Line tool, draw a line the size of the embedded image scale bar. Select Analyze/Set Scale. Enter the proper scale information (Known distance and Unit of length) and click OK. Save this adjusted scaled image.
Select Process/Filters/Gaussian blur (1.5 nm radius) and click OK (Figure 5B).
Select Image/Adjust/Auto Local Threshold (using the Niblack method) and click OK (Figure 5C).
Select Edit/Invert and then Process/Binary/Watershed (Figure 5D).
Draw a border around the region of interest (ROI) with the Freehand Selection tool. Add the selection to the ROI manager by selecting Edit/Selection/Add to Manager or by clicking 'T'.
Select Edit/Clear Outside (Figure 5E).
Select Analyze/Set Measurements, choose the appropriate parameters (e.g., area, fit ellipse).
Select Analyze/Analyze Particles. Enter an appropriate range for the size of particles and mark 'Display results', 'Add to Manager' and, if necessary, the 'Exclude on edges' option and click OK. The result is shown in Figure 5F.
Open the saved scaled imaged file (step 4.2). Select 'Show All' and deselect 'Labels' in the ROI Manager. Review the selected particles laid onto the original image and manually add or remove selections by using the 'Add' or 'Delete' buttons of the ROI Manager. Correct any selections necessary by using the Freehand Selection tool and the 'Update' button of the ROI Manager (Figures 5G and 5H).
Analyze the data using the obtained measurements. In the Results window, click Results/Summarize to obtain, for example, the mean Area, and mean Major and Minor axes of the particles listed in the ROI Manager. Click Results/Distribution, select a Parameter (such as Area) and click OK to view a histogram for this parameter.
Representative Results
Figure 1 shows cryo-SEM images of platelets containing high-pressure frozen, freeze-fractured Craterostigma pumilum leaf pieces. In some samples, large regions of fractured cells are obtained (Figure 1A). In others, the leaf piece stays tightly bound to the upper disc and is knocked off along with it (Figure 1B). However, even in the second case, some leaf tissue may remain attached to the knife grooves on the platelet (Figure 1B, arrowheads) and a fair amount of data can still be collected by imaging the leaf "remnants", provided they were fractured. After successful fracture is found, low magnification images of cells are acquired to identify regions of interest, in this case chloroplasts (Figure 2).
Higher-magnification images of thylakoid membranes in C. pumilum leaves are shown in Figures 3 and 4. The four different fracture faces, EFs, EFu, PFs and PFu, can be distinguished (Figure 3). Photosystem II (PSII), which is the largest protein complex found in the membranes, is located in both the EFs (grana) and EFu (stroma lamellae). At hydrated conditions, PSII density is ~3 times higher in EFs than in EFu (~1,550 complexes µm-2 vs. ~550 complexes µm-2, Figure 3). This is one basis for differentiating between these two continuous faces. Another is the difference in their background. While the EFs background appears smooth, the EFu face is rough and contains holes that are the footprints of detached PSI complexes, which fracture to the complementary face, the PFu10 (Figure 3). An example for the segmentation of PSII complexes from the EFs face of the thylakoid membrane is shown in Figure 5.
During dehydration of C. pumilum, the density of PSII in the grana membranes (EFs) gradually decreases, reaching roughly half (~700 complexes µm-2 at ~15% RWC, Figure 4C) of their density at hydrated conditions (100% RWC, Figure 4A). Notably, upon further dehydration, to 5 - 10% RWC, PSII complexes organize into rows and arrays (arrows, Figures 4D and 4E). For complete dataset, see24. Formation of such PSII arrays necessitates that some of their bound antenna complexes, LHCII, detach from PSII. An example for LHCII complexes organized into rows in the PFs that would be complementary to organized PSII complexes (in the EFs) is shown in Figure 4E (arrowhead). PSII complexes in the arrays, as well as the detached LHCII, are likely to be in a photochemically quenched state, serving a photo-protective role. These structural rearrangements are part of the mechanisms utilized by the resurrection plant C. pumilum to protect itself in the dehydrated state24.
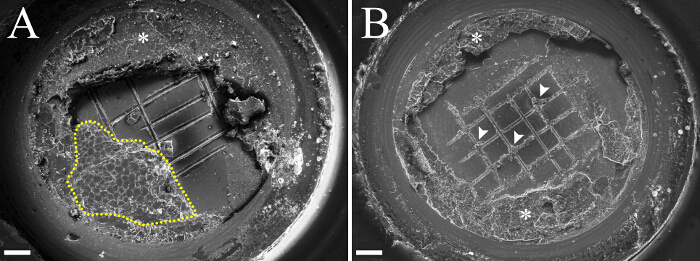 Figure 1. Low Magnification Cryo-SEM Images of Platelets with Freeze-fractured C. pumilum Leaf Pieces. (A) A large region of fractured cells (dashed outline) of a C. pumilum leaf. (B) The leaf piece was knocked off with the top platelet, but some leaf tissue remained attached to the knife marks at the bottom platelet (arrowheads mark some of these). The regions surrounding the leaf pieces are frozen 1-hexadecene (asterisks). Scale bars: 200 µm. Please click here to view a larger version of this figure.
Figure 1. Low Magnification Cryo-SEM Images of Platelets with Freeze-fractured C. pumilum Leaf Pieces. (A) A large region of fractured cells (dashed outline) of a C. pumilum leaf. (B) The leaf piece was knocked off with the top platelet, but some leaf tissue remained attached to the knife marks at the bottom platelet (arrowheads mark some of these). The regions surrounding the leaf pieces are frozen 1-hexadecene (asterisks). Scale bars: 200 µm. Please click here to view a larger version of this figure.
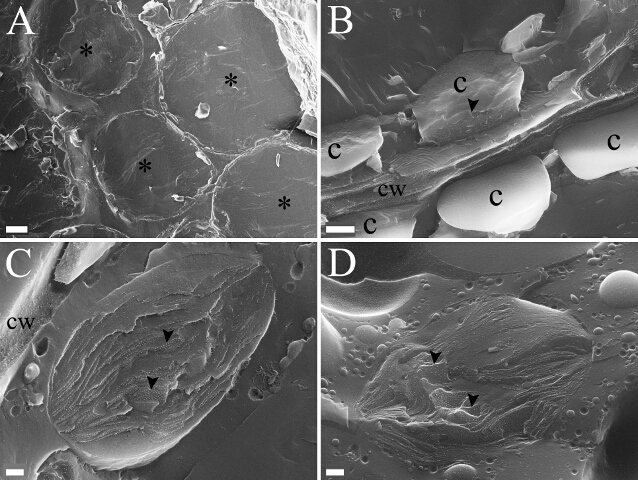 Figure 2. Zooming into Chloroplasts. (A) A group of fractured photosynthetic (mesophyll) cells (asterisks) of hydrated leaf tissue. Most of the cell volume is comprised of a large central vacuole, with all cytoplasmic constituents surrounding the vacuole in a narrow strip. (B) Two neighboring fractured cells - the cell wall (cw) marks the border between the cells. Five chloroplasts (c) are apparent in the image, only one of which has been fractured (fractured plane marked by the arrowhead). (C and D) Examples for single fractured chloroplasts from hydrated (C) and dehydrated (D, ~15% relative water content [RWC]) plants. The small white dots (e.g., arrowheads) are the membrane protein complexes found within the thylakoid membranes of the chloroplast. Note the massive vesiculation surrounding the chloroplast found in the dehydrated leaf (D). Scale bars: 10 µm (A); 1 µm (B); 200 nm (C and D). Please click here to view a larger version of this figure.
Figure 2. Zooming into Chloroplasts. (A) A group of fractured photosynthetic (mesophyll) cells (asterisks) of hydrated leaf tissue. Most of the cell volume is comprised of a large central vacuole, with all cytoplasmic constituents surrounding the vacuole in a narrow strip. (B) Two neighboring fractured cells - the cell wall (cw) marks the border between the cells. Five chloroplasts (c) are apparent in the image, only one of which has been fractured (fractured plane marked by the arrowhead). (C and D) Examples for single fractured chloroplasts from hydrated (C) and dehydrated (D, ~15% relative water content [RWC]) plants. The small white dots (e.g., arrowheads) are the membrane protein complexes found within the thylakoid membranes of the chloroplast. Note the massive vesiculation surrounding the chloroplast found in the dehydrated leaf (D). Scale bars: 10 µm (A); 1 µm (B); 200 nm (C and D). Please click here to view a larger version of this figure.
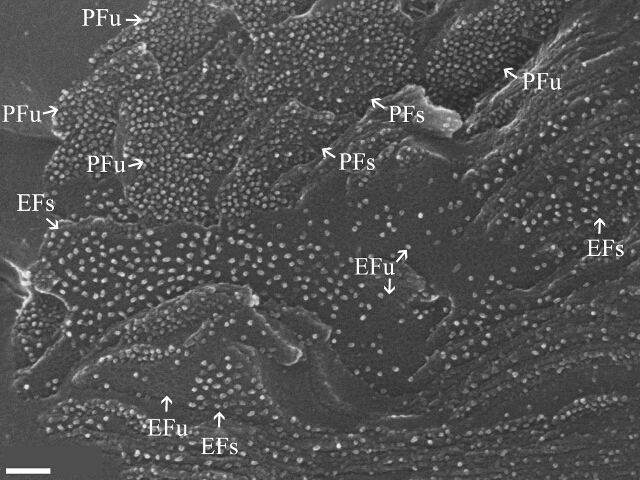 Figure 3. The Supramolecular Organization of Thylakoid Membranes within Plant Tissues. A fractured chloroplast in which the different thylakoid fracture faces can be seen: the exoplasmic fracture faces (EF) of both stacked (EFs) and unstacked (EFu) membrane regions contain photosystem II (PSII) complexes; The protoplasmic fracture face of stacked regions (PFs) contains the peripheral light-harvesting antenna complexes of PSII, LHCII; Photosystem I (PSI) and ATP synthase fracture to the protoplasmic fracture face of unstacked regions (PFu); Cytochrome b6f fractures to both PFs and PFu. Image was scanned at a magnification of 203 kX (horizontal field of view = 1.45 µm). Scale bar: 100 nm. Please click here to view a larger version of this figure.
Figure 3. The Supramolecular Organization of Thylakoid Membranes within Plant Tissues. A fractured chloroplast in which the different thylakoid fracture faces can be seen: the exoplasmic fracture faces (EF) of both stacked (EFs) and unstacked (EFu) membrane regions contain photosystem II (PSII) complexes; The protoplasmic fracture face of stacked regions (PFs) contains the peripheral light-harvesting antenna complexes of PSII, LHCII; Photosystem I (PSI) and ATP synthase fracture to the protoplasmic fracture face of unstacked regions (PFu); Cytochrome b6f fractures to both PFs and PFu. Image was scanned at a magnification of 203 kX (horizontal field of view = 1.45 µm). Scale bar: 100 nm. Please click here to view a larger version of this figure.
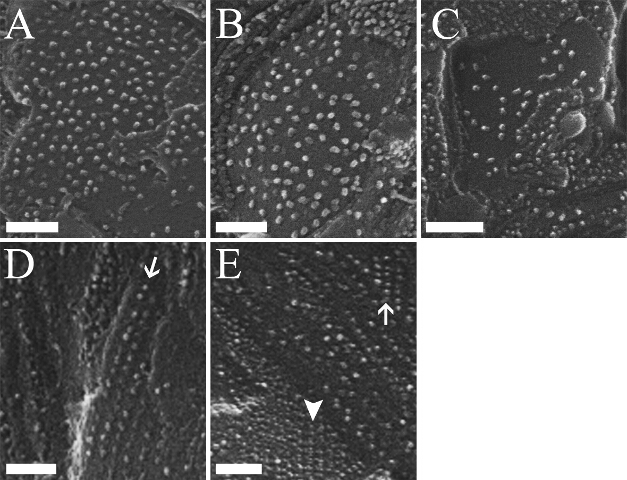 Figure 4. Changes in Photosystem II Organization during Dehydration of C. pumilum. EFs faces of thylakoid membranes of plants at different RWC: (A) 100%, (B) ~40%, (C) ~15%, and (D and E) 5 - 10%. During dehydration, the density of PSII in the EFs face gradually decreases from ~1,500 complexes µm-2 (A) to ~700 complexes µm-2 (~15% RWC, C). Notably, at drier conditions (5 - 10% RWC), some PSII complexes organize into rows and arrays (D and E, arrows). The arrowhead (E) marks the PFs face, with LHCII antenna appearing to also be organized in rows, in between which PSII rows would be found in the complementary EFs. For additional information on this data see24. Scale bars: 100 nm. Please click here to view a larger version of this figure.
Figure 4. Changes in Photosystem II Organization during Dehydration of C. pumilum. EFs faces of thylakoid membranes of plants at different RWC: (A) 100%, (B) ~40%, (C) ~15%, and (D and E) 5 - 10%. During dehydration, the density of PSII in the EFs face gradually decreases from ~1,500 complexes µm-2 (A) to ~700 complexes µm-2 (~15% RWC, C). Notably, at drier conditions (5 - 10% RWC), some PSII complexes organize into rows and arrays (D and E, arrows). The arrowhead (E) marks the PFs face, with LHCII antenna appearing to also be organized in rows, in between which PSII rows would be found in the complementary EFs. For additional information on this data see24. Scale bars: 100 nm. Please click here to view a larger version of this figure.
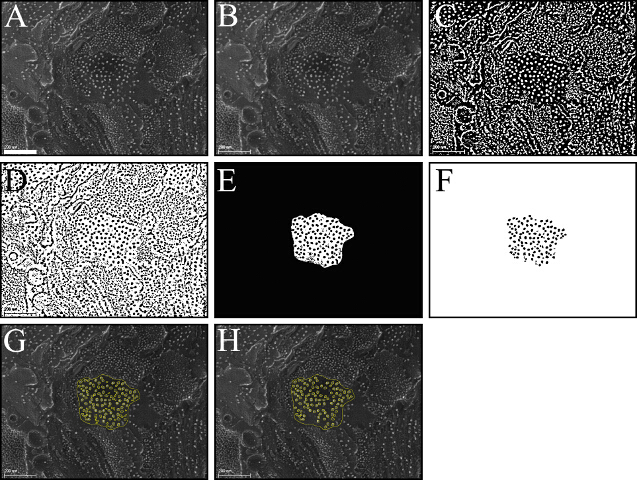 Figure 5. An Example for Segmentation of Photosystem II Particles. Using the Fiji open-source package, protein complexes can be segmented from the images with a few steps (complete detailed procedure found in part 4 of the Protocol section). Images (A, original image) are blurred with the Gaussian filter (B); the image is thresholded using an auto local threshold tool (C); the image is inverted and the watershed algorithm is applied in order to separate objects found in close contact (D); a region of interest is selected and the outside is cleared (E); particles (within a user-defined size range) are selected using the particle analyzer function. The resulting mask is shown in (F). Particles are added to the ROI Manager and overlaid onto the original image (G) to check for errors or for faulty or missed particles. The list of ROIs can be manually edited, replacing, deleting or adding new ROIs, as appropriate. The final user edited selection is shown in (H). Scale bar: 200 nm. Please click here to view a larger version of this figure.
Figure 5. An Example for Segmentation of Photosystem II Particles. Using the Fiji open-source package, protein complexes can be segmented from the images with a few steps (complete detailed procedure found in part 4 of the Protocol section). Images (A, original image) are blurred with the Gaussian filter (B); the image is thresholded using an auto local threshold tool (C); the image is inverted and the watershed algorithm is applied in order to separate objects found in close contact (D); a region of interest is selected and the outside is cleared (E); particles (within a user-defined size range) are selected using the particle analyzer function. The resulting mask is shown in (F). Particles are added to the ROI Manager and overlaid onto the original image (G) to check for errors or for faulty or missed particles. The list of ROIs can be manually edited, replacing, deleting or adding new ROIs, as appropriate. The final user edited selection is shown in (H). Scale bar: 200 nm. Please click here to view a larger version of this figure.
Discussion
The technique described in this paper allows investigation of freeze-fractured membranes within the context of well-preserved high-pressure frozen plant tissues by cryo-scanning electron microscopy. The major advantage of using these procedures is that sample preparation is purely physical; no steps involving chemicals or dehydration are necessary. Thus, it allows studying biological structures at a near-native state26,32. The benefit of using leaf tissues is that one can obtain information on the overall cellular organization, as well as study specific membranes, such as the thylakoid membranes, within their native physiological context, i.e., within the chloroplast. Another advantage, imperative to studying C. pumilum plants at different relative water contents (RWC), is that their hydration state is not altered during preparation. This is as opposed to utilizing isolated chloroplasts or thylakoid membranes as the starting material for a freeze-fracture experiment. In the latter, one also has to take into account that some structural and/or supramolecular alterations likely take place during organelle/membrane isolation.
Working with plant tissue samples, which are relatively thick [>50 - 100 µm, and can be significantly thicker depending on the type of tissue and species], dictates the use of high-pressure freezing (HPF) for sample vitrification. Application of high pressure [2,100 bar], at which the melting temperature of water is about −22 °C, affects the hydrogen bond network of the water such that it slows down the rate of ice crystal formation. Therefore, the chance of obtaining a vitrified sample is high even at relatively slow cooling rates. HPF is currently the only method available for vitrification of thick, yet not exceeding 200 µm, samples. A critical step for HPF of plant tissues is infiltration of their intercellular air spaces with an inert "space filler" prior to freezing, to prevent tissue collapse under the high pressure (see reviews29,33). In the case of C. pumilum leaves, whose thickness can be up to 1 mm, the leaves also have to be trimmed or thinned prior to HPF.
Essentially, the technique of replica preparation18 can also be applied to freeze-fractured leaf tissues. However, in our experience, sufficient yield of intact fractured areas was not obtained, since replicas often fragment to small pieces. Moreover, searching for a specific region of interest such as chloroplasts can be impractical within fragmented replicas of plant tissues. This is mainly because mesophyll (photosynthetic) cells are comprised of a large central vacuole surrounded by a narrow cytoplasmic strip (Figure 2A), which includes the nucleus, chloroplasts and all other organelles. Thus, chloroplasts represent only a very small fraction of the total fractured area of mesophyll tissues. Cryo-SEM imaging of freeze-fractured samples offers a solution to this problem since samples are directly visualized following fracture (and coating/shadowing). However, this method suffers from the limitation that frozen-hydrated samples are beam-sensitive and thus scanning at high magnifications readily damages the sample on the first scan. Double-layer coating (DLC)27,28 offers a solution to this serious drawback.
In DLC, fractured samples are coated in a way similar to how they are for replica preparation. First, a thin layer (1 - 3 nm) of Pt/C is evaporated onto the sample, providing contrast and conductivity. This is followed by a thicker (5 - 10 nm), protective layer of carbon. DLC combined with a high accelerating voltage (10 kV) was used to image samples using the backscattered electron (BSE) signal28. The BSE signal, originating from the platinum layer allows obtaining surface information through the carbon and, possibly, water vapor contaminant layer, which are practically transparent to the BSE at high acceleration voltages (water contamination accumulates in experiments in which a vacuum-cryo transfer unit is not used and the fractured surface is exposed to the atmosphere, even if this is for a fraction of a second). In addition, imaging using the BSE signal minimized the problem of charging effects which are apparent in secondary electron (SE) detection27. With the microscope setup described here, imaging of the SE signal with the In-lens SE detector at 10 kV yielded information equivalent to that obtained when samples were coated solely with 2 - 3 nm of Pt/C and imaged at low accelerating voltages. The difference is that samples prepared by DLC and imaged as described were considerably less beam-sensitive. This enabled us to scan them at high magnifications and in many cases even repeatedly, and allowed obtaining valuable information on photosynthetic protein supramolecular organization and other cellular constituents (Figures 2 - 4)24.
Finally, the procedures described here can be modified to study the structure and/or membrane protein organization in other types of samples. We have successfully employed the technique to study the supramolecular organization of photosynthetic membranes in cyanobacterial and algal cells (Shperberg-Avni et al. unpublished data). The main consideration is finding the balance between having as thin a sample as possible, in order to increase the chance for vitrification, while keeping sample intactness. For different types of samples, which can be suspensions or animal/plant tissues, different packaging should be used (e.g., in terms of the type of platelets utilized for high-pressure freezing). Once successful cryo-immobilization is achieved, the combination of freeze-fracture, DLC and cryo-SEM imaging provides an excellent means for obtaining information on membrane protein organization at high magnification with minimal beam damage.
Disclosures
The authors declare they have no competing financial interests.
Acknowledgments
We thank Andres Kaech (University of Zurich) for his helpful advice on scanning electron microscopy imaging. This work was supported by the United States-Israel Binational Agricultural Research and Development Fund (grant no. US-4334-10, Z.R.), the Israel Science Foundation (grant no. 1034/12, Z.R.), and the Human Frontier Science Program (RGP0005/2013, Z.R.). The electron microscopy studies were conducted at the Irving and Cherna Moskowitz Center for Nano and Bio-Nano Imaging at the Weizmann Institute of Science.
References
- Anderson JM. Insights into the consequences of grana stacking of thylakoid membranes in vascular plants: a personal perspective. Aust. J. Plant Physiol. 1999;26(7):625–639. [Google Scholar]
- Branton D. Fracture Faces of Frozen Membranes. Proc. Natl. Acad. Sci. U. S. A. 1966;55(5):1048–1056. doi: 10.1073/pnas.55.5.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branton D, et al. Freeze-Etching Nomenclature. Science. 1975;190(4209):54–56. doi: 10.1126/science.1166299. [DOI] [PubMed] [Google Scholar]
- Goodenough UW, Staehelin LA. Structural Differentiation of Stacked and Unstacked Chloroplast Membranes - Freeze-Etch Electron Microscopy of Wild-Type and Mutant Strains of Chlamydomonas. J. Cell Biol. 1971;48(3):594–619. doi: 10.1083/jcb.48.3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DJ. Freeze-Fracture Studies on Barley Plastid Membranes .6. Location of the P700-Chlorophyll a-Protein-1. Eur. J. Cell Biol. 1983;31(2):305–314. doi: 10.1007/BF02906493. [DOI] [PubMed] [Google Scholar]
- Staehelin LA. Reversible Particle Movements Associated with Unstacking and Restacking of Chloroplast Membranes. Invitro. J. Cell Biol. 1976;71(1):136–158. doi: 10.1083/jcb.71.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KR. A Chloroplast Membrane Lacking Photosystem-I - Changes in Unstacked Membrane Regions. Biochim. Biophys. Acta. 1980;592(1):143–152. doi: 10.1016/0005-2728(80)90121-8. [DOI] [PubMed] [Google Scholar]
- Miller KR, Cushman RA. Chloroplast Membrane Lacking Photosystem-II - Thylakoid Stacking in the Absence of the Photosystem-II Particle. Biochim. Biophys. Acta. 1979;546(3):481–497. doi: 10.1016/0005-2728(79)90083-5. [DOI] [PubMed] [Google Scholar]
- Miller KR, Staehelin LA. Analysis of Thylakoid Outer Surface - Coupling Factor Is Limited to Unstacked Membrane Regions. J. Cell Biol. 1976;68(1):30–47. doi: 10.1083/jcb.68.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson DJ. Freeze-Fracture Studies on Barley Plastid Membranes .3. Location of the Light-Harvesting Chlorophyll-Protein. Carlsberg Res. Commun. 1979;44(5):305–336. [Google Scholar]
- Olive J, Recouvreur M, Girardbascou J, Wollman FA. Further Identification of the Exoplasmic Face Particles on the Freeze-Fractured Thylakoid Membranes - a Study Using Double and Triple Mutants from Chlamydomonas-Reinhardtii Lacking Various Photosystem-Ii Subunits and the Cytochrome B6/F Complex. Eur. J. Cell Biol. 1992;59(1):176–186. [PubMed] [Google Scholar]
- Olive J, Vallon O, Wollman FA, Recouvreur M, Bennoun P. Studies on the Cytochrome B6/F Complex .2. Localization of the Complex in the Thylakoid Membranes from Spinach and Chlamydomonas-Reinhardtii by Immunocytochemistry and Freeze-Fracture Analysis of B6/F Mutants. Biochim. Biophys. Acta. 1986;851(2):239–248. [Google Scholar]
- Armond PA, Staehelin LA, Arntzen CJ. Spatial Relationship between Light Harvesting Complex and Photosystem-1 and Photosystem-2 in Stacked and Unstacked Chloroplast Membranes. J. Cell Biol. 1976;70(2):400–418. doi: 10.1083/jcb.73.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehelin LA. Chloroplast structure: from chlorophyll granules to supra-molecular architecture of thylakoid membranes. Photosynth. Res. 2003;76(1-3):185–196. doi: 10.1023/A:1024994525586. [DOI] [PubMed] [Google Scholar]
- Nevo R, Charuvi D, Tsabari O, Reich Z. Composition, architecture and dynamics of the photosynthetic apparatus in higher plants. Plant J. 2012;70(1):157–176. doi: 10.1111/j.1365-313X.2011.04876.x. [DOI] [PubMed] [Google Scholar]
- Platt KA, Oliver MJ, Thomson WW. Membranes and Organelles of Dehydrated Selaginella and Tortula Retain Their Normal Configuration and Structural Integrity - Freeze-Fracture Evidence. Protoplasma. 1994;178(1-2):57–65. [Google Scholar]
- Platt-Aloia KA, Thomson WW. Advantages of the use of intact plant tissues in freeze-fracture electron microscopy. J. Electron Microsc. Tech. 1989;13(4):288–299. doi: 10.1002/jemt.1060130404. [DOI] [PubMed] [Google Scholar]
- Carson JL. Fundamental technical elements of freeze-fracture/freeze-etch in biological electron microscopy. J. Vis. Exp. 2014. p. e51694. [DOI] [PMC free article] [PubMed]
- Kirchhoff H, et al. Low-light-induced formation of semicrystalline photosystem II arrays in higher plant chloroplasts. Biochemistry. 2007;46(39):11169–11176. doi: 10.1021/bi700748y. [DOI] [PubMed] [Google Scholar]
- Johnson MP, et al. Photoprotective Energy Dissipation Involves the Reorganization of Photosystem II Light-Harvesting Complexes in the Grana Membranes of Spinach Chloroplasts. Plant Cell. 2011;23(4):1468–1479. doi: 10.1105/tpc.110.081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff H, Tremmel I, Haase W, Kubitscheck U. Supramolecular photosystem II organization in grana thylakoid membranes: evidence for a structured arrangement. Biochemistry. 2004;43(28):9204–9213. doi: 10.1021/bi0494626. [DOI] [PubMed] [Google Scholar]
- Belgio E, Ungerer P, Ruban A. V Light-harvesting superstructures of green plant chloroplasts lacking photosystems. Plant Cell Environ. 2015. [DOI] [PubMed]
- Goral TK, et al. Light-harvesting antenna composition controls the macrostructure and dynamics of thylakoid membranes in Arabidopsis. Plant J. 2012;69(2):289–301. doi: 10.1111/j.1365-313X.2011.04790.x. [DOI] [PubMed] [Google Scholar]
- Charuvi D, et al. Photoprotection Conferred by Changes in Photosynthetic Protein Levels and Organization during Dehydration of a Homoiochlorophyllous Resurrection Plant. Plant Physiol. 2015;167(4):1554–1565. doi: 10.1104/pp.114.255794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant JM, Brandt W, Lindsey GG. An Overview of Mechanisms of Desiccation Tolerance in Selected Angiosperm Resurrection Plants. Plant Stress. 2007;1(1):72–84. [Google Scholar]
- Walther P. High-resolution cryoscanning electron microscopy of biological samples. In: Schatten H, Pawley JB, editors. Biological Low-Voltage Scanning Electron Microscopy. New York: Springer; 2008. pp. 245–261. [Google Scholar]
- Walther P, Müller M. Double-layer coating for field-emission cryo-scanning electron microscopy--present state and applications. Scanning. 1997;19(5):343–348. doi: 10.1002/sca.4950190501. [DOI] [PubMed] [Google Scholar]
- Walther P, Wehrli E, Hermann R, Müller M. Double-layer coating for high-resolution low-temperature scanning electron microscopy. J. Microsc. 1995;179:229–237. doi: 10.1111/j.1365-2818.1995.tb03635.x. (Pt 3) [DOI] [PubMed] [Google Scholar]
- Hess MW. Cryopreparation methodology for plant cell biology. Cell. Electron Microsc. 2007;79:57–100. doi: 10.1016/S0091-679X(06)79003-3. [DOI] [PubMed] [Google Scholar]
- Schertel A, et al. Cryo FIB-SEM: Volume imaging of cellular ultrastructure in native frozen specimens. J. Struct. Biol. 2013;184(2):355–360. doi: 10.1016/j.jsb.2013.09.024. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther P. Recent progress in freeze-fracturing of high-pressure frozen samples. J. Microsc. 2003;212(1):34–43. doi: 10.1046/j.1365-2818.2003.01236.x. [DOI] [PubMed] [Google Scholar]
- Nevo R, et al. Architecture of Thylakoid Membrane Networks. Lipids Photosynth. 2009;30:295–328. [Google Scholar]