

Abstract
Objectives
Research-derived evidence about the impact of sickle cell disease (SCD) on the lives of affected adults is lacking. We conducted formative research to provide the basis for a comprehensive description of how SCD affects the lives of adults, with the goal of developing a SCD-specific quality-of-life measurement system.
Methods
We conducted a comprehensive literature review of patient-reported outcomes, followed by a series of focus groups and structured individual interviews with adults with SCD (n = 122) and their health care providers (n = 15).
Results
We reviewed 473 abstracts and included 86 articles in the final review. The literature revealed broad categories of the impact of SCD and its treatment on the lives of adults—pain; emotional distress; social-role functioning; overall quality-of-life; and quality of care. We classified 1213 incidents from the focus groups and interviews into a taxonomy (16 domains) that met the criterion for saturation and was demonstrated to be reliable for the classification of incidents. The final conceptual model was built upon the taxonomy.
Discussion
Our conceptual model was similar to previous models with the effects of pain predominating, interwoven with emotional distress, quality of care, and stigmatization. We found a broad range of emotions reflected, including positive effects of SCD. Items for the quality-of-life measure were derived from the taxonomy and the conceptual model may be of use in generating hypotheses for clinical research and improving understanding for clinicians of the lived experience of adults with SCD.
Keywords: health-related quality of life, sickle cell disease, quality of care
Sickle cell disease (SCD) is one of the most common genetic disorders in the United States; estimates are that up to 100,000 individuals are affected.1 What was once a disease of childhood is now a chronic health condition that most affected people manage into their fifth decade and beyond. Although the lifespan has increased, adults with SCD face debilitating health problems including multiorgan failure, chronic pain, and neurocognitive deficits.2-4 These individuals can also face social, economic, and academic challenges as well as barriers to accessing quality health care.2,5 Research-derived evidence is lacking about the impact of SCD and related treatments on the lives of affected adults (ie, what these adults are able to do and how they feel)4,6-9 that would be in keeping with a broad definition of health as a combination of physical, mental, and social well-being.10
The increased lifespan and number of adults living with SCD prompted the National Heart, Lung, and Blood Institute (NHLBI) to convene a workshop and working groups of consumers, clinicians, and scientists to examine the unmet needs of adults living with SCD in 2002.5 The workshop participants concluded that the top research priority was the development of a SCD-specific tool to measure health-related quality of life (HRQOL). Consumer workgroups identified the preliminary domains that should be included in this HRQOL tool: pain and its impact, mental health, economic resources, satisfaction with and access to care, education and employment opportunities, burden of treatment, independent living and life skills, discrimination, stigmatization, and coping strategies. The Adult Sickle Cell Quality-of-Life Measurement System (ASCQ-Me) project11 was designed to develop this tool and to conduct the qualitative research to more fully characterize these domains.
To lay the groundwork for the research, we conducted a comprehensive literature review of patient-reported outcomes for adults with SCD. The results of the literature review and initial domains served as the basis for the individual and focus group interview guides. We analyzed the qualitative data from the focus groups with adults, as well as structured individual interviews with both adults and their health care providers, to refine our conceptual model. We then used the results to develop a preliminary conceptual model of the effects of SCD on adults. The objective of the conceptual model is to guide patient-centered assessment and to assist in the design of treatments and future research for adults with SCD.
MATERIALS AND METHODS
Literature Review
Table 1 summarizes the selection and retrieval of articles about the subjective experience of adults with SCD. PubMed and PsychLit databases were searched using keywords chosen because of their relation to patient-centered health domains. The search included publications after 1999 (subsequent to the literature reviews conducted for the NHLBI working group on SCD and quality of life) to June 2010. The selection of keywords was informed by the results of the NHLBI workgroups and reflected reported symptoms (eg, pain, cognitive, psychological), functioning (eg, sexual, work), quality of life, coping, and stigmatization. Ultimately, 473 abstracts were retrieved and read by 2 independent reviewers. Eighty-six articles from this review and from the NHLBI working group review were determined to be relevant. Articles were included if they contained self-reports of the experiences of adults with SCD, whether these reports were obtained by interviews, focus groups, case studies, medical histories, questionnaires, or surveys. Articles containing perceptions of others—particularly health care providers—of the effects of SCD on adults were also included. Articles were excluded if (1) they did not include self-report of the lived/felt experience of adults with SCD and/or their quality of life; (2) they did not include a relevant survey or questionnaire; or (3) the main topic was treatment options. Once an article was read, an abstraction form was completed to summarize essential information on ways in which SCD had been found to affect adults’ lives and symptomatology. We did not attempt to use formal statistical meta-analysis methods to quantitatively pool the data because of the considerable heterogeneity with regard to populations, designs, and outcome measures among the studies.
TABLE 1.
Literature Review Strategies and Result
| Queries | Results |
|---|---|
| Search quality of life OR pain OR coping OR psychological OR social OR cognitive OR sleep OR sex OR sexual OR stigma OR stigmatization OR employment OR work OR focus groups OR qualitative research OR qualitative findings field: all fields | 3,825,407 |
| Search sickle cell AND adult | 12,013 |
| Search #1 AND #2 | 2528 |
| Search #1 AND #2 field: title, limits: all Adult: 19 + years, publication date from 2000 to June 2010, only items with abstracts, English, humans | 473 |
| Total number of manuscripts judged appropriate for inclusion, including those from NHLBI working group | 86 |
NHLBI indicates National Heart, Lung, and Blood Institute.
Results of the Literature Review
Annotations of the articles that were reviewed can be found in the supplemental materials (Supplemental Digital Content 1, http://links.lww.com/CJP/A76). Our analysis of the literature revealed broad categories of the impact of SCD and its treatment on the lives of adults. These categories included pain and its management; emotional distress; functioning in social, family, sexual, and work-related roles; overall quality of life; and quality of care.
Pain and Pain Management
Pain is the hallmark symptom of SCD and the most common reason for seeking treatment.12 Severe pain episodes can occur unpredictably, making coping with them especially difficult and socially disruptive.13 Pain may also undermine the ability to achieve academically and vocationally and may cause negative thoughts about the future.14 Quantitative studies have shown that perceptions of pain severity, its frequency, and the duration of these episodes, can vary across individuals, with younger persons and male individuals reporting greater severity of their symptoms.15-18
There is increasing evidence for the contribution of a number of factors to the pain experience.13 These include social and demographic factors (eg, sex, age, education, socioeconomic status, urban versus rural19-21); psychological factors including depression, anxiety, and coping style1,22-26; and social support.27
Adults with SCD have reported using a variety of skills to manage pain.9,28-30 These methods include: social support31; limiting activity; cognitive strategies, including thinking positively, distraction, relaxation, or guided imagery16,32,33; physical strategies including massage and aquatic rehabilitation28,34-37; denial38; religiosity/spirituality16,17,39-14; and medications.38,43-47 In a cross-cultural study, Nigerians with SCD reported using active coping strategies to deal with pain significantly more often than adults in the United Kingdom did. At the same time, the Nigerians reported a significantly greater inclination toward an external locus of control (belief in forces outside of themselves) with regard to health,25 compared with adults in the United Kingdom. More recently, alcohol use as a possible adjunctive pain-management strategy has been examined.48 However, adults with SCD also report concerns about addiction to pain medications.49,50
Almost half of the manuscripts reviewed (n = 41) addressed the impact of pain or its management on adults with SCD. Study designs ranged from a case report to 2 randomized clinical trials—the majority of the study designs were descriptive and cross-sectional, utilizing convenience sampling from a single site. Measurement strategies varied and were generally appropriate to the research questions, but a few studies were not adequately powered for the number of measures included. Almost half of the studies did not include appropriate comparison groups. However, 9 compared subpopulations of patients, for example, male versus female, high emergency department (ED) utilizers versus less frequent ED utilizers, urban versus rural populations, and 6 studies compared results from the patients with SCD against results from other studies of healthy populations or other chronic illness groups. About one third of the studies adequately controlled for potential confounding variables. Despite methodological issues, results were consistent across qualitative and quantitative studies regarding the challenges that adults with SCD face with pain management, the importance of active pain coping and social support in managing pain, and the relation between psychological distress and poor pain coping.
Of note, 6 papers described results obtained from the Pain in Sickle Cell Epidemiology Study (PiSCES).1,7,21,23,26,48 PiSCES was a cohort study measuring the variability in pain and response to pain in 308 patients with SCD, 16 years of age and older. Data were collected between 2001 and 2006 from patients recruited from multiple clinical settings. Patients completed daily pain diaries for 6 months and standardized assessments. PiSCES was the first large-scale study examining pain in adults with SCD since the lifespan data from the Cooperative Study of Sickle Cell Disease was published in 1991.
PiSCES utilized well-defined outcome variables including mean pain intensity, painful episodes, and various types of utilization episodes—non-opioid analgesic use, opioid use, office visits, ED visits, and hospital admissions. The PiSCES data was tested within a biobehavioral model of pain and its effects, using multivariate statistical methods that evaluated the effect of several classes of explanatory variables (demographic, disease-related, psychosocial, and readiness to utilize care). Although the PiSCES study demonstrated that severe, acute pain is still the hallmark of SCD, results also showed that chronic pain is much more common than previously thought, and the quantity and severity of SCD pain has been vastly underestimated because patients largely manage all types of pain away from the health care setting.
Emotional Distress
The relation between living with SCD and the presence of psychological difficulties has been investigated most often in comparison with either healthy control groups or other chronic illness groups. Some studies have found greater psychiatric morbidity in individuals with SCD compared with the general population,51,52 particularly a higher prevalence of depression and anxiety.22,53-56 Other studies have found no difference in morbidity prevalence.15,30,57 Similarly, in comparison with populations with chronic diseases, such as adults with diabetes, some studies identified higher rates of social disability and psychiatric morbidity among patients with SCD.58 Depressed adults with SCD rated their quality of life on the Short-Form 36 Health Survey (SF-36) as significantly poorer in all areas, as compared with nondepressed adults with SCD.1 In regression models, depression was a stronger predictor of SF-36 scores than demographics, hemoglobin type, and pain measures.1
Untreated sickle cell-related pain has been examined in relation to specific psychological states that are associated with emotional distress—anxiety, stress,15,34,35 lack of control, and depression.59 Lack of understanding of SCD pain and its behavioral effects on the part of health care providers and other individuals in the community (eg, teachers, employers, and neighbors) may lead to stigmatization and stereotyping of individuals with SCD.
In a comparison study of adults with SCD in Jamaica and the United Kingdom, Jamaicans were more likely to see emotional stress as linked to the cause of their illness.17 Feeling a lack of control over symptoms and treatment has been reported.29,60 An association between lack of control and increased pain, poor psychological adjustment, or increased health care utilization has also been documented.35,60 Conversely, higher perceived-control scores positively predicted reported frequency of self-care behaviors.61
Qualitative studies have found that feelings of helplessness and depression can originate from painful episodes and resulting inabilities to locate or maintain a job,38,62 and individuals with frequent hospital admissions for severe vaso-occlusive episodes also suffered from depression.63 Several quantitative studies have found a significant relation between pain episodes and depression64 or negative mood,65 although other studies have not demonstrated this relation.66 Frequent use of opioids has been associated with disruption in daily life, reduced activity levels, and pessimistic mood.67
Qualitative studies have described stigmatization of adults who have SCD and stereotypes that these adults are drug addicts,14,39,68,69 resulting in their feeling shame, hostility, or anger with health care providers.14,39 Perceptions of substance abuse appear to have more to do with problematic pain management versus actual abuse. The prevalence of substance abuse in SCD was found to be only 2%, when controlling for behaviors related to pain.46 Some individuals express dislike of taking pain medication for fear of becoming addicted.47 Reports do indicate that some adults with SCD turn to cocaine,70 marijuana,71 or alcohol to address unrelieved pain.48
Providers’ perceptions of substance abuse among patients with SCD have been studied. In one study, 9% of hematologists and 22% of ED physicians thought that a majority of patients with SCD were addicted to pain medication.72 In another survey, 8% of staff physicians, 17% of residents, and 13% of nurses reported higher estimates of dependence on opioids for adults with SCD (in excess of the documented prevalence) compared with other patients in pain.73 Although 63% of nurses in another survey believed that drug addiction frequently develops during treatment of sickle cell pain episodes, 87% said that addiction should not be a primary nursing concern when caring for patients.74
There was less consistency in the findings from the studies of emotional distress compared with results in relation to pain and its management. This may be in part related to the variability in defining and measuring psychiatric symptoms, making it difficult to make comparisons across studies. There was also a wider range of methodologies, with more qualitative designs utilized, fewer studies including comparison groups, and no clinical trials found. A range of populations were studied, including health care providers. Finally, studies related to emotional distress were as likely as those related to pain and its management to have adequate sample size and controls for potential confounders. The strongest evidence, derived from well- designed studies of emotional distress, was found for a higher prevalence of depression among individuals with SCD compared with the general population, and depression was in turn associated with more severe disease and higher pain rates.
Social, Family, Sexual, and Work Functioning
Adults with SCD face stigma, discrimination, loneliness and isolation, and disruptions in relationships, work, and social activities.13,18,27,53,57,75-77 Pain affects feelings of self-esteem with resulting difficulties in maintaining employment and personal relationships.39 Negative emotional states can affect the ability of adults with SCD to function in social situations. For example, Thompson et al34 reported that adults with poorer psychological adjustment had lower levels of reported support and higher levels of conflict in family functioning. Conversely, social support has been described as important to adherence to treatment and to health promotion behaviors.78
Adults with SCD have described growing up as challenging because of strained relationships with peers due to absences from school, awareness of the potential for a shortened lifespan, and differences in physical appearance.14,39 Adults with SCD have reported feelings of isolation from others and reluctance to disclose SCD status,14,79 especially because the general public knows little about SCD.80 Furthermore, SCD has been found to affect sexual functioning—men with priapism reported dissatisfaction with sexual intercourse and fear of engaging in sexual activity.81 Some were reluctant to disclose priapism; therefore, it went unreported until late in its course.18
Securing and maintaining employment is challenging and has financial and social implications.14,82,83 Employers fail to understand the nature of SCD and its variable course,62 and drowsiness due to pain medication can interfere with driving and working.46 Sickle cell-related pain can interrupt work attendance, creating unstable financial situations that affect self-esteem.14,39 Pain related to SCD can also lead to feelings of helplessness and depression62 that enhance the experience of pain. In one study, men with SCD cited depression due to problems related to employment as a factor in the onset of severe pain episodes.38 People with SCD question whether to disclose SCD status to employers14 and describe employment discrimination and denial of health insurance.59,79
Sickle cell–related pain has a marked effect upon the activities of daily living for adults with SCD. Porter et al84 found that adults in pain were most likely to reduce their household chores, followed by social activities, and infrequently their employment. In this study, stress was the most significant predictor of activity reduction, after controlling for pain intensity, with significant positive correlations with average pain intensity and household and social activity reduction but no relation to medication and health care utilization. Increased stress levels have also been associated with pain severity and absences from work.65
Comparisons with other groups have helped elucidate some of the life activities that are affected by SCD and its symptoms. In one study, adults with SCD identified more difficulties with employment, finances, child care, and leisure-time activities than adults with diabetes reported.58 Individuals with SCD have higher unemployment rates, less education, lower incomes,57 and higher rates of being single29 than healthy comparison groups. Adults with SCD described sleep, eating, and financial difficulties, and the severity of their problems was comparable with adults with other chronic illnesses.82 Individuals with SCD in rural settings may fare more poorly than their urban cohorts with regard to income, educational attainment, limitations in functioning, and access to health care.85
The negative effects of SCD in relation to employment, education, and relationships were consistently described across the 27 studies reviewed in this category, as were the moderating effects of social supports. Methodological issues with the studies included failure to include appropriate control groups or to adequately control for confounding in about two thirds of the studies; inadequate sample sizes in a few studies; and a range of methods and measures utilized.
Quality of Life and Quality of Care
The quality of life of adults with SCD has been described through the use of questionnaires and survey instruments that ask patients to report on their activities, ability to function, health perceptions, and subjective experiences.75,86,87 The use of generic quality-of-life measurements with the SCD population has increased in the past decade, allowing comparison with well and other chronic populations. A recently developed SCD-specific quality-of- life measure found that overall quality of life did not differ between adults with SCD and patients with rheumatoid arthritis.87 Patients with iron overload on iron chelation therapy were found to have lower quality-of-life scores on the SF-36 compared with population norms,88 particularly if they experienced comorbidities.89
In one study using the SF-36, leg ulcers were found to have a significant effect on role functioning, general health, and vitality.76 Patients with SCD who were high users of EDs had more pain, more distress, and lower scores on the SF-36.1 Patients who were followed from the Multicenter Study of Hydroxyurea in Sickle Cell Disease and who responded to hydroxyurea showed improvements in social function and perceptions of health overall90 on the SF-36. Self-care management resources (social support, self-efficacy, assertiveness, and self-care ability) were found to be positively associated with positive health outcomes and quality of life, when controlling for sociodemographic factors.9
Quantitative and qualitative studies report that adults with SCD prefer to manage their pain at home.8,20 Adults who managed their pain at home felt that spending time in the hospital was not in their best interest, and they had a strong sense of self-responsibility for managing their condition.68 Adults with SCD valued positive contact with providers59 and appreciated health care providers who were prompt and efficient.14 Better ratings of provider communication were associated with higher levels of trust toward the medical profession.91 Unfortunately, providers were too often perceived as lacking knowledge about SCD, being insensitive to pain,32,59 failing to involve patients in the decision-making process, and exerting control over treatment regimens.68,92 Patients felt that various needs were neglected, such as assessing pain,92 monitoring of vital signs, psychosocial support,68 and timely intervention.62 Negative reactions of providers have been reported toward patients who ask questions or voice opinions regarding the care provided to them.69 Bobo et al93 found that adults with SCD, compared with adults with asthma, reported more unfavorable ratings of the humaneness of and lower satisfaction with medical care despite comparable access to care. Adults with SCD have characterized their emergency and hospital experiences of pain management as affected negatively by mistrust from providers, stigmatization, and lack of provider knowledge.68,94,95 Such negative interpersonal experiences and lower levels of trust have been associated with hospital self-discharge96 and delays in seeking care.45
Studies with health care providers support these patients’ reports. In a survey of hematologists and ED physicians throughout the United states, 65% of physicians rated their management of pain for people with SCD as only moderately effective.72 Nurses felt that the greatest barrier in management of SCD pain was the lack of adequate pain assessment tools.74 In one study, African American providers, female providers, and providers who served adults were more likely to agree that race issues influence quality of care for individuals with SCD.97 More frequent hospitalizations and prior disputes with staff were associated with providers’ negative attitudes in another study.98 A systematic review found a high level of evidence that negative provider attitudes (and a moderate level of evidence that lack of provider knowledge) pose significant barriers to effective pain management in SCD.95 Thus, individuals with SCD often avoid interacting with the health care system and turn, instead, to other methods for coping with pain. The PiSCES study provided a comprehensive picture of the adult’s experience of pain related to SCD,8,20 including both health care utilization patterns and patient responses to pain.
The majority of studies on quality of life and quality of care were adequately powered and most appropriately controlled for potential confounding variables. Measurement of quality of life for the patients with SCD was consistent across the majority of the studies, using established generic quality-of-life instruments (eg, SF-36). However, the goals of the studies were wide-ranging, from evaluating the validity and reliability of existing quality-of-life measures with SCD populations to evaluating such theoretical models as the theory of self-care management. Given the focus on quality of care, more studies in this group utilized populations of providers, with findings that were consistent with results from patient populations.
Summary of Findings
Many of the life effects uncovered in the literature review relate to the topic of HRQOL because they are typical of domains assessed by HRQOL measures: pain, fatigue, emotional distress, social relationships, and ability to carry out daily activities and social roles (employee, student, parent, etc.). Other classes of life effects covered were access to treatment and interactions with providers, specific side effects or discomfort associated with treatments, and methods of coping with and managing a chronic condition. Finally, many of the effects of SCD uncovered by the literature review are connected to the way individuals feel or perceive themselves in relation to society, including perceptions of stigmatization, feelings of isolation and alienation, and consequent concerns about disclosure of SCD status. We next describe the methods and results of our qualitative data collection that were integrated with the results of the literature review to refine our conceptual model.
MATERIALS AND METHODS
Qualitative Data Collection
We conducted individual and group interviews with adults with SCD and with health care providers across the United States to identify ways that SCD can positively or negatively influence the quality of life of adults with SCD. The interviews and classification of the obtained information into the initial taxonomy followed a rigorous, systematic protocol for qualitative data collection, using the Critical Incident Technique.99-101 The methods and results of the Critical Incident Technique are detailed by Levine and Shore102 and will be touched on briefly here. All materials, methods, and procedures received approval from the Institutional Review Boards of both the American Institutes for Research and Children’s Hospital & Research Center Oakland.
Participant Recruitment
Participants in the data collection were adults with SCD as well as their health care providers. Study personnel invited health care providers listed in the rosters of the American Society of Hematology and the NHLBI National Sickle Cell Disease Program to participate. We used multiple strategies to maximize demographic and geographic diversity, including distributing flyers describing the study to sites providing clinical care for adults with SCD, working through member organizations of the Sickle Cell Disease Association of America (SCDAA), and other SCD community-based organizations, and posting information about the study on the SCDAA website.
Interviews
We used multiple interview methods, including focus groups and individual interviews with adults with SCD and their health care providers. The focus groups were facilitated discussions led by moderators trained for the study (nurses, psychologists, and social workers) who had experience working with individuals with SCD. The groups were designed to elicit participants’ responses in relation to specific questions. The focus groups were homogenous with regard to sex, and moderators were matched to the sex of the group to facilitate candid discussion about sex-related effects (eg, romantic or sexual experiences).
The majority of the focus group protocol involved structured discussion to elicit critical incidents (ie, the effects of SCD on the participant’s lives). After each focus group generated its own critical incidents, participants were asked to “vote” on the importance of topics identified from the literature review and the 2002 working group as being affected by SCD. The topics were listed on a large sheet of paper, and each participant was given 7 sticky dots (votes) to place beside the life effects of SCD that they felt were most important. Participants were allowed to give any topic as many of the 7 votes as they deemed appropriate.
The patient and provider interviews were conducted individually for 2 hours, usually by telephone. The patient interviews were conducted by project staff who were matched to the sex of the adults with SCD and trained in the interview protocol. The participants reported on events that were due to SCD and that affected their lives. Providers reported on events due to SCD that had affected their patients’ lives. All individual and group interviews were audio taped.
Analysis
Project staff trained in qualitative research strategies reviewed the audiotapes to extract from the narratives all individual instances that illustrated behaviors resulting in positive or negative effects on the patient that were related to SCD. These abstracted incidents were “anonymized” to remove any information that could identify specific individuals. A taxonomy of SCD life effects was generated on the basis of a review of these data. The project team was divided into 2 teams of 2 members each, which separately categorized and grouped 200 incidents into domains based on the similarity of content. The coding teams met to compare preliminary domains from the initial grouping to create a draft taxonomy. Discrepancies in the classification of incidents were resolved through discussion to consensus. The teams then individually classified an additional 600 incidents until no further categories could be identified, indicating saturation of the topic. The final taxonomy met the criterion for saturation (the final 100 incidents identified 3 or fewer new behaviors99) and was demonstrated to be reliable for the classification of incidents at the domain level (κ = 0.623, P < 0.0001) and at the category level (κ = 0.606, P < 0.0001).
Results of the Qualitative Data Collection
Participants were 122 adults with SCD and 15 clinicians with expertise in caring for individuals with SCD (Table 2). All of the clinicians and 37 adults were interviewed individually, whereas 85 adults participated in 11 focus group interviews. Women were 54% (n = 67) of the adults with SCD and 60% (n = 9) of the providers. Consistent with the population primarily affected by SCD in the United States, 94% (n = 114) of the adults with SCD were African American/Black. Five percent of the adults (n = 6) self-identified as Hispanic/Latino ethnicity. Over one half of the providers were African American/Black (n = 8), no providers self-identified as of Flispanic/Latino ethnicity and 46% (n = 7) reported they were Caucasian/white and “Other.” Providers were therefore significantly different on race/ethnicity from the adults with SCD (Fisher exact = 0.001). Adults with SCD ranged from 18 to 64 years of age (median, 35 to 44 y). Men and women differed significantly on education, with 59% of women completing at least some college compared with 33% of men, with the median education level for men being high school or GED (Fisher exact = 0.001). Finally, it can be seen in Table 2 that 47% of the health care providers were physicians.
TABLE 2.
Demographic Characteristics of All Participants
| n (%)
|
||||||
|---|---|---|---|---|---|---|
| Adults With Sickle Cell Disease
|
||||||
| Focus Group Participants
|
Individual Interviewees
|
Health Care Providers
|
||||
| Men | Women | Men | Women | Men | Women | |
| Race/ethnicity*† | ||||||
| Hispanic/Latino‡ | 5 (13) | 1 (2) | 0 | 0 | 0 | 0 |
| Black or African American | 35 (90) | 43 (96) | 14 (93) | 22 (100) | 1 (17) | 7 (78) |
| White | 1 (3) | 0 | 0 | 0 | 4 (67) | 0 |
| Mixed or other | 3 (8) | 2 (4) | 1 (7) | 0 | 1 (17) | 2 (22) |
| Age group | ||||||
| 18-24 | 5 (13) | 12 (27) | 2 (15) | 4 (19) | 0 | 0 |
| 25-34 | 16 (41) | 12 (27) | 1 (8) | 6 (29) | 1 (25) | 1 (13) |
| 35-44 | 9 (23) | 10 (22) | 5 (38) | 6 (29) | 2 (50) | 2 (25) |
| 45-54 | 8 (21) | 8 (18) | 4 (31) | 2 (10) | 1 (25) | 3 (38) |
| 55-64 | 1 (3) | 3 (7) | 1 (8) | 3 (14) | 0 | 2 (25) |
| Education level (adults with sickle cell disease)§* | ||||||
| High school or less | 18 (46) | 4 (9) | 8 (62) | 1 (5) | ||
| Some college | 15 (38) | 27 (60) | 2 (15) | 12 (57) | ||
| College graduate | 5 (13) | 11 (2) | 2 (15) | 5 (24) | ||
| Post college | 1 (3) | 3 (7) | 1 (8) | 3 (14) | ||
| Profession (clinicians) | ||||||
| Physician (including hematologist) | 3 (50) | 4 (44) | ||||
| Nurse (RN, PNP) | 0 | 3 (33) | ||||
| Physician assistant | 1 (17) | 0 | ||||
| Social worker | 1 (17) | 2 (22) | ||||
| Other | 1 (17) | 0 | ||||
Fisher exact = 0.001.
Differences on race/ethnicity for men compared with women with sickle cell disease were not significant; differences on race/ethnicity between, adults with sickle cell disease and their clinicians were significant using the Fisher exact test.
Percentages for race/ethnicity sum to > 100% because participants can indicate Hispanic/Latino ethnicity in addition to race.
Differences between men and women with sickle cell disease on education were significant using the Fisher exact test.
PNP indicates pediatric nurse practitioner; RN, registered nurse.
Adults with SCD and their providers were recruited from 22 states across the United States, representing a diversity of urban, suburban, and rural populations as well as medical center and community-based settings. Table 3 depicts the geographic diversity among participants, across all of the qualitative data collection strategies.
TABLE 3.
Geographic Diversity Across All Participants
| Individuals With Sickle Cell Disease, n (%)
|
|||
|---|---|---|---|
| Individual Interviewees | Focus Group Participants | Clinicians, n (%) | |
| Region of the United States | |||
| Northeast | 4 (11) | 29 (34) | 2 (13) |
| Southeast | 18 (49) | 17 (20) | 6 (40) |
| Middle West/Central | 6 (16) | 9 (11) | 3 (20) |
| Southwest/West | 9 (24) | 30 (35) | 4(27) |
| Total | 37 | 85 | 15 |
Men and women also differed significantly in the number of pain episodes experienced during the previous 12 months (Table 4—Fisher exact = 0.001), with all men reporting that they had experienced at least 1 episode of pain, whereas 22% of women reported that they had not experienced any pain during the previous 12 months. Eighty-two percent of men reported that they had ≥ 4 pain episodes in the previous year, compared with 53% of women.
TABLE 4.
Number of Pain Episodes Reported in Previous 12 Months—Adults With SCD
| n (%)
|
||
|---|---|---|
| Men | Women | |
| No. pain episodes* | ||
| None | 0 | 14 (22) |
| 1-3 | 9 (18) | 17 (26) |
| 4-7 | 19 (38) | 14 (22) |
| 8 or more | 22 (44) | 20 (31) |
| Total reporting | 50 | 65 |
Differences between men and women with sickle cell disease on the number of pain episodes reported in the previous 12 months were significant, Fisher exact = 0.001.
Table 5 shows the number of votes cast by the focus group participants regarding the importance of the health and quality-of-life domains identified by the 2002 working groups and the literature review. It can be seen that pain was the first priority, followed by fatigue/tiredness, work, and physical activities. Relationships were voted on relatively infrequently, whereas career, sleep, emotional issues, stress, and interactions with the health care system each received 5% to 6% of the votes. Childbearing and sexuality also received relatively smaller percentages of the votes, despite arising as critical incidents from the focus groups and individual interviews. The pattern of votes cast by men and women were similar across domains, with the exception that women chose issues of tiredness and fatigue significantly more often than men (12% vs. 4%, χ2 [1 df] = 9.98, P = 0.002) and men chose emotional issues (8% vs. 4%, χ2 [l df] = 5.00, P = 0.025) significantly more often than women.
TABLE 5.
Focus Group Members’ Prioritization of Domains
| n (%)
|
|||
|---|---|---|---|
| Domains | Men | Women | Total |
| Pain issues | 25 (12) | 45 (15) | 70 (14) |
| Tiredness/fatigue issues (problems during the daytime)** | 8 (4) | 36 (12) | 44 (9) |
| Work issues (day-to-day employment issues, etc.) | 20 (10) | 19 (6) | 39 (8) |
| Physical activities (walking, carrying things, etc.) | 15 (7) | 22 (7) | 37 (7) |
| Career issues (choosing a profession, | 11 (5) | 19 (6) | 30 (6) |
| pursuing career goals, etc.) | |||
| Sleeping issues (problems during the nighttime) | 12 (6) | 17 (6) | 29 (6) |
| Emotional issues (worry, anger, sadness, etc.)* | 17 (8) | 11 (4) | 28 (5) |
| Interactions with the health care system | 9 (4) | 19 (6) | 28 (5) |
| Stress | 12 (6) | 12 (4) | 24 (5) |
| Other health issues | 11 (5) | 10 (3) | 21 (4) |
| Childbearing issues | 6 (3) | 12 (4) | 18 (4) |
| School/training issues (attending classes, doing homework, etc.) | 6 (3) | 12 (4) | 18 (4) |
| Mental activities (concentrating, remembering, etc.) | 10 (5) | 7 (2) | 17 (3) |
| Recreational activities | 8 (4) | 8 (3) | 16 (3) |
| Romantic relationships | 6 (3) | 9 (3) | 15 (3) |
| Religious issues (church attendance, prayer, spirituality, etc.) | 5 (2) | 10 (3) | 5 (3) |
| Housework issues (chores, etc.) | 5 (2) | 9 (3) | 14 (3) |
| Relationships with other people | 6 (3) | 7 (2) | 13 (3) |
| Sexual issues | 6 (3) | 6 (2) | 12 (2) |
| Relationships with friends | 6 (3) | 5 (2) | 11 (2) |
| Relationships with family (including parenting) | 2 (1) | 6 (2) | 8 (2) |
| Volunteer work issues | 2 (1) | 4 (1) | 6 (1) |
| Total number of votes | 208 | 305 | 513 |
P < 0.05.
P < 0.01.
The individual interviews and the focus groups yielded 1213 incidents. These incidents were used to develop a taxonomy that comprised 16 major domains, which included 94 categories describing different ways through which SCD had positive or negative impacts on the lives of individuals with this condition.
We summarized the literature review and qualitative study results in a conceptual model of the impact of SCD on the lives of adults (Fig. 1). Our model acknowledges the primary manifestation of SCD, that is, pain. In illustrative critical incidents, respondents stated:
When I am in pain, I can’t do anything …. (Female, Focus Group)
I woke up (Mother’s Day) morning with a crisis in my lower back. It was (my mom’s) day and I couldn ’t step up and be a son to her because of my crisis. (Male, Focus Group)
I have an 8 year old son and I can’t do anything with this hip. It’s breaking my heart because I want my son to play basketball and baseball like I did, but I can’t show him anything because of my sickle cell. (Male, Focus Group)
FIGURE 1.
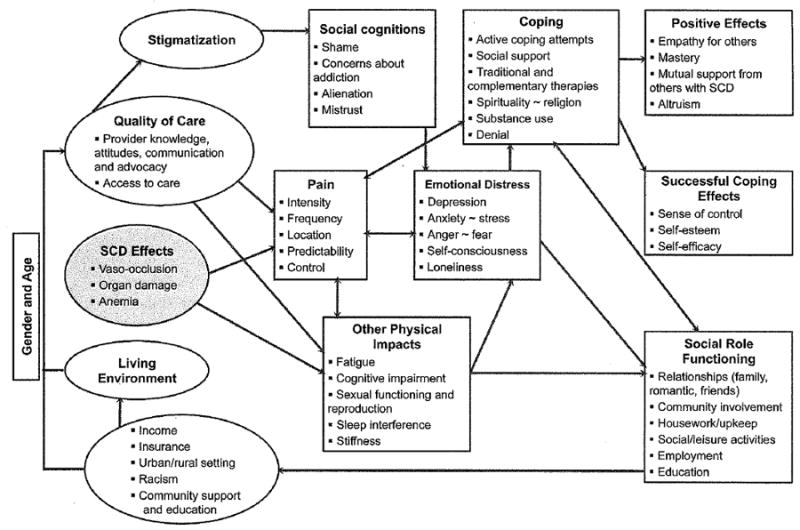
Conceptual model of clinical and life effects associated with sickle cell disease for adults providing basis for Adult Sickle Cell Quality of Life Measurement development. SCD indicates sickle cell disease.
The effects of pain are related to intensity, frequency, location, and unpredictability (and thus lack of control).
I went with a friend, sitting on the beach … watching the sunset … just sitting there having a good time. I suddenly didn’t know what was happening, I thought I was having a heart attack. My whole chest just hurt, hurt real bad. I told (my friend) … I hate to be a spoilsport, but I gotta get out of here, and I left. This is one of those evenings you want to last or 3 days, and I had to cut it short. (Male, Individual Interview)
Physical impacts can result from pain, and/or are directly experienced as a result of the diagnosis of the disease, for example, in the fatigue experienced in relation to anemia, neurocognitive impairment, reproductive issues, and vaso-occlusive episodes experienced in relation to sexual arousal.
When I have sex, and I get excited, I have a crisis. The pain from those crises is worse than from normal crises (so) I’ve learned that I can’t get excited like that. (Female, Focus Group)
Other physical impacts include sleep interference and stiffness.
(This morning) it was rough trying to get up because of the stiffness of the hip replacement and the crisis that I was having in my right arm. I lay for an hour in the bed trying to just move my joints. (Male, Focus Group)
The physical distress accompanying the diagnosis of SCD impacts social and role functioning in all areas: relationships, ability to be involved with family, in the community, social/leisure activities, and household upkeep.
I feel like they use it as an excuse, for why I can’t help them out. They (family) won’t ask me to baby-sit, or help do something around the house. (Female, Individual Interview)
I missed when my baby first crawled. I had to watch it on the camcorder from my hospital bed. That’s the hardest part for me, not being with my kids. (Female, Focus Group)
Employment and education were found to be strongly negatively impacted by SCD in the literature and in the qualitative data.
I’m pursuing a (nursing degree), and I have to miss classes when I’m in the hospital. I have to re-do the work that I missed. And all my friends all move ahead but I’m stuck in the back and have to make up the work on the weekends. (Female, Focus Group)
The influence of SCD on relationships was not always negative:
My dad is always there by my side when I am in the hospital for a pain crisis. He will come right to the hospital after work to be with me. (Female, Individual Interview)
Pain and other physical impacts can operate bidirectionally with the experience of emotional distress, including mental health symptoms. Depression was the most prevalent mental health issue, both from the literature and the data collection:
I get completely pooped, just going to the store. It makes me depressed … (that I don’t have) more energy. (Female, Individual Interview)
Emotionally I don’t feel fine most of the time … half the month I feel depressed or am in pain. I was in school and ended up crying in the bathroom, which isn’t good because it’s a public place. (Female, Individual Interview)
My total hip replacement caused me to become really depressed. All the career goals I had seemed to be over as I can’t work any more. I am disabled before I even turned 40. (Male, Individual Interview)
Coping strategies also influence and are influenced by physical and emotional distress. Successful coping efforts were highlighted in the literature, including active coping attempts and spirituality. Other coping strategies reflected both in the literature and our qualitative data collection included the use of traditional and complementary therapies and more general social support. Substance use and denial were less successful coping strategies described in the literature. Positive effects of SCD that emerged from our qualitative data collection included increased empathy for others and a sense of mastery, including being motivated to achieve and having a sense of accomplishment with those achievements.
People told my mother that she would waste money by sending me to college, because I’d be sick all the time. But I went … was number one in my business class … and I joined a sorority. (Female, Individual Interview)
Mutual support from others with SCD was described both in the literature and the data, whereas altruism was revealed most strongly in the data collection.
I went to a sickle cell convention (and told other people) about my experiences with sickle cell. It made me feel good to know that I was helping somebody. (Female, Individual Interview)
I am a counselor at the sickle cell camp that I went to when I was a kid. One of the camper’s mothers told me the other day that her daughter always speaks highly of me. That makes me feel good, knowing that I can be an inspiration to other people. (Male, Individual Interview)
Quality of care emerged as a critical feature in the experience of adults with SCD in the qualitative data finding, for example, that stigmatization was experienced mostly in the medical treatment context. Medical mal-treatment and neglect was frequently described:
I was in the emergency room and in so much pain. I needed pain medication immediately (but) the nurse refused to access my port. She just kept sticking me and blowing up my vein. (Female, Focus Group)
I was having kidney problems (and) they said they would remove a kidney, so I had surgery. When I came out of surgery, they told me that they didn’t remove a kidney but instead removed my spleen. I was in the hospital for 3 months, with lots of complications. (Twenty years later) I had my first hip surgery, and afterwards found out that I still had a spleen. Those surgeons had done nothing. I went into the archives to see my medical records—and lo and behold, they had done nothing but open me up. (Female, Focus Group)
The local doctor I was seeing wasn’t helping me when I was in a pain crisis—when I would go to the local emergency room in pain, they would call him and if my blood work looked okay to him he would tell them I didn’t need to be there. I was still in lots of pain though despite what my blood work was showing. I would be discharged and would go home in pain and treat myself at home. (Female, Focus Group)
Conversely, advocacy from SCD providers was cited by participants as an important source of support.
I was in so much pain one weekend that I called my doctor at his home. I didn’t want to go to the emergency room. My doctor told me that he would go with me and call ahead with my pain prescription. When he arrived and found that they didn’t have my pain medication ready, he yelled at the staff and demanded that they get it ready immediately. In five minutes, the nurses got my IV ready for me. (Female, Focus Group)
One time the phone company was going to turn off my phone service. I would have the money but not until a few days after the bill was due … My doctor called them and told them that I needed my phone for possible medical emergencies because I have sickle cell. The phone company gave me an extension on paying the phone bill. (Female, Focus Group)
I thought I would never know a doctor who cares about patients with sickle cell … but Dr. X looks for ways to help sickle cell patients get better rather than treating us like drug addicts. (Male, Focus Group)
Recent findings about hospital self-discharge as associated with patient perceptions of the quality of interpersonal experiences with care100 support the importance of the interplay between medical care received (or not) and the experience of pain and emotional distress. Our model also acknowledges the context of the adult with SCD, with some impacts experienced differently depending on sex and age and urban versus rural setting. Racism has been an important contextual factor for adults with SCD in the literature. Our respondents commented on the lack of community support as evidenced by prejudice and lack of knowledge.
There’s very little knowledge of sickle cell (in the community). People just don’t know about it. (Male, Focus Group)
(People) can be really insensitive and ignorant so I try to not tell them about sickle cell or how it affects me. I feel frustrated because (my teammates) don’t understand how sickle cell affects me and sometimes they think I’m being weak when it’s just that I am affected by sickle cell. (Male, Individual Interview)
Finally, inadequate income and/or insurance can affect the living environment for adults with SCD as they are unable to cover the cost of medications of treatments, or access appropriate health services.
About a month ago … I went to the hospital in pain, but I wasn’t bad enough to be admitted. They gave me a prescription for 10 pain pills, but I couldn’t fill the prescription because I had no money. I ended up having to go back to the hospital 3 days in a row. Each time I would go home and try to deal with the pain, and it would get worse, so I had to go back to the hospital. (Male, Individual Interview)
DISCUSSION
We engaged in 3 activities to define and validate a comprehensive conceptual framework for the impacts of SCD on adults: (1) a literature review, (2) focus groups with adults living with SCD, and (3) a critical incident study with individuals living with SCD and providers who care for them. We aligned our approach to the development of ASCQ-Me with the wide range of qualitative and quantitative research methods used in the development of the Patient-Reported Outcomes Measurement Information System (PROMIS), an NIH Common Fund initiative. Our goal was to create in ASCQ-Me a detailed supplement to the “generic core” provided by PROMIS. In parallel to the approach used in the development of PROMIS, the purpose of this formative research and method of sampling was to generate a comprehensive taxonomy of potential impacts of SCD on the health-related quality of life for adult patients.
Previous conceptual models that have been developed and evaluated for SCD include a transactional model of psychological adjustment34,35; the theory of self-care management9; and a model of pain and utilization from the PiSCES project.8 Our model is broader than each of these, incorporating a wider range of the potential impacts of SCD on the lives of adults derived from a comprehensive literature review that includes cross-cultural considerations. Our model was informed with consumer input from its beginning and then refined with qualitative data collection methods. Data collection efforts were wide-reaching, representing a diversity of clinic and community settings; urban, rural and suburban populations; and a range of regions within the United States.
As in previous models, the issue of pain is prominent in our model and interwoven with emotional distress, quality of care, and stigmatization. For some incidents that described limitations in daily activities due to pain, one could surmise that the limiting pain was chronic. However, because coding of critical incidents requires that no assumptions are made beyond the actual statement made by the interviewee, the data that we gathered does not allow us to definitively state what the experience/effects of chronic pain versus acute pain are for SCD.
In contrast with previous reviews and the results of the 2002 working group, we found a broader range of emotions reflected. These included positive effects of SCD including altruism and mutual support experienced among adults with SCD. Results from the qualitative data collection emphasized “missing out” on relationships and activities, medical complications and side effects of treatments, family and social relationships, concerns about disclosure, and discrimination occurring in education and employment. Issues related to faith/ spirituality and housing did not emerge as strongly as anticipated, based on the 2002 working group recommendations. Previously, sleep problems have rarely been noted as interfering with quality of life,89 but sleep interference emerged strongly as an issue through our data collection. This finding is consistent with research with other pain populations that highlights the reciprocal influence between pain and sleep, with sleep impaired by pain103,104 and pain exacerbated by sleep disorders.105 Issues of stigmatization were found primarily in relation to medical care providers, although some issues were found with denial of employment and health insurance.
Our approach to developing ASCQ-Me was very different from the approach used to create the Sickle Cell Impact Measurement Scale (SIMS), the only other sickle cell health- related quality-of-life instrument of which we are aware.87 The SIMS was developed from 4 existing instruments—3 of these instruments are general quality-of-life instruments and the fourth is an arthritis quality-of-life scale. After the SIMS was developed and psychometrically evaluated, it was modified based on a single focus group with 15 people with SCD. The scores of people with SCD were compared with those of a convenience sample of individuals with arthritis. Subsequent psychometric testing was restricted to test-retest reliability measures with these 15 focus group participants.
In contrast to the SIMS, the ASCQ-Me instrument was developed specifically as an instrument to assess the quality of life of people with SCD.11 We derived items for the instrument directly from the formative research that identified the myriad ways SCD affects the lives of adults. We evaluated items using cognitive testing strategies and field tested the instrument. Over 800 individuals with SCD and their health care providers participated in all of these efforts, and psychometric evaluation was restricted to individuals with SCD.
Future Directions for Research
The field testing phase of the ASCQ-Me development will provide not only information on the psychometric properties of the instrument but will allow us to correlate genotype with reported disease severity. Data from the field test will allow us to investigate differential impacts of SCD in relation to such variables as age and sex. Our literature review revealed a number of important gaps that should be addressed in future research. Research in all of the broad categories of the impacts of SCD (pain and its management; emotional distress; social, family, sexual, and work functioning; quality of life and quality of care) would benefit from more attention to the inclusion of appropriate comparison groups, control for potentially confounding variables, and consistency in measurement. More cross-cultural studies of SCD impacts and responses, including positive effects, are required. Emerging issues in need of further study include the use of substances such as alcohol or marijuana to address unrelieved pain and the impacts of chronic versus acute pain on the lived experiences of adults with SCD. Our qualitative data collection highlighted such gaps in the SCD literature as the poorly understood relation between pain, sleep problems, and HRQOL, and the impacts on sexuality for this population. There is a wealth of information from research with other pain disorders that could be drawn upon in developing effective multidimensional treatments for sickle cell–related difficulties with sleep and sexuality, for example, while furthering our understanding of the unique aspects of sickle cell-related pain. There is a critical need to address the pervasive medical maltreatment and neglect of SCD described by our study participants.
CONCLUSIONS
Our formative research provides the basis for a comprehensive model of issues that affect adults with SCD. The model includes well-being and functioning, patient preferences, satisfaction with care, and access to care. Such a model may be of use in generating hypotheses for clinical research with adults with SCD. This model may also provide clinicians with an improved understanding of the lived experience of adults with SCD upon which they can draw to improve their practice. A conclusion of the 2002 workshop on adults with SCD was that there was a need to develop a disease-specific HRQOL measurement system to assess the impact of SCD on the lives and experiences of affected adults. Establishing a conceptual framework of the impact of SCD on patients’ lives was the first major step in constructing the measure. In the context of developing a measure, the conceptual framework can be thought of as the set of domains and subdomains that must be measured to provide a comprehensive assessment of the impact of a disease on a person’s HRQOL. We used a variety of strategies to ensure that important constructs were not overlooked.
Supplementary Material
Acknowledgments
The authors thank Ellen Werner, PhD, Division of Blood Diseases and Resources, National Heart, Lung and Blood Institutes, National Institutes of Health, Bethesda, MD; for her support and contributions to previous versions of this manuscript. The authors also thank Maureen Maurer, MPH, American Institutes for Research, Raleigh-Durham, NC; Corrina Moucheraud, MPH, Tamika Cowans, BA, American Institutes for Research, Washington, DC; and Eileen Murray, BA, Department of Hematology/Oncology, Children’s Hospital & Research Center Oakland, Oakland, CA; for their contributions to the literature review; qualitative data collection and analyses; and manuscript preparation.
Funded by a contract from the National Heart, Lung, and Blood Institute; National Institutes of Health, to the American Institutes for Research, Bethesda, MD, Contract No. HHSN-268-2005-74264C.
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Website, www.elinicalpain.com.
The authors declare no conflict of interest.
References
- 1.Aisilcu IP, Smith WR, McClish DK, et al. Comparisons of high versus low emergency department utilizers in sickle cell disease. Ann Emerg Med. 2009;53:587–593. doi: 10.1016/j.annemergmed.2008.07.050. [DOI] [PubMed] [Google Scholar]
- 2.Brawley OW, Cornelius LJ, Edwards LR, et al. National Institutes of Health Consensus Development Conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148:932–938. doi: 10.7326/0003-4819-148-12-200806170-00220. [DOI] [PubMed] [Google Scholar]
- 3.Vichinsky EP, Neumayr LD, Gold JI, et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. 2010;303:1823–1831. doi: 10.1001/jama.2010.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claster S, Vichinsky EP. Managing sickle cell disease. BMJ. 2003;327:1151–1155. doi: 10.1136/bmj.327.7424.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Heart, Lung and Blood Institute, National Institutes of Health. Workshop on adults with sickle cell disease: meeting unmet needs. 2002 Available at http://www.nhlbi.nih.gov/meetings/scdmtg/execsum.htm.
- 6.Asnani MR, Lipps GE, Reid ME. Utility of WHOQOL- BREF in measuring quality of life in sickle cell disease. Health Qual Life Outcomes. 2009;7:75. doi: 10.1186/1477-7525-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClish DK, Penberthy LT, Bovbjerg VE, et al. Health related quality of life in sickle cell patients: the PiSCES project. Health Qual Life Outcomes. 2005;3:50. doi: 10.1186/1477-7525-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith WR, Bovbjerg VE, Penberthy LT, et al. Understanding pain and improving management of sickle cell disease: the PiSCES study. J Natl Med Assoc. 2005;97:183–193. [PMC free article] [PubMed] [Google Scholar]
- 9.Jenerette CM, Murdaugh C. Testing the theory of self-care management for sickle cell disease. Res Nurs Health. 2008;31:355–369. doi: 10.1002/nur.20261. [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization. Constitution of the World Health Organization—Basic Documents. (45) 2006;(Supplement) [Google Scholar]
- 11.Keller S, Evensen C, Yang M, et al. Adult Sickle Cell Quality of Life Measurement Information System: ASCQ Me User’s Manual. Bethesda, MD: National Heart, Lung and Blood Institute; 2011. [Google Scholar]
- 12.Balias SK. Current issues in sickle cell pain and its management. Hematology Am Soc Hematol Educ Program. 2007:97–105. doi: 10.1182/asheducation-2007.1.97. [DOI] [PubMed] [Google Scholar]
- 13.Mann-Jiles V, Morris DL. Quality of life of adult patients with sickle cell disease. J Am Acad Nurse Pract. 2009;21:340–349. doi: 10.1111/j.1745-7599.2009.00416.x. [DOI] [PubMed] [Google Scholar]
- 14.Thomas VJ, Taylor LM. The psychosocial experience of people with sickle cell disease and its impact on quality of life: qualitative findings from focus groups. Br J Health Psychol. 2005;7(part 3):345–363. doi: 10.1348/135910702760213724. [DOI] [PubMed] [Google Scholar]
- 15.Leavell SR, Ford CV. Psychopathology in patients with sickle cell disease. Psychosomatics. 1983;24:23–25. 28–29. doi: 10.1016/S0033-3182(83)73256-1. passim. [DOI] [PubMed] [Google Scholar]
- 16.Gil KM, Abrams MR, Phillips G, et al. Sickle cell disease pain: relation of coping strategies to adjustment. J Consult Clin Psychol. 1989;57:725–731. doi: 10.1037//0022-006x.57.6.725. [DOI] [PubMed] [Google Scholar]
- 17.Thomas VJ, Hambleton I, Serjeant G. Psychological distress and coping in sickle cell disease: comparison of British and Jamaican attitudes. Ethn Health. 2001;6:129–136. doi: 10.1080/13557850120068450. [DOI] [PubMed] [Google Scholar]
- 18.Addis G, Spector R, Shaw E, et al. The physical, social and psychological impact of priapism on adult males with sickle cell disorder. Chronic Illn. 2007;3:145–154. doi: 10.1177/1742395307081505. [DOI] [PubMed] [Google Scholar]
- 19.Asnani MR, Reid ME, Ali SB, et al. Quality of life in patients with sickle cell disease in Jamaica: rural-urban differences. Rural Remote Health. 2008;8:890. [PubMed] [Google Scholar]
- 20.McClish DK, Smith WR, Dahman BA, et al. Pain site frequency and location in sickle cell disease: the PiSCES project. Pain. 2009;145:246–251. doi: 10.1016/j.pain.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClish DK, Levenson JL, Penberthy LT, et al. Gender differences in pain and healthcare utilization for adult sickle cell patients: the PiSCES Project. J Womens Health. 2006;15:146–154. doi: 10.1089/jwh.2006.15.146. [DOI] [PubMed] [Google Scholar]
- 22.Wilson Schaeffer JJ, Gil KM, Burchinal M, et al. Depression, disease severity, and sickle cell disease. J Behav Med. 1999;22:115–126. doi: 10.1023/a:1018755831101. [DOI] [PubMed] [Google Scholar]
- 23.Citero VA, Levenson JL, McClish DK, et al. The role of catastrophizing in sickle cell disease—the PiSCES project. Pain. 2007;133:39–46. doi: 10.1016/j.pain.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Anie KA, Steptoe A, Bevan DH. Sickle cell disease: pain, coping and quality of life in a study of adults in the UK. Br J Health Psychol. 2002;7:331–344. doi: 10.1348/135910702760213715. [DOI] [PubMed] [Google Scholar]
- 25.Anie KA, Dasgupta T, Ezenduka P, et al. A cross-cultural study of psychosocial aspects of sickle cell disease in the UK and Nigeria. Psychol Health Med. 2007;12:299–304. doi: 10.1080/13548500600984034. [DOI] [PubMed] [Google Scholar]
- 26.Levenson JL, McClish DK, Dahman BA, et al. Depression and anxiety in adults with sickle cell disease: the PiSCES project. Psychosom Med. 2008;70:192–196. doi: 10.1097/PSY.0b013e31815ff5c5. [DOI] [PubMed] [Google Scholar]
- 27.Edwards CL, Scales MT, Loughlin C, et al. A brief review of the pathophysiology, associated pain, and psychosocial issues in sickle cell disease. Int J Behav Med. 2005;12:171–179. doi: 10.1207/s15327558ijbm1203_6. [DOI] [PubMed] [Google Scholar]
- 28.Thomas V. Cognitive behavioural therapy in pain management for sickle cell disease. Int J Palliat Nurs. 2000;6:434–442. doi: 10.12968/ijpn.2000.6.9.9055. [DOI] [PubMed] [Google Scholar]
- 29.Abrams MR, Phillips G, Jr, Whitworth E. Adaptation and coping: a look at a sickle cell patient population over age 30—an integral phase of the life long developmental process. J Health Soc Policy. 1994;5:141–160. doi: 10.1300/J045v05n03_09. [DOI] [PubMed] [Google Scholar]
- 30.Ohaeri JU, Shokunbi WA, Shokunbi KS, et al. The psychosocial problems of sickle cell disease sufferers and their methods of coping. Soc Sci Med. 1995;40:955–960. doi: 10.1016/0277-9536(94)00154-l. [DOI] [PubMed] [Google Scholar]
- 31.Kramer KD, Nash KB. The Sickle Cell Mutual Assistance Movement. In: Nash KB, editor. Psychosocial Aspects of Sickle Cell Disease: Past, Present, and Future Directions of Research. New York: The Haworth Press Inc; 1994. pp. 203–214. [Google Scholar]
- 32.Murray N, May A. Painful crises in sickle cell disease—patients’ perspectives. BMJ. 1988;297:452–454. doi: 10.1136/bmj.297.6646.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gil KM, Carson JW, Sedway JA, et al. Follow-up of coping skills training in adults with sickle cell disease: analysis of daily pain and coping practice diaries. Health Psychol. 2000;19:85–90. doi: 10.1037//0278-6133.19.1.85. [DOI] [PubMed] [Google Scholar]
- 34.Thompson RJ, Jr, Gil KM, Abrams MR, et al. Stress coping, and psychological adjustment of adults with sickle cell disease. J Consult Clin Psychol. 1992;60:433–440. doi: 10.1037//0022-006x.60.3.433. [DOI] [PubMed] [Google Scholar]
- 35.Thompson RJ, Jr, Gil KM, Abrams MR, et al. Psychological adjustment of adults with sickle cell anemia: stability over 20 months, correlates, and predictors. J Clin Psychol. 1996;52:253–261. doi: 10.1002/(SICI)1097-4679(199605)52:3<253::AID-JCLP2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 36.Anie KA, Green J. Psychological therapies for sickle cell disease and pain. Cochrane Database Syst Rev. 2002 doi: 10.1002/14651858.CD001916. CD001916. [DOI] [PubMed] [Google Scholar]
- 37.Tinti G, Somera R, Jr, Valente FM, et al. Benefits of kinesiotherapy and aquatic rehabilitation on sickle cell anemia. A case report. Genet Mol Res. 2010;9:360–364. doi: 10.4238/vol9-1gmr722. [DOI] [PubMed] [Google Scholar]
- 38.Nadel C, Portadin G. Sickle cell crises: psychological factors associated with onset. N Y State J Med. 1977;77:1075–1078. [PubMed] [Google Scholar]
- 39.Strickland OL, Jackson G, Gilead M, et al. Use of focus groups for pain and quality of life assessment in adults with sickle cell disease. J Natl Black Nurses Assoc. 2001;12:36–43. [PubMed] [Google Scholar]
- 40.Harrison MO, Edwards CL, Koenig HG, et al. Religiosity/spirituality and pain in patients with sickle cell disease. J Nerv Ment Dis. 2005;193:250–257. doi: 10.1097/01.nmd.0000158375.73779.50. [DOI] [PubMed] [Google Scholar]
- 41.Cooper-Effa M, Blount W, Kaslow N, et al. Role of spirituality in patients with sickle cell disease. J Am Board Fam Pract. 2001;14:116–122. [PubMed] [Google Scholar]
- 42.McCrae JD, Lumley MA. Health status in sickle cell disease: examining the roles of pain coping strategies, somatic awareness, and negative affectivity. J Behav Med. 1998;21:35–55. doi: 10.1023/a:1018763404868. [DOI] [PubMed] [Google Scholar]
- 43.Gil KM, Abrams MR, Phillips G, et al. Sickle cell disease pain: 2. Predicting health care use and activity level at 9- month follow-up. J Consult Clin Psychol. 1992;60:267–273. doi: 10.1037//0022-006x.60.2.267. [DOI] [PubMed] [Google Scholar]
- 44.Gil KM, Wilson JJ, Edens JL. The stability of pain coping strategies in young children adolescents, and adults with sickle cell disease over an 18-month period. Clin J Pain. 1997;13:110–115. doi: 10.1097/00002508-199706000-00005. [DOI] [PubMed] [Google Scholar]
- 45.Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med. 1998;3:171–179. [Google Scholar]
- 46.Elander J, Lusher J, Bevan D, et al. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med. 2003;57:1683–1696. doi: 10.1016/s0277-9536(02)00553-1. [DOI] [PubMed] [Google Scholar]
- 47.Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Symptom Manage. 2004;27:156–169. doi: 10.1016/j.jpainsymman.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Levenson JL, McClish DK, Dahman BA, et al. Alcohol abuse in sickle cell disease: the Pisces Project. Am J Addict. 2007;16:383–388. doi: 10.1080/10550490701525434. [DOI] [PubMed] [Google Scholar]
- 49.Wright K, Adeosum O. Barriers to effective pain management in sickle cell disease. Br J Nurs. 2009;18:158–161. doi: 10.12968/bjon.2009.18.3.39043. [DOI] [PubMed] [Google Scholar]
- 50.Wilkie DJ, Molokie R, Boyd-Seal D, et al. Patient-reported outcomes: descriptors of nociceptive and neuropathic pain and barriers to effective pain management in adult outpatients with sickle cell disease. J Natl Med Assoc. 2010;102:18–27. doi: 10.1016/s0027-9684(15)30471-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hilton C, Osborn M, Knight S, et al. Psychiatric complications of homozygous sickle cell disease among young adults in the Jamaican Cohort Study. Br J Psychiatry. 1997;170:69–76. doi: 10.1192/bjp.170.1.69. [DOI] [PubMed] [Google Scholar]
- 52.Udofia O, Oseikhuemen AE. Psychiatric morbidity in patients with sickle cell anaemia. West Afr J Med. 1996;15:196–200. [PubMed] [Google Scholar]
- 53.Asnani MR, Fraser R, Lewis NA, et al. Depression and loneliness in Jamaicans with sickle cell disease. BMC Psychiatry. 2010;10:40. doi: 10.1186/1471-244X-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hasan SP, Hashmi S, Alhassen M, et al. Depression in sickle cell disease. J Natl Med Assoc. 2003;95:533–537. [PMC free article] [PubMed] [Google Scholar]
- 55.Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol. 1982;4:328–333. [PubMed] [Google Scholar]
- 56.Belgrave FZ, Molock SD. The role of depression in hospital admissions and emergency treatment of patients with sickle cell disease. J Natl Med Assoc. 1991;83:777–781. [PMC free article] [PubMed] [Google Scholar]
- 57.Laurence BD, George D, Woods D. Association between elevated depressive symptoms and clinical disease severity in African-American adults with sickle cell disease. J Natl Med Assoc. 2006;98:365–369. [PMC free article] [PubMed] [Google Scholar]
- 58.Damlouji NF, Kevess-Cohen R, Charache S, et al. Social disability and psychiatric morbidity in sickle cell anemia and diabetes patients. Psychosomatics. 1982;23:925–931. doi: 10.1016/S0033-3182(82)73062-2. [DOI] [PubMed] [Google Scholar]
- 59.Atkin K, Ahmad WI. Living a ‘normal’ life: young people coping with thalassaemia major or sickle cell disorder. Soc Sei Med. 2001;53:615–626. doi: 10.1016/s0277-9536(00)00364-6. [DOI] [PubMed] [Google Scholar]
- 60.Edwards R, Telfair J, Cecil H, et al. Self-efficacy as a predictor of adult adjustment to sickle cell disease: one-year outcomes. Psychosom Med. 2001;63:850–858. doi: 10.1097/00006842-200109000-00020. [DOI] [PubMed] [Google Scholar]
- 61.Lenoci JM, Telfair J, Cecil H, et al. Self-care in adults with sickle cell disease. West J Nurs Res. 2002;24:228–245. doi: 10.1177/01939450222045879. [DOI] [PubMed] [Google Scholar]
- 62.Collins R. Psychosocial variables and interventions in the patient populations. In: Hurtig A, Viera CT, editors. Sickle Cell Disease: Psychological and Psychosocial Issues. Chicago, IL: University of Illinois Press; 1986. pp. 62–74. [Google Scholar]
- 63.Morin C, Waring EM. Depression and sickle cell anemia. South Med J. 1981;74:766–768. doi: 10.1097/00007611-198106000-00035. [DOI] [PubMed] [Google Scholar]
- 64.Jenerette C, Funk M, Murdaugh C. Sickle cell disease: a stigmatizing condition that may lead to depression. Issues Ment Health Nurs. 2005;26:1081–1101. doi: 10.1080/01612840500280745. [DOI] [PubMed] [Google Scholar]
- 65.Gil KM, Carson JW, Porter LS. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol. 2004;23:267–274. doi: 10.1037/0278-6133.23.3.267. [DOI] [PubMed] [Google Scholar]
- 66.Grant MM, Gil KM, Floyd MY, et al. Depression and functioning in relation to health care use in sickle cell disease. Ann Behav Med. 2000;22:149–157. doi: 10.1007/BF02895779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anie KA, Steptoe A. Pain, mood and opioid medication use in sickle cell disease. Hematol J. 2003;4:71–73. doi: 10.1038/sj.thj.6200227. [DOI] [PubMed] [Google Scholar]
- 68.Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ. 1999;318:1585–1590. doi: 10.1136/bmj.318.7198.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shelley B, Kramer KD, Nash KB. Sickle cell mutual assistance groups and the health services delivery system. J Health Soc Policy. 1994;5:243–259. doi: 10.1300/J045v05n03_14. [DOI] [PubMed] [Google Scholar]
- 70.Alao AO, Westmoreland N, Jindal S. Drug addiction in sickle cell disease: case report. Int J Psychiatry Med. 2003;33:97–101. doi: 10.2190/7XMD-L45D-47DH-7MEC. [DOI] [PubMed] [Google Scholar]
- 71.Howard J, Anie KA, Holdcroft A, et al. Cannabis use in sickle cell disease: a questionnaire study. Br J Haematol. 2005;131:123–128. doi: 10.1111/j.1365-2141.2005.05723.x. [DOI] [PubMed] [Google Scholar]
- 72.Shapiro BS, Benjamin LJ, Payne R, et al. Sickle cell-related pain: perceptions of medical practitioners. J Pain Symptom Manage. 1997;14:168–174. doi: 10.1016/S0885-3924(97)00019-5. [DOI] [PubMed] [Google Scholar]
- 73.Waldrop RD, Mandry C. Health professional perceptions of opioid dependence among patients with pain. Am J Emerg Med. 1995;13:529–531. doi: 10.1016/0735-6757(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 74.Pack-Mabien A, Labbe E, Herbert D, et al. Nurses’ attitudes and practices in sickle cell pain management. Appl Nurs Res. 2001;14:187–192. doi: 10.1053/apnr.2001.26783. [DOI] [PubMed] [Google Scholar]
- 75.Asnani MR, Lipips GE, Reid ME. Validation of the SF-36® in Jamaicans with sickle-cell disease. Psychol Health Med. 2009;14:606–618. doi: 10.1080/13548500903016567. [DOI] [PubMed] [Google Scholar]
- 76.Halabi-Tawil M, Lionnet F, Girot R, et al. Sickle cell leg ulcers: a frequently disabling complication and a marker of severity. Br J Dermatol. 2008;158:339–344. doi: 10.1111/j.1365-2133.2007.08323.x. [DOI] [PubMed] [Google Scholar]
- 77.Anderson LP, Rehm LP. The relationship between strategies of coping and perception of pain in three chronic pain groups. J Clin Psychol. 1984;40:1170–1177. doi: 10.1002/1097-4679(198409)40:5<1170::aid-jclp2270400508>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 78.Belgrave FZ, Lewis DM. The role of social support in compliance and other health behaviors for African Americans with chronic illnesses. J Health Soc Policy. 1994;5:55–68. doi: 10.1300/J045v05n03_05. [DOI] [PubMed] [Google Scholar]
- 79.Kass NE, Hull SC, Natowicz MR, et al. Medical privacy and the disclosure of personal medical information: the beliefs and experiences of those with genetic and other clinical conditions. Am J Med Genet A. 2004;128A:261–270. doi: 10.1002/ajmg.a.30057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boyd JH, Watkins AR, Price CL, et al. Inadequate community knowledge about sickle cell disease among African-American women. J Natl Med Assoc. 2005;97:62–67. [PMC free article] [PubMed] [Google Scholar]
- 81.Adeyoju AB, Olujohungbe AB, Morris J, et al. Priapism in sickle-cell disease; incidence, risk factors and complications—an international multicentre study. BJU Int. 2002;90:898–902. doi: 10.1046/j.1464-410x.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 82.Barrett DH, Wisotzek IE, Abel GG, et al. Assessment of psychosocial functioning of patients with sickle cell disease. South Med J. 1988;81:745–750. doi: 10.1097/00007611-198806000-00015. [DOI] [PubMed] [Google Scholar]
- 83.Ohaeri JU, Shokunbi WA. Psychosocial burden of sickle cell disease on caregivers in a Nigerian setting. J Natl Med Assoc. 2002;94:1058–1070. [PMC free article] [PubMed] [Google Scholar]
- 84.Porter LS, Gil KM, Sedway JA, et al. Pain and stress in sickle cell disease: an analysis of daily pain records. Int J Behav Med. 1998;5:185–203. doi: 10.1207/s15327558ijbm0503_1. [DOI] [PubMed] [Google Scholar]
- 85.Telfair J, Haque A, Etienne M, et al. Rural/urban differences in access to and utilization of services among people in Alabama with sickle cell disease. Public Health Rep. 2003;118:27–36. doi: 10.1093/phr/118.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Asnani M, Lipps G, Reid M. Component structure of the SF-36® in Jamaicans with sickle cell disease. West Indian Med J. 2007;56:491–497. [PubMed] [Google Scholar]
- 87.Adams-Graves P, Lamar K, Johnson C, et al. Development and validation of SIMS: an instrument for measuring quality of life of adults with sickle cell disease. Am J Hematol. 2008;83:558–562. doi: 10.1002/ajh.21146. [DOI] [PubMed] [Google Scholar]
- 88.Payne KA, Rofail D, Baladi JF, et al. Iron chelation therapy: clinical effectiveness, economic burden and quality of life in patients with iron overload. Adv Ther. 2008;25:725–742. doi: 10.1007/s12325-008-0085-z. [DOI] [PubMed] [Google Scholar]
- 89.Thuret I, Hacini M, Pegourie-Bandelier B, et al. Socio-psychological impact of infused iron chelation therapy with deferoxamine in metropolitan France: ISOSFER study results. Hematology. 2009;14:315–322. doi: 10.1179/102453309X12473408860424. [DOI] [PubMed] [Google Scholar]
- 90.Balias SK, Barton FB, Waclawiw MA, et al. Hydroxyurea and sickle cell anemia: effect on quality of life. Health Qual Life Outcomes. 2006;4:59. doi: 10.1186/1477-7525-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haywood C, Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med. 2010;25:543–548. doi: 10.1007/s11606-009-1247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alleyne J, Thomas VJ. The management of sickle cell crisis pain as experienced by patients and their carers. J Adv Nurs. 1994;19:725–732. doi: 10.1111/j.1365-2648.1994.tb01144.x. [DOI] [PubMed] [Google Scholar]
- 93.Bobo L, Miller ST, Smith WR, et al. Health perceptions and medical care opinions of inner-city adults with sickle cell disease or asthma compared with those of their siblings. South MedJ. 1989;82:9–12. doi: 10.1097/00007611-198901000-00004. [DOI] [PubMed] [Google Scholar]
- 94.Booker M, Blethyn K, Wright C, et al. Pain management in sickle cell disease. Chronic Illn. 2006;2:39–50. doi: 10.1177/17423953060020011101. [DOI] [PubMed] [Google Scholar]
- 95.Haywood C, Jr, Beach MC, Lanzkron S, et al. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. J Natl Med Assoc. 2009;101:1022–1033. doi: 10.1016/s0027-9684(15)31069-5. [DOI] [PubMed] [Google Scholar]
- 96.Haywood C, Jr, Lanzlcron S, Ratanawongsa N, et al. Hospital self-discharge among adults with sickle-cell disease (SCD): associations with trust and interpersonal experiences with care. J Hosp Med. 2010;5:289–294. doi: 10.1002/jhm.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Telfair J, Myers J, Drezner S. Does race influence the provision of care to persons with sickle cell disease? Perceptions of multidisciplinary providers. J Health Care Poor Underserved. 1998;9:184–195. doi: 10.1353/hpu.2010.0127. [DOI] [PubMed] [Google Scholar]
- 98.Ratanawongsa N, Haywood C, Jr, Bediako S, et al. Health care provider attitudes toward patients with acute vaso- occlusive crisis due to sickle cell disease: development of a scale. Patient Educ Couns. 2009;76:272–278. doi: 10.1016/j.pec.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Flanagan JC. The critical incident technique. Psychol Bull. 1954;51:327–358. doi: 10.1037/h0061470. [DOI] [PubMed] [Google Scholar]
- 100.Butterfield LD, Borgen WA, Amundsen NE, et al. Fifty years of the critical incident technique 1954-2004 and beyond. Quant Res. 2005;5:475–497. [Google Scholar]
- 101.Fitzgerald K, Seale NS, Kerins CA, et al. The critical incident technique: a useful tool for conducting qualitative research. J Dent Educ. 2008;72:299–304. [PubMed] [Google Scholar]
- 102.Levine R, Shore K. Use of the Critical Incident Technique to Develop Survey Items Measuring Patient Experiences of Ambulatory Care; Baltimore, MD. Proceedings of CAHPS (R) Ninth National User Group Meeting; 2006. [Google Scholar]
- 103.Tang NK, Goodchild CE, Hester J, et al. Pain-related insomnia versus primary insomnia: a comparison study of sleep pattern, psychological characteristics, and cognitive- behavioral processes. Clin J Pain. 2012;28:428–436. doi: 10.1097/AJP.0b013e31823711bc. [DOI] [PubMed] [Google Scholar]
- 104.Menefee LA, Frank ED, Doghramji K, et al. Self-reported sleep quality and quality of life for individuals with chronic pain conditions. Clin J Pain. 2000;16:290–297. doi: 10.1097/00002508-200012000-00003. [DOI] [PubMed] [Google Scholar]
- 105.Chiu YH, Silman AJ, Macfarlane GJ, et al. Poor sleep and depression are independently associated with a reduced pain threshold. Results of a population based study. Pain. 2005;115:316–321. doi: 10.1016/j.pain.2005.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.